
Emerging Drug Targets for Endometriosis


{kind=link}
Abstract
1. Introduction
1.1. Concept of Progesterone Resistance
1.2. Heterogeneity of Lesions
1.3. Crucial Role of Inflammation and Inflammatory Molecules
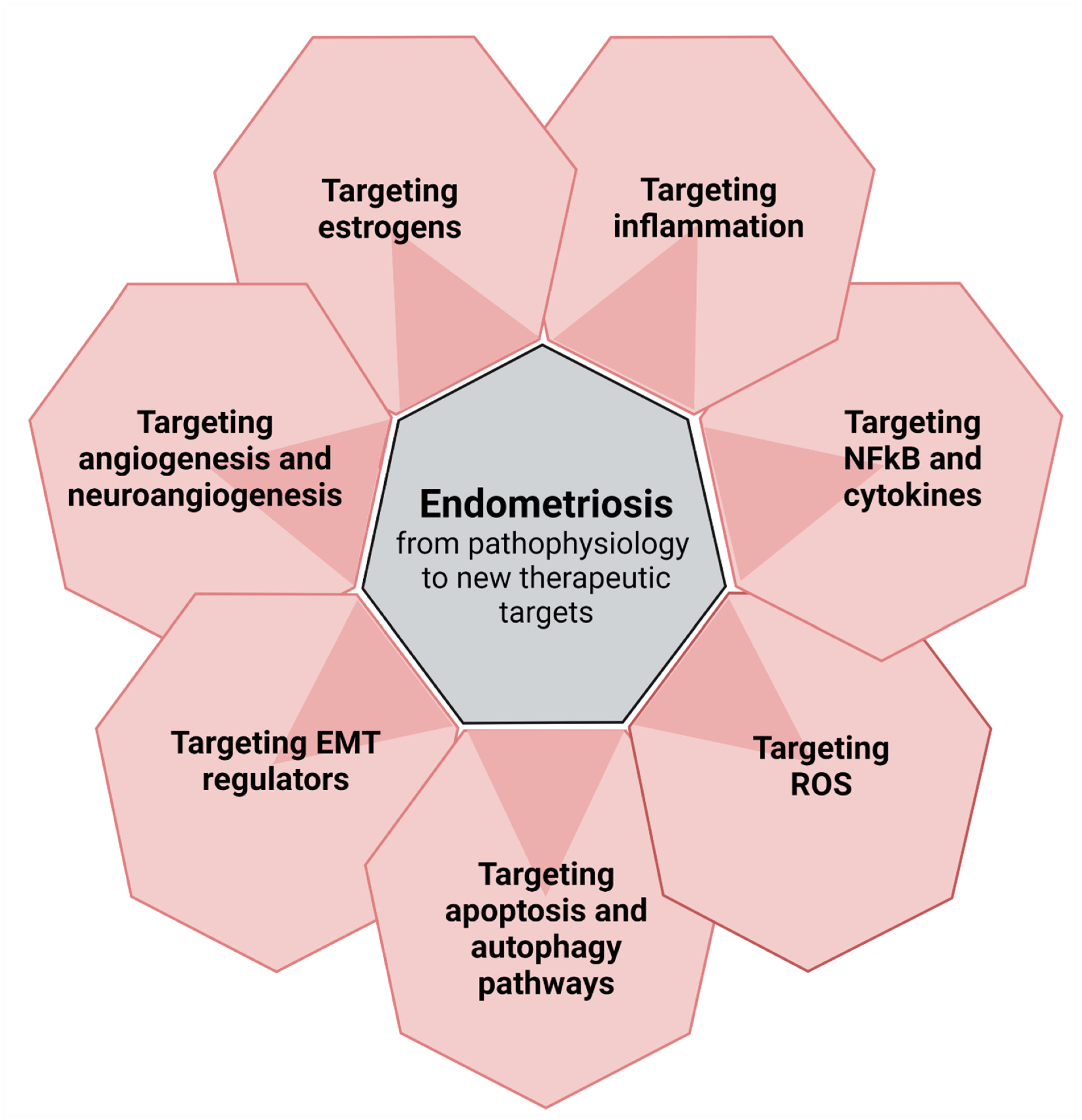
2. From Pathophysiology to New Perspectives (Figure 1)

2.1. Targeting Inflammation
2.1.1. Prostaglandin E2, Cyclooxygenase-2 and Tumor Necrosis Factor-α
2.1.2. Targeting NFκB and Cytokines
2.1.3. Chronic Inflammation and Epigenetics
2.2. Targeting Reactive Oxygen Species
2.3. Targeting Apoptotic and Autophagic Pathways and Tumor-Promoting Genes/Proteins
2.4. Targeting Regulators of Epithelial-Mesenchymal Transition
2.5. Targeting Angiogenesis and Neuroangiogenesis
2.6. Targeting Estrogens
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Donnez, J.; Chantraine, F.; Nisolle, M. The efficacy of medical and surgical treatment of endometriosis-associated infertility: Arguments in favour of a medico-surgical approach. Hum. Reprod. Update 2002, 8, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Nisolle, M.; Donnez, J. Peritoneal endometriosis, ovarian endometriosis, and adenomyotic nodules of the rectovaginal septum are three different entities. Fertil. Steril. 1997, 68, 585–596. [Google Scholar] [CrossRef]
- Giudice, L.C. Clinical practice endometriosis. N. Engl. J. Med. 2010, 362, 2389–2398. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, S.; Evangelisti, G.; Barra, F. Current and emerging treatment options for endometriosis. Expert Opin. Pharm. 2018, 19, 1109–1125. [Google Scholar] [CrossRef]
- Vercellini, P. Are combined hormonal contraceptives the neglected treatment for symptomatic endometriosis? Fertil. Steril. 2018, 110, 61–62. [Google Scholar] [CrossRef] [PubMed]
- Vercellini, P.; Buggio, L.; Berlanda, N.; Barbara, G.; Somigliana, E.; Bosari, S. Estrogen-progestins and progestins for the management of endometriosis. Fertil. Steril. 2016, 106, 1552–1571. [Google Scholar] [CrossRef] [PubMed]
- Casper, R.F. Progestin-only pills may be a better first-line treatment for endometriosis than combined estrogen-progestin contraceptive pills. Fertil. Steril. 2017, 107, 533–536. [Google Scholar] [CrossRef]
- Surrey, E.S.; Soliman, A.M.; Johns, B.; Vora, J.B.; Taylor, H.S.; Agarwal, S.K. Real-World Characterization of Women with Diagnosed Endometriosis Initiating Therapy with Elagolix Using a US Claims Database. Clin. Outcomes Res. 2020, 12, 473–479. [Google Scholar] [CrossRef]
- Soliman, A.M.; Coyne, K.S.; Zaiser, E.; Castelli-Haley, J.; Fuldeore, M.J. The burden of endometriosis symptoms on health-related quality of life in women in the United States: A cross-sectional study. J. Psychosom. Obstet. Gynecol. 2017, 38, 238–248. [Google Scholar] [CrossRef]
- Soliman, A.M.; Yang, H.; Du, E.X.; Kelkar, S.S.; Winkel, C. The direct and indirect costs associated with endometriosis: A systematic literature review. Hum. Reprod. 2016, 31, 712–722. [Google Scholar] [CrossRef]
- Donnez, J.; Dolmans, M.M. Endometriosis and Medical Therapy: From Progestogens to Progesterone Resistance to GnRH Antagonists: A Review. J. Clin. Med. 2021, 10, 1085. [Google Scholar] [CrossRef] [PubMed]
- Bulun, S.E.; Yilmaz, B.D.; Sison, C.; Miyazaki, K.; Bernardi, L.; Liu, S.; Kohlmeier, A.; Yin, P.; Milad, M.; Wei, J. Endometriosis. Endocr. Rev. 2019, 40, 1048–1079. [Google Scholar] [CrossRef] [PubMed]
- Bulun, S.E.; Cheng, Y.H.; Pavone, M.E.; Yin, P.; Imir, G.; Utsunomiya, H.; Thung, S.; Xue, Q.; Marsh, E.E.; Tokunaga, H.; et al. 17Beta-hydroxysteroid dehydrogenase-2 deficiency and progesterone resistance in endometriosis. Semin. Reprod. Med. 2010, 28, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, B.D.; Bulun, S.E. Endometriosis and nuclear receptors. Hum. Reprod. Update 2019, 25, 473–485. [Google Scholar] [CrossRef]
- Flores, V.A.; Vanhie, A.; Dang, T.; Taylor, H.S. Progesterone Receptor Status Predicts Response to Progestin Therapy in Endometriosis. J. Clin. Endocrinol. Metab. 2018, 103, 4561–4568. [Google Scholar] [CrossRef]
- Reis, F.M.; Coutinho, L.M.; Vannuccini, S.; Batteux, F.; Chapron, C.; Petraglia, F. Progesterone receptor ligands for the treatment of endometriosis: The mechanisms behind therapeutic success and failure. Hum. Reprod Update 2020, 26, 565–585. [Google Scholar] [CrossRef]
- Cacciottola, L.; Donnez, J.; Dolmans, M.M. Can Endometriosis-Related Oxidative Stress Pave the Way for New Treatment Targets? Int. J. Mol. Sci. 2021, 22, 7138. [Google Scholar] [CrossRef]
- Donnez, J.; Binda, M.M.; Donnez, O.; Dolmans, M.M. Oxidative stress in the pelvic cavity and its role in the pathogenesis of endometriosis. Fertil. Steril. 2016, 106, 1011–1017. [Google Scholar] [CrossRef]
- Van Langendonckt, A.; Casanas-Roux, F.; Donnez, J. Iron overload in the peritoneal cavity of women with pelvic endometriosis. Fertil. Steril. 2002, 78, 712–718. [Google Scholar] [CrossRef]
- Kapoor, R.; Stratopoulou, C.A.; Dolmans, M.M. Pathogenesis of Endometriosis: New Insights into Prospective Therapies. Int. J. Mol. Sci. 2021, 22, 11700. [Google Scholar] [CrossRef]
- Simpson, J.L.; Bischoff, F.Z.; Kamat, A.; Buster, J.E.; Carson, S.A. Genetics of endometriosis. Obstet. Gynecol. Clin. N. Am. 2003, 30, 21–40. [Google Scholar] [CrossRef]
- Koninckx, P.R.; Ussia, A.; Adamyan, L.; Wattiez, A.; Gomel, V.; Martin, D.C. Pathogenesis of endometriosis: The genetic/epigenetic theory. Fertil. Steril. 2019, 111, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.S.; Kotlyar, A.M.; Flores, V.A. Endometriosis is a chronic systemic disease: Clinical challenges and novel innovations. Lancet 2021, 397, 839–852. [Google Scholar] [CrossRef]
- Patel, B.G.; Rudnicki, M.; Yu, J.; Shu, Y.; Taylor, R.N. Progesterone resistance in endometriosis: Origins, consequences and interventions. Acta Obstet. Gynecol. Scand. 2017, 96, 623–632. [Google Scholar] [CrossRef]
- Koninckx, P.R.; Ussia, A.; Adamyan, L.; Gomel, V.; Martin, D.C. Peritoneal fluid progesterone and progesterone resistance in superficial endometriosis lesions. Hum. Reprod. 2022, 37, 203–211. [Google Scholar] [CrossRef]
- Brichant, G.; Laraki, I.; Henry, L.; Munaut, C.; Nisolle, M. New Therapeutics in Endometriosis: A Review of Hormonal, Non-Hormonal, and Non-Coding RNA Treatments. Int. J. Mol. Sci. 2021, 22, 10498. [Google Scholar] [CrossRef]
- Hogg, C.; Horne, A.W.; Greaves, E. Endometriosis-associated macrophages: Origin, phenotype, and function. Front. Endocrinol. 2020, 11, 7. [Google Scholar] [CrossRef]
- Donnez, J.; Nisolle, M.; Casanas-Roux, F.; Brion, P.; Da Costa Ferreira, N. Stereometric evaluation of peritoneal endometriosis and endometriotic nodules of the rectovaginal septum. Hum. Reprod. 1996, 11, 224–228. [Google Scholar] [CrossRef]
- Donnez, J.; Smoes, P.; Gillerot, S.; Casanas-Roux, F.; Nisolle, M. Vascular endothelial growth factor (VEGF) in endometriosis. Hum. Reprod. 1998, 13, 1686–1690. [Google Scholar] [CrossRef]
- Redwine, D.B. Was Sampson wrong? Fertil. Steril. 2002, 78, 686–693. [Google Scholar] [CrossRef]
- Colgrave, E.M.; Bittinger, S.; Healey, M.; Dior, U.P.; Rogers, P.A.W.; Keast, J.R.; Girling, J.E.; Holdsworth-Carson, S.J. Superficial peritoneal endometriotic lesions are histologically diverse and rarely demonstrate menstrual cycle synchronicity with matched eutopic endometrium. Hum. Reprod. 2020, 35, 2701–2714. [Google Scholar] [CrossRef] [PubMed]
- Donnez, J. The heterogeneity of endometriotic lesions could be explained by their progesterone resistance. Hum. Reprod. 2021, 36, 2624–2625. [Google Scholar] [CrossRef] [PubMed]
- Forster, R.; Sarginson, A.; Velichkova, A.; Hogg, C.; Dorning, A.; Horne, A.W.; Saunders, P.T.K.; Greaves, E. Macrophage-derived insulin-like growth factor-1 is a key neurotrophic and nerve-sensitizing factor in pain associated with endometriosis. FASEB J. 2019, 33, 11210–11222. [Google Scholar] [CrossRef] [PubMed]
- Delbandi, A.A.; Mahmoudi, M.; Shervin, A.; Heidari, S.; Kolahdouz-Mohammadi, R.; Zarnani, A.H. Evaluation of apoptosis and angiogenesis in ectopic and eutopic stromal cells of patients with endometriosis compared to non-endometriotic controls. BMC Womens Health 2020, 20, 3. [Google Scholar] [CrossRef]
- Yu, X.; Wang, Y.; Tan, X.; Li, M. Upregulation of fibroblast growth factor 2 contributes to endometriosis through SPRYs/DUSP6/ERK signaling pathway. Acta Histochem. 2021, 123, 151749. [Google Scholar] [CrossRef]
- Akoum, A.; Kong, J.; Metz, C.; Beaumont, M.C. Spontaneous and stimulated secretion of monocyte chemotactic protein-1 and macrophage migration inhibitory factor by peritoneal macrophages in women with and without endometriosis. Fertil. Steril. 2002, 77, 989–994. [Google Scholar] [CrossRef]
- Akoum, A.; Metz, C.N.; Al-Akoum, M.; Kats, R. Macrophage migration inhibitory factor expression in the intrauterine endometrium of women with endometriosis varies with disease stage, infertility status, and pelvic pain. Fertil. Steril. 2006, 85, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Vanhie, A.; Tomassetti, C.; Peeraer, K.; Meuleman, C.; D’Hooghe, T. Challenges in the development of novel therapeutic strategies for treatment of endometriosis. Expert Opin Targets 2016, 20, 593–600. [Google Scholar] [CrossRef]
- Yu, P.H.; Chou, P.Y.; Li, W.N.; Tsai, S.J.; Wu, M.H. The pro-inflammatory and anti-inflammatory role of hyaluronic acid in endometriosis. Taiwan J. Obs. Gynecol. 2021, 60, 711–717. [Google Scholar] [CrossRef]
- Wu, M.H.; Wang, C.A.; Lin, C.C.; Chen, L.C.; Chang, W.C.; Tsai, S.J. Distinct regulation of cyclooxygenase-2 by interleukin-1beta in normal and endometriotic stromal cells. J. Clin. Endocrinol Metab. 2005, 90, 286–295. [Google Scholar] [CrossRef]
- Lai, Z.Z.; Yang, H.L.; Ha, S.Y.; Chang, K.K.; Mei, J.; Zhou, W.J.; Qiu, X.M.; Wang, X.Q.; Zhu, R.; Li, D.J.; et al. Cyclooxygenase-2 in Endometriosis. Int. J. Biol. Sci. 2019, 15, 2783–2797. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Lao, X.; Zheng, H. Influencing COX-2 Activity by COX Related Pathways in Inflammation and Cancer. Mini Rev. Med. Chem. 2016, 16, 1230–1243. [Google Scholar] [CrossRef] [PubMed]
- Nandakishore, R.; Yalavarthi, P.R.; Kiran, Y.R.; Rajapranathi, M. Selective cyclooxygenase inhibitors: Current status. Curr. Drug Discov. Technol. 2014, 11, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Olivares, C.N.; Bilotas, M.A.; Ricci, A.G.; Barañao, R.I.; Meresman, G.F. Anastrozole and celecoxib for endometriosis treatment, good to keep them apart? Reproduction 2013, 145, 119–126. [Google Scholar] [CrossRef]
- D’Hooghe, T.M.; Nugent, N.P.; Cuneo, S.; Chai, D.C.; Deer, F.; Debrock, S.; Kyama, C.M.; Mihalyi, A.; Mwenda, J.M. Recombinant human TNFRSF1A (r-h-TBP1) inhibits the development of endometriosis in baboons: A prospective, randomized, placebo- and drug-controlled study. Biol. Reprod. 2006, 74, 131–136. [Google Scholar] [CrossRef]
- D’Antonio, M.; Martelli, F.; Peano, S.; Papoian, R.; Borrelli, F. Ability of recombinant human TNF binding protein-1 (r-hTBP-1) to inhibit the development of experimentally-induced endometriosis in rats. J. Reprod Immunol. 2000, 48, 81–98. [Google Scholar] [CrossRef]
- Koninckx, P.R.; Craessaerts, M.; Timmerman, D.; Cornillie, F.; Kennedy, S. Anti-TNF-alpha treatment for deep endometriosis-associated pain: A randomized placebo controlled-trial. Human Reprod. 2008, 23, 2017–2023. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J.; Zhang, X. An Update on the Multifaceted Role of NF-kappaB in Endometriosis. Int. J. Biol. Sci. 2022, 18, 4400–4413. [Google Scholar] [CrossRef]
- Wieser, F.; Vigne, J.L.; Ryan, I.; Hornung, D.; Djalali, S.; Taylor, R.N. Sulindac suppresses nuclear factor-kappaB activation and RANTES gene and protein expression in endometrial stromal cells from women with endometriosis. J. Clin. Endocrinol. Metab. 2005, 90, 6441–6447. [Google Scholar] [CrossRef]
- Alvarado-Díaz, C.P.; Núñez, M.T.; Devoto, L.; González-Ramos, R. Iron overload-modulated nuclear factor kappa-B activation in human endometrial stromal cells as a mechanism postulated in endometriosis pathogenesis. Fertil. Steril. 2015, 103, 439–447. [Google Scholar] [CrossRef]
- El-Zayadi, A.A.; Mohamed, S.A.; Arafa, M.; Mohammed, S.M.; Zayed, A.; Abdelhafez, M.S.; Badawy, A.M. Anti-IL-6 receptor monoclonal antibody as a new treatment of endometriosis. Immunol. Res. 2020, 68, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Karamian, A.; Paktinat, S.; Esfandyari, S.; Nazarian, H.; Ziai, S.A.; Zarnani, A.H.; Salehpour, S.; Hosseinirad, H.; Karamian, A.; Novin, M.G. Pyrvinium pamoate induces in-vitro suppression of IL-6 and IL-8 produced by human endometriotic stromal cells. Hum. Exp. Toxicol. 2021, 40, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Shao, X. Nobiletin alleviates endometriosis via down-regulating NF-κB activity in endometriosis mouse model. Biosci. Rep. 2018, 38, BSR20180470. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.W. Epigenetics of endometriosis. Mol. Hum. Reprod. 2009, 15, 587–607. [Google Scholar] [CrossRef]
- Wu, Y.; Halverson, G.; Basir, Z.; Strawn, E.; Yan, P.; Guo, S.W. Aberrant methylation at HOXA10 may be responsible for its aberrant expression in the endometrium of patients with endometriosis. Am. J. Obs. Gynecol. 2005, 193, 371–380. [Google Scholar] [CrossRef]
- Wang, L.; Tan, Y.J.; Wang, M.; Chen, Y.F.; Li, X.Y. DNA Methylation Inhibitor 5-Aza-2’-Deoxycytidine Modulates Endometrial Receptivity Through Upregulating HOXA10 Expression. Reprod. Sci. 2019, 26, 839–846. [Google Scholar] [CrossRef]
- Cacciottola, L.; Donnez, J.; Dolmans, M.M. Oxidative stress, mitochondria, and infertility: Is the relationship fully established? Fertil. Steril. 2021, 17, 320–324. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Lahair, M.M.; Franklin, R.A. Reactive oxygen species induced activation of the MAP kinase signaling pathways. Antioxid. Redox. Signal. 2006, 8, 1775–1789. [Google Scholar] [CrossRef]
- Ngô, C.; Nicco, C.; Leconte, M.; Chéreau, C.; Arkwright, S.; Vacher-Lavenu, M.C.; Weill, B.; Chapron, C.; Batteux, F. Protein kinase inhibitors can control the progression of endometriosis in vitro and in vivo. J. Pathol. 2010, 222, 148–157. [Google Scholar] [CrossRef]
- Breedveld, F.C.; Dayer, J.M. Leflunomide: Mode of action in the treatment of rheumatoid arthritis. Ann. Rheum. Dis. 2000, 59, 841–849. [Google Scholar] [CrossRef]
- Aytan, H.; Caglar, P.; Uygur, D.; Zergeroglu, S.; Batioglu, S. Effect of the immunomodulator leflunomide on the induction of endometriosis in an experimental rat model. Fertil. Steril. 2007, 87, 698–701. [Google Scholar] [CrossRef] [PubMed]
- Ozer, H.; Boztosun, A.; Acmaz, G.; Atilgan, R.; Akkar, O.B.; Kosar, M. The efficacy of bevacizumab, sorafenib, and retinoic acid on rat endometriosis model. Reprod. Sci. 2013, 20, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, C.; Kacan, T.; Akkar, O.B.; Karakus, S.; Kacan, S.B.; Ozer, H.; Cetin, A. Effects of Pazopanib, Sunitinib, and Sorafenib, Anti-VEGF Agents, on the Growth of Experimental Endometriosis in Rats. Reprod. Sci. 2015, 22, 1445–1451. [Google Scholar] [CrossRef] [PubMed]
- Leconte, M.; Nicco, C.; Ngô, C.; Chéreau, C.; Chouzenoux, S.; Marut, W.; Guibourdenche, J.; Arkwright, S.; Weill, B.; Chapron, C.; et al. The mTOR/AKT inhibitor temsirolimus prevents deep infiltrating endometriosis in mice. Am. J. Pathol. 2011, 179, 880–889. [Google Scholar] [CrossRef]
- Yagyu, T.; Tsuji, Y.; Haruta, S.; Kitanaka, T.; Yamada, Y.; Kawaguchi, R.; Kanayama, S.; Tanase, Y.; Kurita, N.; Kobayashi, H. Activation of mammalian target of rapamycin in postmenopausal ovarian endometriosis. Int. J. Gynecol. Cancer 2006, 16, 1545–1551. [Google Scholar] [CrossRef]
- Leconte, M.; Nicco, C.; Ngô, C.; Arkwright, S.; Chéreau, C.; Guibourdenche, J.; Batteux, F. Antiproliferative effects of cannabinoid agonists on deep infiltrating endometriosis. Am. J. Pathol. 2010, 177, 2963–2970. [Google Scholar] [CrossRef]
- Rudzitis-Auth, J.; Menger, M.D.; Laschke, M.W. Resveratrol is a potent inhibitor of vascularization and cell proliferation in experimental endometriosis. Hum. Reprod. 2013, 28, 1339–1347. [Google Scholar] [CrossRef]
- Taguchi, A.; Wada-Hiraike, O.; Kawana, K.; Koga, K.; Yamashita, A.; Shirane, A.; Urata, Y.; Kozuma, S.; Osuga, Y.; Fujii, T. Resveratrol suppresses inflammatory responses in endometrial stromal cells derived from endometriosis: A possible role of the sirtuin 1 pathway. J. Obstet. Gynaecol. Res. 2014, 40, 770–778. [Google Scholar] [CrossRef]
- Park, S.; Lim, W.; Bazer, F.W.; Song, G. Naringenin induces mitochondria-mediated apoptosis and endoplasmic reticulum stress by regulating MAPK and AKT signal transduction pathways in endometriosis cells. Mol. Hum. Reprod. 2017, 23, 842–854. [Google Scholar] [CrossRef]
- Kapoor, R.; Sirohi, V.K.; Gupta, K.; Dwivedi, A. Naringenin ameliorates progression of endometriosis by modulating Nrf2/Keap1/HO1 axis and inducing apoptosis in rats. J. Nutr. Biochem. 2019, 70, 215–226. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y. Curcumin and Endometriosis. Int. J. Mol. Sci. 2020, 21, 2440. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, E.N.; Park, J.K.; Lee, J.R.; Kim, J.H.; Choi, H.J.; Yoon, S. Curcumin attenuates TNF-α-induced expression of intercellular adhesion molecule-1, vascular cell adhesion molecule-1 and proinflammatory cytokines in human endometriotic stromal cells. Phytother. Res. 2012, 26, 1037–1047. [Google Scholar] [CrossRef]
- Parasassi, T.; Brunelli, R.; Bracci-Laudiero, L.; Greco, G.; Gustafsson, A.C.; Krasnowska, E.K.; Lundeberg, J.; Lundeberg, T.; Pittaluga, E.; Romano, M.C.; et al. Differentiation of normal and cancer cells induced by sulfhydryl reduction: Biochemical and molecular mechanisms. Cell Death Differ. 2005, 12, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Pittaluga, E.; Costa, G.; Krasnowska, E.; Brunelli, R.; Lundeberg, T.; Porpora, M.G.; Santucci, D.; Parasassi, T. More than antioxidant: N-acetyl-L-cysteine in a murine model of endometriosis. Fertil. Steril. 2010, 94, 2905–2908. [Google Scholar] [CrossRef]
- Porpora, M.G.; Brunelli, R.; Costa, G.; Imperiale, L.; Krasnowska, E.K.; Lundeberg, T.; Nofroni, I.; Piccioni, M.G.; Pittaluga, E.; Ticino, A.; et al. A promise in the treatment of endometriosis: An observational cohort study on ovarian endometrioma reduction by N-acetylcysteine. Evid. Based Complement Alternat. Med. 2013, 2013, 240702. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Hu, T.; Hu, P.; Qi, C.; Qian, L. miR-143-3p inhibits endometriotic stromal cell proliferation and invasion by inactivating autophagy in endometriosis. Mol. Med. Rep. 2021, 23, 356. [Google Scholar] [CrossRef]
- Mao, H.; Zhang, X.; Yin, L.; Ji, X.; Huang, C.; Wu, Q. Silencing of circ_0007299 suppresses proliferation, migration, and invasiveness and promotes apoptosis of ectopic endometrial stromal cells in endometriosis via miR-424-5p-dependent modulation of CREB1. Arch. Gynecol. Obstet. 2022, 1–13. [Google Scholar] [CrossRef]
- Sapmaz, T.; Coskun, G.; Saker, D.; Pence, H.H.; Keles, P.; Hayretdag, C.; Kuras, S.; Topkaraoglu, S.; Erdem, E.; Efendic, F.; et al. Effects of metformin, letrozole and atorvastatin on inflammation and apoptosis in experimental peritoneal and ovarian endometriosis in the rat. Pathol. Res. Pract. 2022, 235, 153951. [Google Scholar] [CrossRef]
- Lin, Y.K.; Li, Y.Y.; Li, Y.; Li, D.J.; Wang, X.L.; Wang, L.; Yu, M.; Zhu, Y.Z.; Cheng, J.J.; Du, M.R. SCM-198 Prevents Endometriosis by Reversing Low Autophagy of Endometrial Stromal Cell via Balancing ERα and PR Signals. Front. Endocrinol. 2022, 13, 858176. [Google Scholar] [CrossRef]
- Konrad, L.; Dietze, R.; Riaz, M.A.; Scheiner-Bobis, G.; Behnke, J.; Horné, F.; Hoerscher, A.; Reising, C.; Meinhold-Heerlein, I. Epithelial-Mesenchymal Transition in Endometriosis-When Does It Happen? J. Clin. Med. 2020, 9, 1915. [Google Scholar] [CrossRef]
- Suda, K.; Nakaoka, H.; Yoshihara, K.; Ishiguro, T.; Adachi, S.; Kase, H.; Motoyama, T.; Inoue, I.; Enomoto, T. Different mutation profiles between epithelium and stroma in endometriosis and normal endometrium. Hum. Reprod. 2019, 34, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- Noë, M.; Ayhan, A.; Wang, T.L.; Shih, I.M. Independent development of endometrial epithelium and stroma within the same endometriosis. J. Pathol. 2018, 245, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Orellana, R.; García-Solares, J.; Donnez, J.; van Kerk, O.; Dolmans, M.M.; Donnez, O. Important role of collective cell migration and nerve fiber density in the development of deep nodular endometriosis. Fertil. Steril. 2017, 107, 987–995. [Google Scholar] [CrossRef] [PubMed]
- García-Solares, J.; Dolmans, M.M.; Squifflet, J.L.; Donnez, J.; Donnez, O. Invasion of human deep nodular endometriotic lesions is associated with collective cell migration and nerve development. Fertil. Steril. 2018, 110, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Zhu, Z.Y.; Chen, X.M.; Lu, J.Q.; Song, Y.; Xia, W. A review of the effects of estrogen and epithelial-mesenchymal transformation on intrauterine adhesion and endometriosis. Transpl. Immunol. 2022, 101679. [Google Scholar] [CrossRef]
- Hsu, Y.W.; Chen, H.Y.; Chiang, Y.F.; Chang, L.C.; Lin, P.H.; Hsia, S.M. The effects of isoliquiritigenin on endometriosis in vivo and in vitro study. Phytomedicine 2020, 77, 153214. [Google Scholar] [CrossRef]
- Chang, L.C.; Chiang, Y.F.; Chen, H.Y.; Huang, Y.J.; Liu, A.C.; Hsia, S.M. The Potential Effect of Fucoidan on Inhibiting Epithelial-to-Mesenchymal Transition, Proliferation, and Increase in Apoptosis for Endometriosis Treatment: In Vivo and In Vitro Study. Biomedicines 2020, 8, 528. [Google Scholar] [CrossRef]
- Qi, S.; Yan, L.; Liu, Z.; Mu, Y.L.; Li, M.; Zhao, X.; Chen, Z.J.; Zhang, H. Melatonin inhibits 17β-estradiol-induced migration, invasion and epithelial-mesenchymal transition in normal and endometriotic endometrial epithelial cells. Reprod. Biol. Endocrinol. 2018, 16, 62. [Google Scholar] [CrossRef]
- Yu, M.M.; Zhou, Q.M. 3,6-dihydroxyflavone suppresses the epithelial-mesenchymal transition, migration and invasion in endometrial stromal cells by inhibiting the Notch signaling pathway. Eur. Rev. Med. Pharm. Sci. 2018, 22, 4009–4017. [Google Scholar]
- Conway, G.E.; Zizyte, D.; Mondala, J.R.M.; He, Z.; Lynam, L.; Lecourt, M.; Barcia, C.; Howe, O.; Curtin, J.F. Ursolic Acid Inhibits Collective Cell Migration and Promotes JNK-Dependent Lysosomal Associated Cell Death in Glioblastoma Multiforme Cells. Pharmaceuticals 2021, 14, 91. [Google Scholar] [CrossRef]
- Wu, Y.; Ali, M.R.K.; Dong, B.; Han, T.; Chen, K.; Chen, J.; Tang, Y.; Fang, N.; Wang, F.; El-Sayed, M.A. Gold Nanorod Photothermal Therapy Alters Cell Junctions and Actin Network in Inhibiting Cancer Cell Collective Migration. ACS Nano 2018, 12, 9279–9290. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, V.E.; Varshney, G.K.; Lee, M.; Bupp, S.; Xu, L.; Shinn, P.; Crawford, N.P.; Inglese, J.; Burgess, S.M. Phenotype-driven chemical screening in zebrafish for compounds that inhibit collective cell migration identifies multiple pathways potentially involved in metastatic invasion. Dis. Model. Mech. 2015, 8, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Mendel, D.B.; Laird, A.D.; Xin, X.; Louie, S.G.; Christensen, J.G.; Li, G.; Schreck, R.E.; Abrams, T.J.; Ngai, T.J.; Lee, L.B.; et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin. Cancer Res. 2003, 9, 327–337. [Google Scholar] [PubMed]
- Abbas, M.A.; Disi, A.M.; Taha, M.O. Sunitinib as an anti-endometriotic agent. Eur. J. Pharm. Sci. 2013, 49, 732–736. [Google Scholar] [CrossRef]
- Gómez, R.; Abad, A.; Delgado, F.; Tamarit, S.; Simón, C.; Pellicer, A. Effects of hyperprolactinemia treatment with the dopamine agonist quinagolide on endometriotic lesions in patients with endometriosis-associated hyperprolactinemia. Fertil. Steril. 2011, 95, 882–888. [Google Scholar] [CrossRef]
- Pellicer, N.; Galliano, D.; Herraiz, S.; Bagger, Y.Z.; Arce, J.C.; Pellicer, A.A. Use of dopamine agonists to target angiogenesis in women with endometriosis. Human Reprod. 2021, 36, 850–858. [Google Scholar] [CrossRef]
- Tejada, M.Á.; Santos-Llamas, A.I.; Fernández-Ramírez, M.J.; Tarín, J.J.; Cano, A.; Gómez, R. A Reassessment of the Therapeutic Potential of a Dopamine Receptor 2 Agonist (D2-AG) in Endometriosis by Comparison against a Standardized Antiangiogenic Treatment. Biomedicines 2021, 9, 269. [Google Scholar] [CrossRef]
- Donnez, O.; Orellana, R.; Van Kerk, O.; Dehoux, J.P.; Donnez, J.; Dolmans, M.M. Invasion process of induced deep nodular endometriosis in an experimental baboon model: Similarities with collective cell migration? Fertil. Steril. 2015, 104, 491–497. [Google Scholar] [CrossRef]
- Donnez, O.; Soares, M.; Defrère, S.; Dehoux, J.P.; van Langendonckt, A.; Donnez, J.; Dolmans, M.M.; Colette, S. Nerve fiber density in deep nodular endometriotic lesions induce.ed in a baboon experimental model. Fertil. Steril. 2013, 100, 1144–1150. [Google Scholar] [CrossRef]
- Sun, H.; Li, D.; Yuan, M.; Li, Q.; Zhen, Q.; Li, N.; Wang, G. Macrophages alternatively activated by endometriosis-exosomes contribute to the development of lesions in mice. Mol. Hum. Reprod. 2019, 25, 5–16. [Google Scholar] [CrossRef]
- Saunders, P.T.K.; Horne, A.W. Endometriosis: Etiology, pathobiology, and therapeutic prospects. Cell 2021, 184, 2807–2824. [Google Scholar] [CrossRef] [PubMed]
- Vannuccini, S.; Clemenza, S.; Rossi, M.; Petraglia, F. Hormonal treatments for endometriosis: The endocrine background. Rev. Endocr. Metab. Disord. 2022, 23, 333–355. [Google Scholar] [CrossRef] [PubMed]
- Donnez, J.; Dolmans, M.M. GnRH Antagonists with or without Add-Back Therapy: A New Alternative in the Management of Endometriosis? Int. J. Mol. Sci. 2021, 22, 11342. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, R.L. Hormone treatment of endometriosis: The estrogen threshold hypothesis. Am. J. Obtset. Gynecol. 1992, 166, 740–745. [Google Scholar] [CrossRef]
- Donnez, J.; Taylor, R.N.; Taylor, H.S. Partial suppression of estradiol: A new strategy in endometriosis management? Fertil. Steril. 2017, 107, 568–570. [Google Scholar] [CrossRef]
- Taylor, H.S.; Giudice, L.C.; Lessey, B.A.; Abrao, M.S.; Kotarski, J.; Archer, D.F.; Diamond, M.P.; Surrey, E.; Johnson, N.P.; Watts, N.B.; et al. Treatment of Endometriosis-Associated Pain with Elagolix, an Oral GnRH Antagonist. N. Engl. J. Med. 2017, 377, 28–40. [Google Scholar] [CrossRef]
- Taylor, H.S.; Dun, E.C.; Chwalisz, K. Clinical evaluation of the oral gonadotropin-releasing hormone-antagonist elagolix for the management of endometriosis-associated pain. Pain Manag. 2019, 9, 497–515. [Google Scholar] [CrossRef]
- Taylor, H.S.; Soliman, A.M.; Johns, B.; Pokrzywinski, R.M.; Snabes, M.; Coyne, K.S. Health-Related Quality of Life Improvements in Patients with Endometriosis Treated with Elagolix. Obstet. Gynecol. 2020, 136, 501–509. [Google Scholar] [CrossRef]
- Donnez, J.; Taylor, H.S.; Taylor, R.N.; Akin, M.D.; Tatarchuk, T.F.; Wilk, K.; Gotteland, J.P.; Lecomte, V.; Bestel, E. Treatment of endometriosis-associated pain with linzagolix, an oral gonadotropin-releasing hormone-antagonist: A randomized clinical trial. Fertil. Steril. 2020, 114, 44–55. [Google Scholar] [CrossRef]
- Pohl, O.; Marchand, L.; Bell, D.; Gotteland, J.P. Effects of combined GnRH receptor antagonist linzagolix and hormonal add-back therapy on vaginal bleeding-delayed add-back onset does not improve bleeding pattern. Reprod. Sci. 2020, 27, 988–995. [Google Scholar] [CrossRef]
- Osuga, Y.; Seki, Y.; Tanimoto, M.; Kusumoto, T.; Kudou, K.; Terakawa, N. Relugolix, an oral gonadotropin-releasing hormone receptor antagonist, reduces endometriosis-associated pain in a dose-response manner: A randomized, double-blind, placebocontrolled study. Fertil. Steril. 2021, 115, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Giudice, L.C.; As-Sanie, S.; Arjona Ferreira, J.C.; Becker, C.M.; Abrao, M.S.; Lessey, B.A.; Brown, E.; Dynowski, K.; Wilk, K.; Li, Y.; et al. Once daily oral relugolix combination therapy versus placebo in patients with endometriosis-associated pain: Two replicate phase 3, randomised, double-blind, studies (SPIRIT 1 and 2). Lancet 2022, 399, 2267–2279. [Google Scholar] [CrossRef]
- Esfandiari, F.; Heidari Khoei, H.; Saber, M.; Favaedi, R.; Piryaei, A.; Moini, A.; Shahhoseini, M.; Ramezanali, F.; Ghaffari, F.; Baharvand, H. Disturbed progesterone signalling in an advanced preclinical model of endometriosis. Reprod. Biomed. Online 2021, 43, 139–147. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dolmans, M.-M.; Donnez, J. Emerging Drug Targets for Endometriosis. Biomolecules 2022, 12, 1654. https://doi.org/10.3390/biom12111654
Dolmans M-M, Donnez J. Emerging Drug Targets for Endometriosis. Biomolecules. 2022; 12(11):1654. https://doi.org/10.3390/biom12111654
Chicago/Turabian StyleDolmans, Marie-Madeleine, and Jacques Donnez. 2022. "Emerging Drug Targets for Endometriosis" Biomolecules 12, no. 11: 1654. https://doi.org/10.3390/biom12111654
APA StyleDolmans, M.-M., & Donnez, J. (2022). Emerging Drug Targets for Endometriosis. Biomolecules, 12(11), 1654. https://doi.org/10.3390/biom12111654