
Suppressing Hepatic UGT1A1 Increases Plasma Bilirubin, Lowers Plasma Urobilin, Reorganizes Kinase Signaling Pathways and Lipid Species and Improves Fatty Liver Disease

 ,
,  , , ,
, , ,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Synthesis of Oligonucleotides
2.3. Liver Triglyceride Measurement
2.4. Plasma Measurements
2.5. Liver Histology
2.6. Lipidomics
2.7. Lipidomics Data Analysis
2.8. PamGene PamStation Sample Preparations
2.9. PamGene PamStation Kinome Data Analysis
2.10. Quantitative Real-Time PCR Analysis
2.11. Western Blotting
2.12. Statistics
3. Results
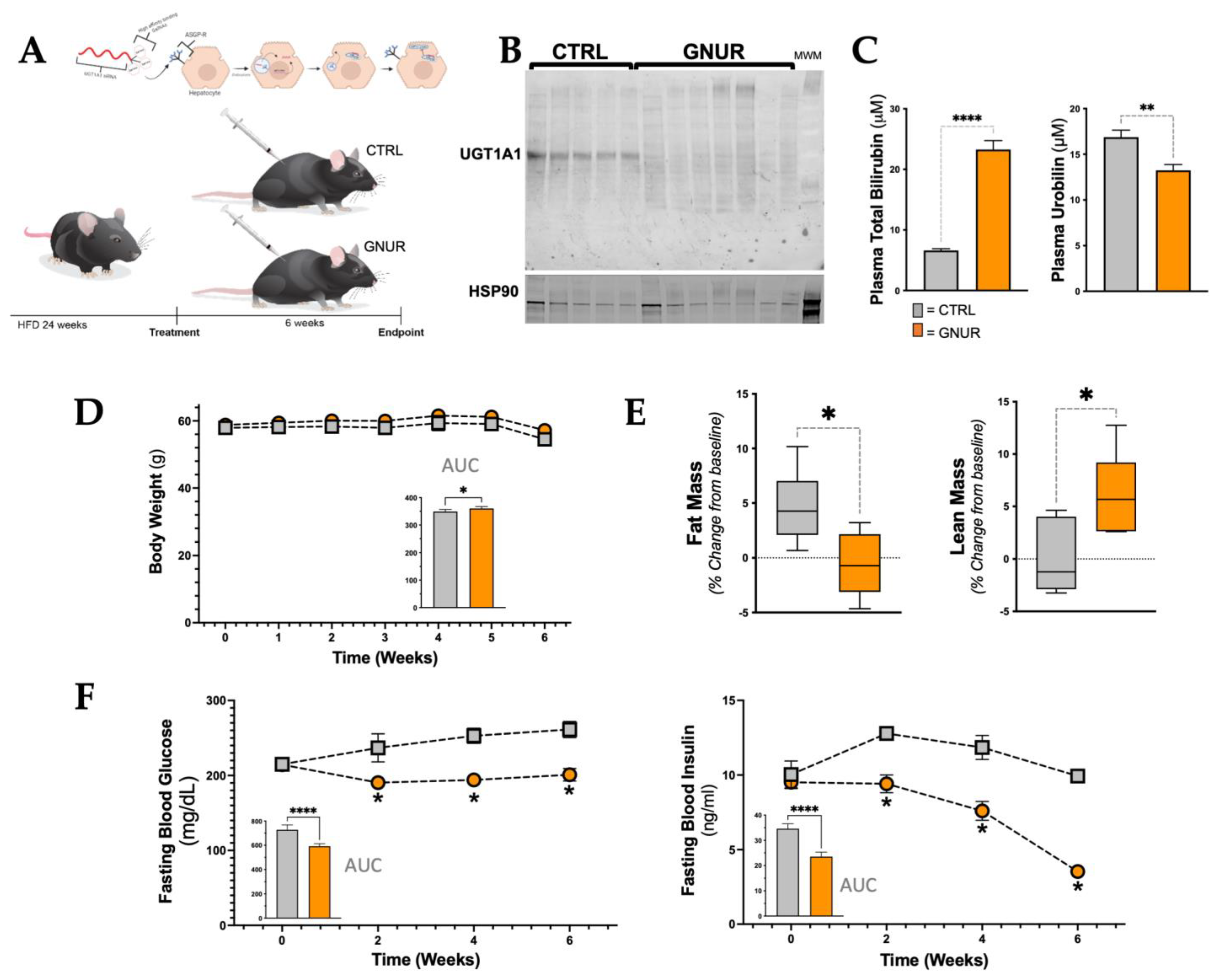
3.1. Suppression of Hepatic UGT1A1 Increases Bilirubin Levels and Lean Mass and Lowers Blood Glucose and Insulin Levels
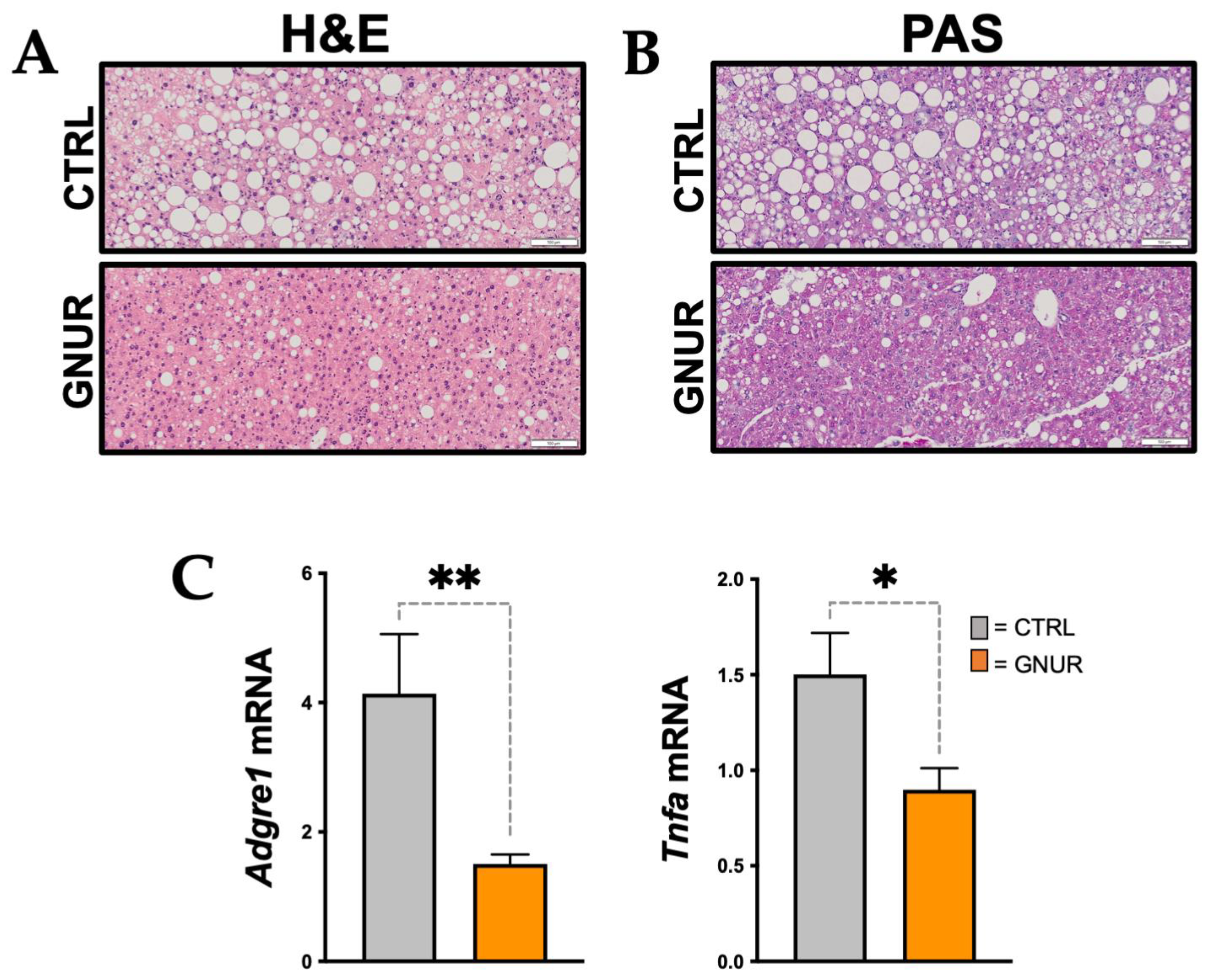
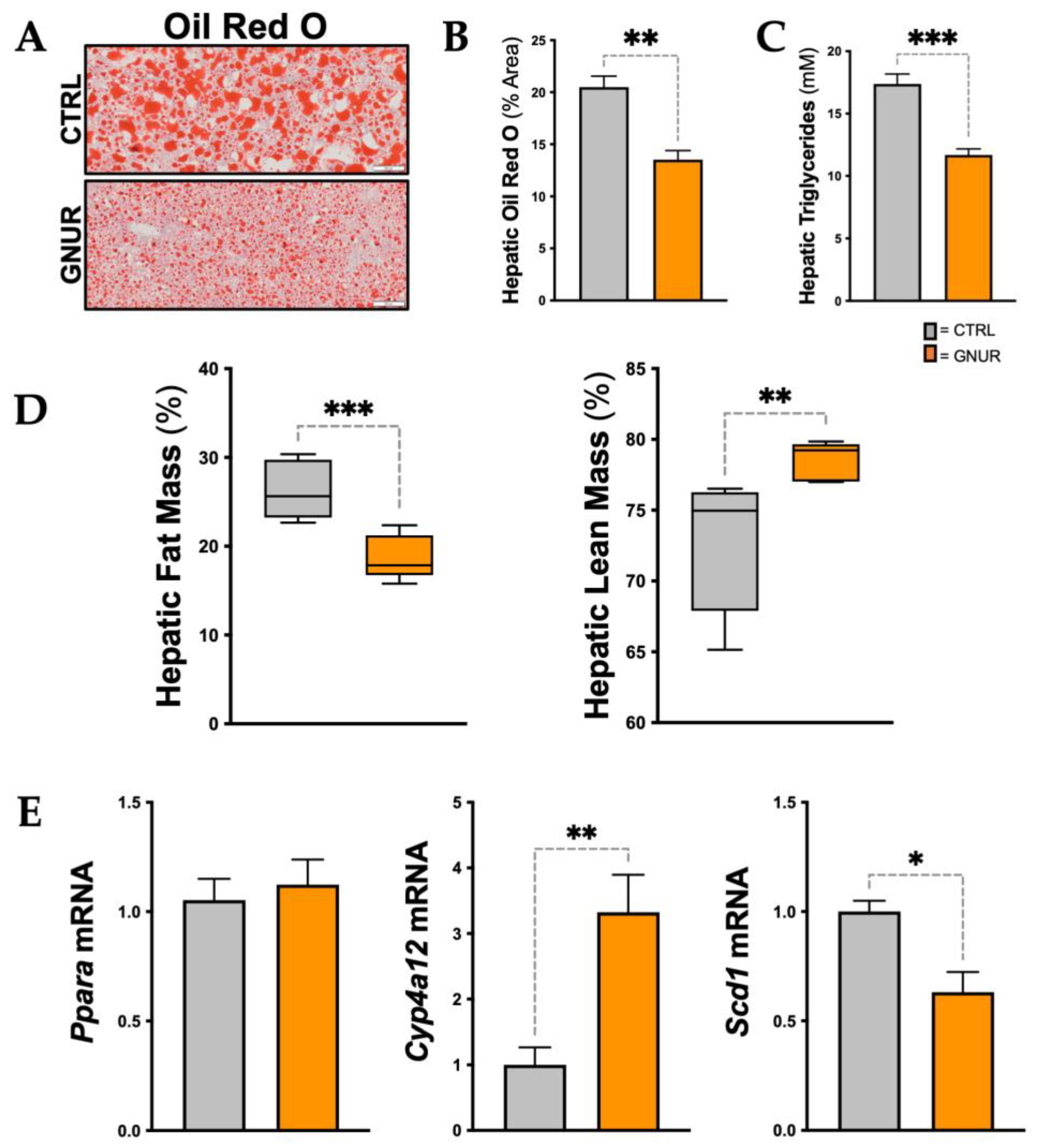
3.2. Suppression of UGT1A1 Decreases Hepatic Steatosis, Inflammation, and Triglyceride Levels
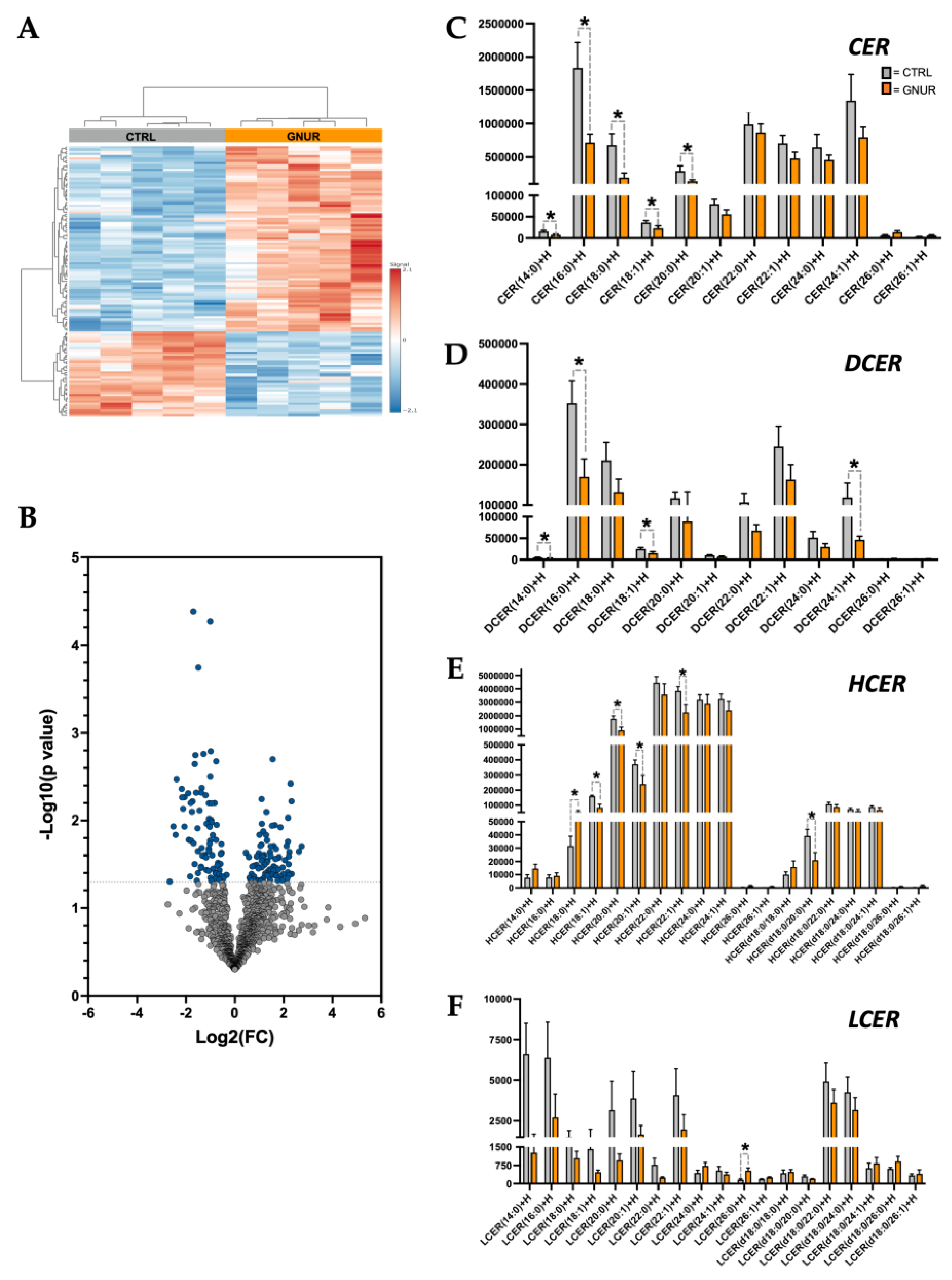
3.3. Lipidomics Reveals Decreased Ceramide Lipid Accumulation in the Liver of GNUR-Treated Mice
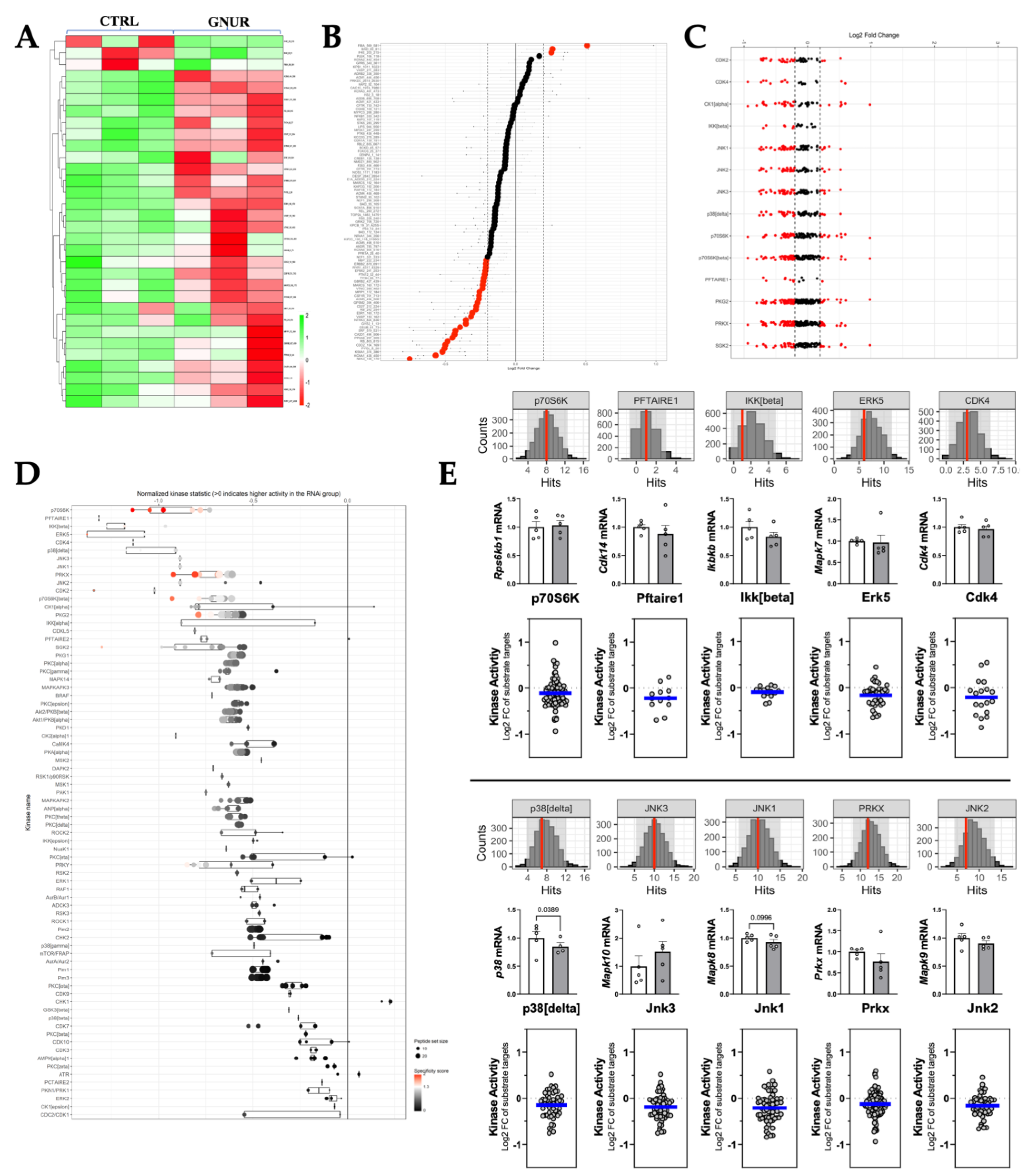
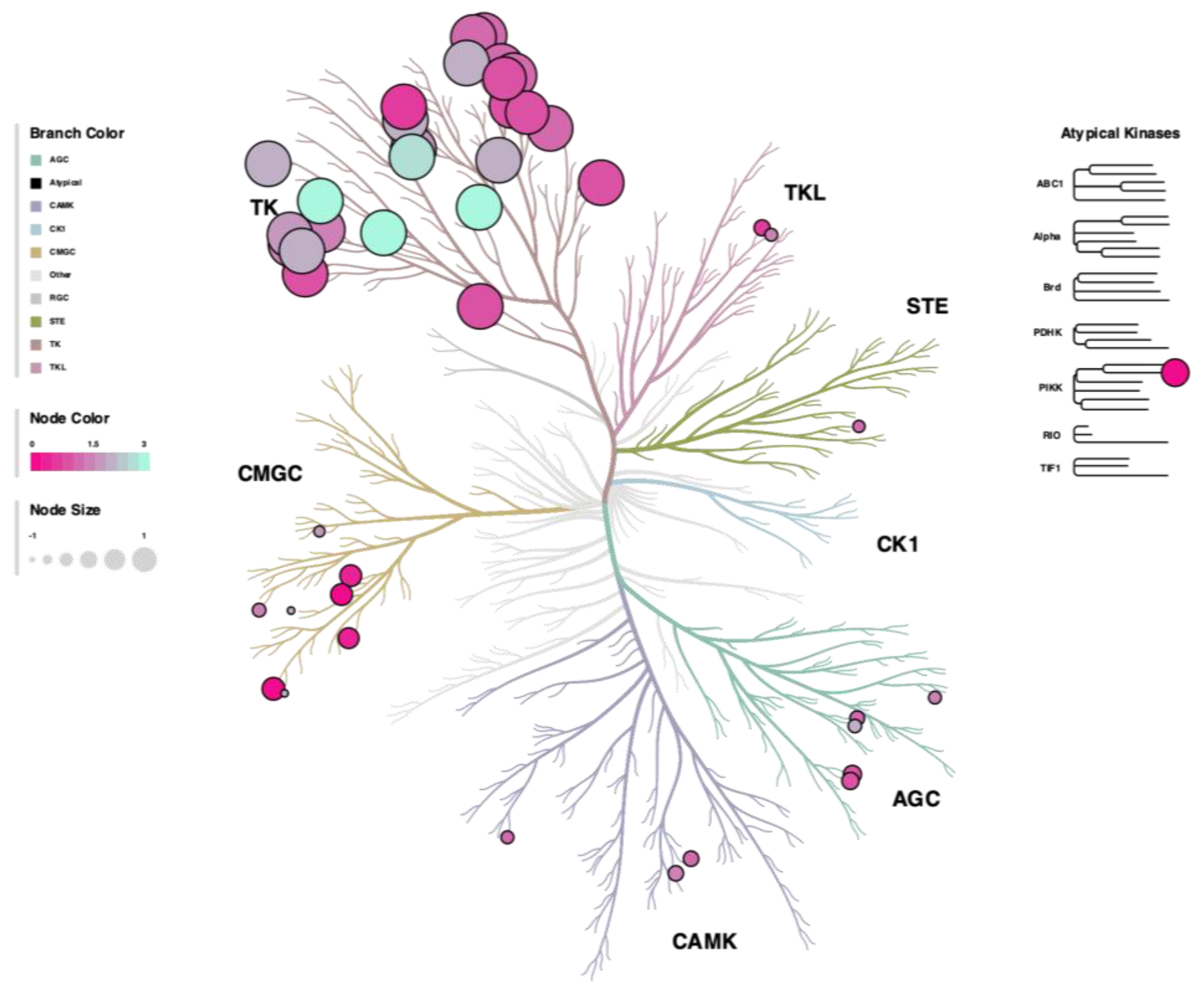
3.4. Kinome Signaling Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Badmus, O.O.; Hillhouse, S.A.; Anderson, C.D.; Hinds, T.D.; Stec, D.E. Molecular mechanisms of metabolic associated fatty liver disease (MAFLD): Functional analysis of lipid metabolism pathways. Clin. Sci. 2022, 136, 1347–1366. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Muthiah, M.D.; Sanyal, A.J. Burden of Disease due to Nonalcoholic Fatty Liver Disease. Gastroenterol. Clin. N. Am. 2020, 49, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Xanthakos, S.A.; Lavine, J.E.; Yates, K.P.; Schwimmer, J.B.; Molleston, J.P.; Rosenthal, P.; Murray, K.F.; Vos, M.B.; Jain, A.K.; Scheimann, A.O.; et al. Progression of Fatty Liver Disease in Children Receiving Standard of Care Lifestyle Advice. Gastroenterology 2020, 159, 1731–1751. [Google Scholar] [CrossRef]
- Baratta, F.; Pastori, D.; Angelico, F.; Balla, A.; Paganini, A.M.; Cocomello, N.; Ferro, D.; Violi, F.; Sanyal, A.J.; Del Ben, M. Nonalcoholic Fatty Liver Disease and Fibrosis Associated With Increased Risk of Cardiovascular Events in a Prospective Study. Clin. Gastroenterol. Hepatol. 2020, 18, 2324–2331. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Weaver, L.; Hamoud, A.R.; Stec, D.E.; Hinds, T.D., Jr. Biliverdin reductase and bilirubin in hepatic disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G668–G676. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Schwertner, H.A.; Vitek, L. Gilbert syndrome, UGT1A1*28 allele, and cardiovascular disease risk: Possible protective effects and therapeutic applications of bilirubin. Atherosclerosis 2008, 198, 1–11. [Google Scholar] [CrossRef]
- Benton, M.C.; Lea, R.A.; Macartney-Coxson, D.; Bellis, C.; Carless, M.A.; Curran, J.E.; Hanna, M.; Eccles, D.; Chambers, G.K.; Blangero, J.; et al. Serum bilirubin concentration is modified by UGT1A1 haplotypes and influences risk of type-2 diabetes in the Norfolk Island genetic isolate. BMC Genet. 2015, 16, 136. [Google Scholar] [CrossRef] [PubMed]
- Underwood, M.D.; Kassir, S.A.; Bakalian, M.J.; Galfalvy, H.; Dwork, A.J.; Mann, J.J.; Arango, V. Serotonin receptors and suicide, major depression, alcohol use disorder and reported early life adversity. Transl. Psychiatry 2018, 8, 279. [Google Scholar] [CrossRef] [PubMed]
- Sundararaghavan, V.L.; Binepal, S.; Stec, D.E.; Sindhwani, P.; Hinds, T.D., Jr. Bilirubin, a new therapeutic for kidney transplant? Transplant. Rev. 2018, 32, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Stec, D.E. Bilirubin, a Cardiometabolic Signaling Molecule. Hypertension 2018, 72, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Hamoud, A.R.; Weaver, L.; Stec, D.E.; Hinds, T.D., Jr. Bilirubin in the Liver-Gut Signaling Axis. Trends Endocrinol. Metab. 2018, 29, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Kipp, Z.A.; Xu, M.; Bates, E.A.; Lee, W.-H.; Kern, P.A.; Hinds, T.D. Bilirubin Levels Are Negatively Correlated with Adiposity in Obese Men and Women, and Its Catabolized Product, Urobilin, Is Positively Associated with Insulin Resistance. Antioxidants 2023, 12, 170. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.; Pfitzner, B.; Neschen, S.; Kahle, M.; Harir, M.; Lucio, M.; Moritz, F.; Tziotis, D.; Witting, M.; Rothballer, M.; et al. Distinct signatures of host-microbial meta-metabolome and gut microbiome in two C57BL/6 strains under high-fat diet. ISME J. 2014, 8, 2380–2396. [Google Scholar] [CrossRef]
- Yao, J.K.; Dougherty, G.G.; Reddy, R.D.; Matson, W.R.; Kaddurah-Daouk, R.; Keshavan, M.S. Associations between purine metabolites and monoamine neurotransmitters in first-episode psychosis. Front. Cell Neurosci. 2013, 7, 90. [Google Scholar] [CrossRef]
- Kwak, M.S.; Kim, D.; Chung, G.E.; Kang, S.J.; Park, M.J.; Kim, Y.J.; Yoon, J.H.; Lee, H.S. Serum bilirubin levels are inversely associated with nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2012, 18, 383–390. [Google Scholar] [CrossRef]
- Liang, X.; Zhang, L.; Natarajan, S.K.; Becker, D.F. Proline mechanisms of stress survival. Antioxid. Redox Signal. 2013, 19, 998–1011. [Google Scholar] [CrossRef]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Vogel, M.E.; Zucker, S.D. Bilirubin acts as an endogenous regulator of inflammation by disrupting adhesion molecule-mediated leukocyte migration. Inflamm. Cell Signal. 2016, 3, e1178. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Huang, H.; Yun, X.; Kim, D.S.; Yue, Y.; Wu, H.; Sutter, A.; Chavin, K.D.; Otterbein, L.E.; Adams, D.B.; et al. Bilirubin increases insulin sensitivity in leptin-receptor deficient and diet-induced obese mice through suppression of ER stress and chronic inflammation. Endocrinology 2014, 155, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.T.; DelCimmuto, N.R.; Flack, K.D.; Stec, D.E.; Hinds, T.D., Jr. Reactive Oxygen Species (ROS) and Antioxidants as Immunomodulators in Exercise: Implications for Heme Oxygenase and Bilirubin. Antioxidants 2022, 11, 179. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.M.; Hong, S.H.; Kipp, Z.A.; Hinds, T.D., Jr. Identification of Binding Regions of Bilirubin in the Ligand-Binding Pocket of the Peroxisome Proliferator-Activated Receptor-A (PPARalpha). Molecules 2021, 26, 2975. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.M.; Neifer, K.L.; Hamoud, A.A.; Hawk, C.F.; Nestor-Kalinoski, A.L.; Miruzzi, S.A.; Morran, M.P.; Adeosun, S.O.; Sarver, J.G.; Erhardt, P.W.; et al. Bilirubin remodels murine white adipose tissue by reshaping mitochondrial activity and the coregulator profile of peroxisome proliferator-activated receptor alpha. J. Biol. Chem. 2020, 295, 9804–9822. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.M.; Blomquist, T.M.; Miruzzi, S.A.; McCullumsmith, R.; Stec, D.E.; Hinds, T.D., Jr. RNA sequencing in human HepG2 hepatocytes reveals PPAR-alpha mediates transcriptome responsiveness of bilirubin. Physiol. Genom. 2019, 51, 234–240. [Google Scholar] [CrossRef]
- Stec, D.E.; John, K.; Trabbic, C.J.; Luniwal, A.; Hankins, M.W.; Baum, J.; Hinds, T.D., Jr. Bilirubin Binding to PPARalpha Inhibits Lipid Accumulation. PLoS ONE 2016, 11, e0153427. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Adeosun, S.O.; Alamodi, A.A.; Stec, D.E. Does bilirubin prevent hepatic steatosis through activation of the PPARalpha nuclear receptor? Med. Hypotheses 2016, 95, 54–57. [Google Scholar] [CrossRef]
- Creeden, J.F.; Gordon, D.M.; Stec, D.E.; Hinds, T.D., Jr. Bilirubin as a metabolic hormone: The physiological relevance of low levels. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E191–E207. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Hosick, P.A.; Hankins, M.W.; Nestor-Kalinoski, A.; Stec, D.E. Mice with hyperbilirubinemia due to Gilbert’s Syndrome polymorphism are resistant to hepatic steatosis by decreased serine 73 phosphorylation of PPARalpha. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E244–E252. [Google Scholar] [CrossRef]
- Seyed Khoei, N.; Grindel, A.; Wallner, M.; Molzer, C.; Doberer, D.; Marculescu, R.; Bulmer, A.; Wagner, K.H. Mild hyperbilirubinaemia as an endogenous mitigator of overweight and obesity: Implications for improved metabolic health. Atherosclerosis 2018, 269, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Molzer, C.; Wallner, M.; Kern, C.; Tosevska, A.; Schwarz, U.; Zadnikar, R.; Doberer, D.; Marculescu, R.; Wagner, K.H. Features of an altered AMPK metabolic pathway in Gilbert’s Syndrome, and its role in metabolic health. Sci. Rep. 2016, 6, 30051. [Google Scholar] [CrossRef] [PubMed]
- Wallner, M.; Marculescu, R.; Doberer, D.; Wolzt, M.; Wagner, O.; Vitek, L.; Bulmer, A.C.; Wagner, K.H. Protection from age-related increase in lipid biomarkers and inflammation contributes to cardiovascular protection in Gilbert’s syndrome. Clin. Sci. 2013, 125, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Bulmer, A.C.; Blanchfield, J.T.; Toth, I.; Fassett, R.G.; Coombes, J.S. Improved resistance to serum oxidation in Gilbert’s syndrome: A mechanism for cardiovascular protection. Atherosclerosis 2008, 199, 390–396. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Burns, K.A.; Hosick, P.A.; McBeth, L.; Nestor-Kalinoski, A.; Drummond, H.A.; AlAmodi, A.A.; Hankins, M.W.; Vanden Heuvel, J.P.; Stec, D.E. Biliverdin reductase A attenuates hepatic steatosis by inhibition of glycogen synthase kinase (GSK) 3beta phosphorylation of serine 73 of peroxisome proliferator-activated receptor (PPAR) alpha. J. Biol. Chem. 2016, 291, 25179–25191. [Google Scholar] [CrossRef] [PubMed]
- Stec, D.E.; Gordon, D.M.; Hipp, J.A.; Hong, S.; Mitchell, Z.L.; Franco, N.R.; Robison, J.W.; Anderson, C.D.; Stec, D.F.; Hinds, T.D., Jr. The loss of hepatic PPARalpha promotes inflammation and serum hyperlipidemia in diet-induced obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R733–R745. [Google Scholar] [CrossRef]
- Nair, J.K.; Willoughby, J.L.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef]
- Rajeev, K.G.; Nair, J.K.; Jayaraman, M.; Charisse, K.; Taneja, N.; O’Shea, J.; Willoughby, J.L.; Yucius, K.; Nguyen, T.; Shulga-Morskaya, S.; et al. Hepatocyte-specific delivery of siRNAs conjugated to novel non-nucleosidic trivalent N-acetylgalactosamine elicits robust gene silencing in vivo. Chembiochem 2015, 16, 903–908. [Google Scholar] [CrossRef]
- Culliton, K.; Louati, H.; Laneuville, O.; Ramsay, T.; Trudel, G.J.n.M. Six degrees head-down tilt bed rest caused low-grade hemolysis: A prospective randomized clinical trial. Npj Microgravity 2021, 7, 1–9. [Google Scholar] [CrossRef]
- Kotal, P.; Fevery, J. (absorbance508) Quantitation of urobilinogen in feces, urine, bile and serum by direct spectrophotometry of zinc complex. Clin. Chim. Acta 1991, 202, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Trudel, G.; Uhthoff, H.K.; Laneuville, O.J.P.R. Hemolysis during and after 21 days of head-down-tilt bed rest. Physiol. Rep. 2017, 5, e13469. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Creeden, J.F.; Gordon, D.M.; Stec, D.F.; Donald, M.C.; Stec, D.E. Bilirubin Nanoparticles Reduce Diet-Induced Hepatic Steatosis, Improve Fat Utilization, and Increase Plasma beta-Hydroxybutyrate. Front. Pharmacol. 2020, 11, 594574. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Creeden, J.F.; Gordon, D.M.; Spegele, A.C.; Britton, S.L.; Koch, L.G.; Stec, D.E. Rats Genetically Selected for High Aerobic Exercise Capacity Have Elevated Plasma Bilirubin by Upregulation of Hepatic Biliverdin Reductase-A (BVRA) and Suppression of UGT1A1. Antioxidants 2020, 9, 889. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Sodhi, K.; Meadows, C.; Fedorova, L.; Puri, N.; Kim, D.H.; Peterson, S.J.; Shapiro, J.; Abraham, N.G.; Kappas, A. Increased HO-1 levels ameliorate fatty liver development through a reduction of heme and recruitment of FGF21. Obesity 2014, 22, 705–712. [Google Scholar] [CrossRef]
- Mueller, P.A.; Yang, L.; Ubele, M.; Mao, G.; Brandon, J.; Vandra, J.; Nichols, T.C.; Escalante-Alcalde, D.; Morris, A.J.; Smyth, S.S. Coronary artery disease risk-associated Plpp3 gene and its product lipid phosphate phosphatase 3 regulate experimental atherosclerosis. ATVB 2019, 39, 2261–2272. [Google Scholar] [CrossRef]
- Kraemer, M.P.; Mao, G.; Hammill, C.; Yan, B.; Li, Y.; Onono, F.; Smyth, S.S.; Morris, A.J. Effects of diet and hyperlipidemia on levels and distribution of circulating lysophosphatidic acid. J. Lipid Res. 2019, 60, 1818–1828. [Google Scholar] [CrossRef]
- Khan, M.J.; Codreanu, S.G.; Goyal, S.; Wages, P.A.; Gorti, S.K.; Pearson, M.J.; Uribe, I.; Sherrod, S.D.; McLean, J.A.; Porter, N.A. Evaluating a targeted multiple reaction monitoring approach to global untargeted lipidomic analyses of human plasma. RCM 2020, 34, e8911. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.J.; Shen, P.C.; Liu, H.C.; Cho, Y.C.; Hsu, M.K.; Lin, I.C.; Chen, F.H.; Yang, J.C.; Ma, W.L.; Cheng, W.C. LipidSig: A web-based tool for lipidomic data analysis. Nucleic Acids Res. 2021, 49, W336–W345. [Google Scholar] [CrossRef]
- Creeden, J.F.; Kipp, Z.A.; Xu, M.; Flight, R.M.; Moseley, H.N.B.; Martinez, G.J.; Lee, W.H.; Alganem, K.; Imami, A.S.; McMullen, M.R.; et al. Hepatic Kinome Atlas: An In-Depth Identification of Kinase Pathways in Liver Fibrosis of Humans and Rodents. Hepatology 2022, 76, 1376–1388. [Google Scholar] [CrossRef]
- McGuire, J.L.; Depasquale, E.A.; Funk, A.J.; O’Donnovan, S.M.; Hasselfeld, K.; Marwaha, S.; Hammond, J.H.; Hartounian, V.; Meador-Woodruff, J.H.; Meller, J.; et al. Abnormalities of signal transduction networks in chronic schizophrenia. NPJ Schizophr. 2017, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Appuhamy, J.A.; Nayananjalie, W.A.; England, E.M.; Gerrard, D.E.; Akers, R.M.; Hanigan, M.D. Effects of AMP-activated protein kinase (AMPK) signaling and essential amino acids on mammalian target of rapamycin (mTOR) signaling and protein synthesis rates in mammary cells. J. Dairy Sci. 2014, 97, 419–429. [Google Scholar] [CrossRef]
- Dorsett, C.R.; McGuire, J.L.; Niedzielko, T.L.; DePasquale, E.A.; Meller, J.; Floyd, C.L.; McCullumsmith, R.E. Traumatic Brain Injury Induces Alterations in Cortical Glutamate Uptake without a Reduction in Glutamate Transporter-1 Protein Expression. J. Neurotrauma 2017, 34, 220–234. [Google Scholar] [CrossRef] [PubMed]
- DePasquale, E.A.K.; Alganem, K.; Bentea, E.; Nawreen, N.; McGuire, J.L.; Naji, F.; Hilhorst, R.; Meller, J.; McCullumsmith, R.E. KRSA: Network-based Prediction of Differential Kinase Activity from Kinome Array Data. bioRxiv 2020. [Google Scholar] [CrossRef]
- Chirumamilla, C.S.; Fazil, M.; Perez-Novo, C.; Rangarajan, S.; de Wijn, R.; Ramireddy, P.; Verma, N.K.; Vanden Berghe, W. Profiling Activity of Cellular Kinases in Migrating T-Cells. Methods Mol. Biol. 2019, 1930, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Creeden, J.F.; Alganem, K.; Imami, A.S.; Brunicardi, F.C.; Liu, S.H.; Shukla, R.; Tomar, T.; Naji, F.; McCullumsmith, R.E. Kinome Array Profiling of Patient-Derived Pancreatic Ductal Adenocarcinoma Identifies Differentially Active Protein Tyrosine Kinases. Int. J. Mol. Sci. 2020, 21, 8679. [Google Scholar] [CrossRef]
- Metz, K.S.; Deoudes, E.M.; Berginski, M.E.; Jimenez-Ruiz, I.; Aksoy, B.A.; Hammerbacher, J.; Gomez, S.M.; Phanstiel, D.H. Coral: Clear and Customizable Visualization of Human Kinome Data. Cell Syst. 2018, 7, 347–350.e341. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Kipp, Z.A.; Xu, M.; Yiannikouris, F.B.; Morris, A.J.; Stec, D.F.; Wahli, W.; Stec, D.E. Adipose-Specific PPARalpha Knockout Mice Have Increased Lipogenesis by PASK-SREBP1 Signaling and a Polarity Shift to Inflammatory Macrophages in White Adipose Tissue. Cells 2021, 11, 4. [Google Scholar] [CrossRef]
- Kumar, R.; Rastogi, A.; Maras, J.S.; Sarin, S.K. Unconjugated hyperbilirubinemia in patients with non-alcoholic fatty liver disease: A favorable endogenous response. Clin. Biochem. 2012, 45, 272–274. [Google Scholar] [CrossRef]
- Tian, J.; Zhong, R.; Liu, C.; Tang, Y.; Gong, J.; Chang, J.; Lou, J.; Ke, J.; Li, J.; Zhang, Y.; et al. Association between bilirubin and risk of Non-Alcoholic Fatty Liver Disease based on a prospective cohort study. Sci. Rep. 2016, 6, 31006. [Google Scholar] [CrossRef]
- Hjelkrem, M.; Morales, A.; Williams, C.D.; Harrison, S.A. Unconjugated hyperbilirubinemia is inversely associated with non-alcoholic steatohepatitis (NASH). Aliment. Pharmacol. Ther. 2012, 35, 1416–1423. [Google Scholar] [CrossRef]
- Stec, D.E.; Hinds, T.D., Jr. Natural Product Heme Oxygenase Inducers as Treatment for Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 9493. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Stec, D.E.; Tiribelli, C. Powering the powerhouse: Heme oxygenase-1 regulates mitochondrial function in non-alcoholic fatty liver disease (NAFLD). Acta. Physiol. 2023, e13931. [Google Scholar] [CrossRef]
- Steventon, G. Uridine diphosphate glucuronosyltransferase 1A1. Xenobiotica 2020, 50, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Dong, H.S.; Zhang, Y.; Cao, M.J.; Song, L.L.; Pan, Q.J.; Bulmer, A.; Adams, D.B.; Dong, X.; Wang, H.J. Bilirubin Increases Insulin Sensitivity by Regulating Cholesterol Metabolism, Adipokines and PPAR gamma Levels. Sci. Rep. 2015, 28, 9886. [Google Scholar] [CrossRef] [PubMed]
- Stec, D.E.; Gordon, D.M.; Nestor-Kalinoski, A.L.; Donald, M.C.; Mitchell, Z.L.; Creeden, J.F.; Hinds, T.D., Jr. Biliverdin Reductase A (BVRA) Knockout in Adipocytes Induces Hypertrophy and Reduces Mitochondria in White Fat of Obese Mice. Biomolecules 2020, 10, 387. [Google Scholar] [CrossRef]
- Gordon, D.M.; Adeosun, S.O.; Ngwudike, S.I.; Anderson, C.D.; Hall, J.E.; Hinds, T.D., Jr.; Stec, D.E. CRISPR Cas9-mediated deletion of biliverdin reductase A (BVRA) in mouse liver cells induces oxidative stress and lipid accumulation. Arch. Biochem. Biophys. 2019, 672, 108072. [Google Scholar] [CrossRef]
- Adeosun, S.O.; Moore, K.H.; Lang, D.M.; Nwaneri, A.C.; Hinds, T.D., Jr.; Stec, D.E. A Novel Fluorescence-Based Assay for the Measurement of Biliverdin Reductase Activity. React. Oxyg. Species 2018, 5, 35–45. [Google Scholar] [CrossRef]
- Adeosun, S.O.; Gordon, D.M.; Weeks, M.F.; Moore, K.H.; Hall, J.E.; Hinds, T.D.; Stec, D.E. Loss of biliverdin reductase-A (BVRA) promotes lipid accumulation and lipotoxicity in mouse proximal tubule cells. Am. J. Physiol. Renal. Physiol. 2018, 315, F323–F331. [Google Scholar] [CrossRef]
- Chen, W.; Tumanov, S.; Fazakerley, D.J.; Cantley, J.; James, D.E.; Dunn, L.L.; Shaik, T.; Suarna, C.; Stocker, R. Bilirubin deficiency renders mice susceptible to hepatic steatosis in the absence of insulin resistance. Redox Biol. 2021, 47, 102152. [Google Scholar] [CrossRef]
- Cimini, F.A.; Barchetta, I.; Zuliani, I.; Pagnotta, S.; Bertoccini, L.; Dule, S.; Zampieri, M.; Reale, A.; Baroni, M.G.; Cavallo, M.G.; et al. Biliverdin reductase-A protein levels are reduced in type 2 diabetes and are associated with poor glycometabolic control. Life Sci. 2021, 284, 119913. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, V.; Barchetta, I.; Cimini, F.A.; Bertoccini, L.; Chiappetta, C.; Capoccia, D.; Carletti, R.; Di Cristofano, C.; Silecchia, G.; Fontana, M.; et al. Reduced Biliverdin Reductase-A Expression in Visceral Adipose Tissue is Associated with Adipocyte Dysfunction and NAFLD in Human Obesity. Int. J. Mol. Sci. 2020, 21, 9091. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Tramutola, A.; Lanzillotta, C.; Arena, A.; Blarzino, C.; Cassano, T.; Butterfield, D.A.; Di Domenico, F.; Perluigi, M.; Barone, E. Loss of biliverdin reductase-A favors Tau hyper-phosphorylation in Alzheimer’s disease. Neurobiol. Dis. 2019, 125, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Cimini, F.A.; Arena, A.; Barchetta, I.; Tramutola, A.; Ceccarelli, V.; Lanzillotta, C.; Fontana, M.; Bertoccini, L.; Leonetti, F.; Capoccia, D.; et al. Reduced biliverdin reductase-A levels are associated with early alterations of insulin signaling in obesity. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1490–1501. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Tramutola, A.; Triani, F.; Calcagnini, S.; Di Domenico, F.; Ripoli, C.; Gaetani, S.; Grassi, C.; Butterfield, D.A.; Cassano, T.; et al. Biliverdin Reductase-A Mediates the Beneficial Effects of Intranasal Insulin in Alzheimer Disease. Mol. Neurobiol. 2019, 56, 2922–2943. [Google Scholar] [CrossRef]
- Triani, F.; Tramutola, A.; Di Domenico, F.; Sharma, N.; Butterfield, D.A.; Head, E.; Perluigi, M.; Barone, E. Biliverdin reductase-A impairment links brain insulin resistance with increased Abeta production in an animal model of aging: Implications for Alzheimer disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3181–3194. [Google Scholar] [CrossRef] [PubMed]
- Kyle, D.; Flack, L.V.; Fry, C.; Stec, D.E.; Hinds, T.D., Jr. Cutting Edge Concepts: Does Bilirubin Enhance Exercise Performance? Front. Sports Act. Living 2023, 4, 1040687. [Google Scholar] [CrossRef]
- Yang, M.; Ni, C.L.; Chang, B.C.; Jiang, Z.H.; Zhu, Y.J.; Tang, Y.Z.; Li, Z.; Li, C.G.; Li, B. Association between serum total bilirubin levels and the risk of type 2 diabetes mellitus. J. Diabetes Res. 2019, 152, 23–28. [Google Scholar] [CrossRef]
- Wu, Y.; Li, M.; Xu, M.; Bi, Y.; Li, X.; Chen, Y.; Ning, G.; Wang, W. Low serum total bilirubin concentrations are associated with increased prevalence of metabolic syndrome in Chinese. J. Diabetes 2011, 3, 217–224. [Google Scholar] [CrossRef]
- Mianowska, B.; Kaminska, A.; Fendler, W.; Szadkowska, A.; Mlynarski, W. Bilirubin is an independent factor inversely associated with glycated hemoglobin level in pediatric patients with type 1 diabetes. Pediatr. Diabetes 2014, 15, 389–393. [Google Scholar] [CrossRef]
- Cheriyath, P.; Gorrepati, V.S.; Peters, I.; Nookala, V.; Murphy, M.E.; Srouji, N.; Fischman, D. High Total Bilirubin as a Protective Factor for Diabetes Mellitus: An Analysis of NHANES Data From 1. J. Clin. Med. Res. 2010, 2, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Fang, X.; Shi, D.; Yao, S.; Wu, W.; Fang, Q.; Yao, H. MSP-RON Pathway: Potential Regulator of Inflammation and Innate Immunity. Front. Immunol. 2020, 11, 569082. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.S.; Lindberg, R.A.; et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 2009, 58, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, J.; Jiang, H.; Ma, Y.; Pan, S.; Reddy, S.; Sun, X. Bilirubin protects grafts against nonspecific inflammation-induced injury in syngeneic intraportal islet transplantation. Exp. Mol. Med. 2010, 42, 739–748. [Google Scholar] [CrossRef]
- Fu, Y.Y.; Kang, K.J.; Ahn, J.M.; Kim, H.R.; Na, K.Y.; Chae, D.W.; Kim, S.; Chin, H.J. Hyperbilirubinemia reduces the streptozotocin-induced pancreatic damage through attenuating the oxidative stress in the Gunn rat. Tohoku J. Exp. Med. 2010, 222, 265–273. [Google Scholar] [CrossRef]
- O’Brien, L.; Hosick, P.A.; John, K.; Stec, D.E.; Hinds, T.D., Jr. Biliverdin reductase isozymes in metabolism. Trends Endocrinol. Metab. 2015, 26, 212–220. [Google Scholar] [CrossRef]
- Sundararaghavan, V.L.; Sindhwani, P.; Hinds, T.D., Jr. Glucuronidation and UGT isozymes in bladder: New targets for the treatment of uroepithelial carcinomas? Oncotarget 2016, 8, 3640–3648. [Google Scholar] [CrossRef]
- Roche, S.P.; Kobos, R. Jaundice in the adult patient. Am. Fam. Physician 2004, 69, 299–304. [Google Scholar]
- Vera, T.; Granger, J.P.; Stec, D.E. Inhibition of bilirubin metabolism induces moderate hyperbilirubinemia and attenuates ANG II-dependent hypertension in mice. Am. J. Physiol Regul. Integr. Comp. Physiol. 2009, 297, R738–R743. [Google Scholar] [CrossRef]
- Vera, T.; Stec, D.E. Moderate hyperbilirubinemia improves renal hemodynamics in ANG II-dependent hypertension. Am. J. Physiol Regul. Integr. Comp. Physiol 2010, 299, R1044–R1049. [Google Scholar] [CrossRef]
- Meixiong, J.; Vasavda, C.; Green, D.; Zheng, Q.; Qi, L.; Kwatra, S.G.; Hamilton, J.P.; Snyder, S.H.; Dong, X. Identification of a bilirubin receptor that may mediate a component of cholestatic itch. Elife 2019, 21, e44116. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bates, E.A.; Kipp, Z.A.; Martinez, G.J.; Badmus, O.O.; Soundarapandian, M.M.; Foster, D.; Xu, M.; Creeden, J.F.; Greer, J.R.; Morris, A.J.; et al. Suppressing Hepatic UGT1A1 Increases Plasma Bilirubin, Lowers Plasma Urobilin, Reorganizes Kinase Signaling Pathways and Lipid Species and Improves Fatty Liver Disease. Biomolecules 2023, 13, 252. https://doi.org/10.3390/biom13020252
Bates EA, Kipp ZA, Martinez GJ, Badmus OO, Soundarapandian MM, Foster D, Xu M, Creeden JF, Greer JR, Morris AJ, et al. Suppressing Hepatic UGT1A1 Increases Plasma Bilirubin, Lowers Plasma Urobilin, Reorganizes Kinase Signaling Pathways and Lipid Species and Improves Fatty Liver Disease. Biomolecules. 2023; 13(2):252. https://doi.org/10.3390/biom13020252
Chicago/Turabian StyleBates, Evelyn A., Zachary A. Kipp, Genesee J. Martinez, Olufunto O. Badmus, Mangala M. Soundarapandian, Donald Foster, Mei Xu, Justin F. Creeden, Jennifer R. Greer, Andrew J. Morris, and et al. 2023. "Suppressing Hepatic UGT1A1 Increases Plasma Bilirubin, Lowers Plasma Urobilin, Reorganizes Kinase Signaling Pathways and Lipid Species and Improves Fatty Liver Disease" Biomolecules 13, no. 2: 252. https://doi.org/10.3390/biom13020252
APA StyleBates, E. A., Kipp, Z. A., Martinez, G. J., Badmus, O. O., Soundarapandian, M. M., Foster, D., Xu, M., Creeden, J. F., Greer, J. R., Morris, A. J., Stec, D. E., & Hinds, T. D., Jr. (2023). Suppressing Hepatic UGT1A1 Increases Plasma Bilirubin, Lowers Plasma Urobilin, Reorganizes Kinase Signaling Pathways and Lipid Species and Improves Fatty Liver Disease. Biomolecules, 13(2), 252. https://doi.org/10.3390/biom13020252