
Post-Translational Modifications by Lipid Metabolites during the DNA Damage Response and Their Role in Cancer

Abstract
1. Introduction
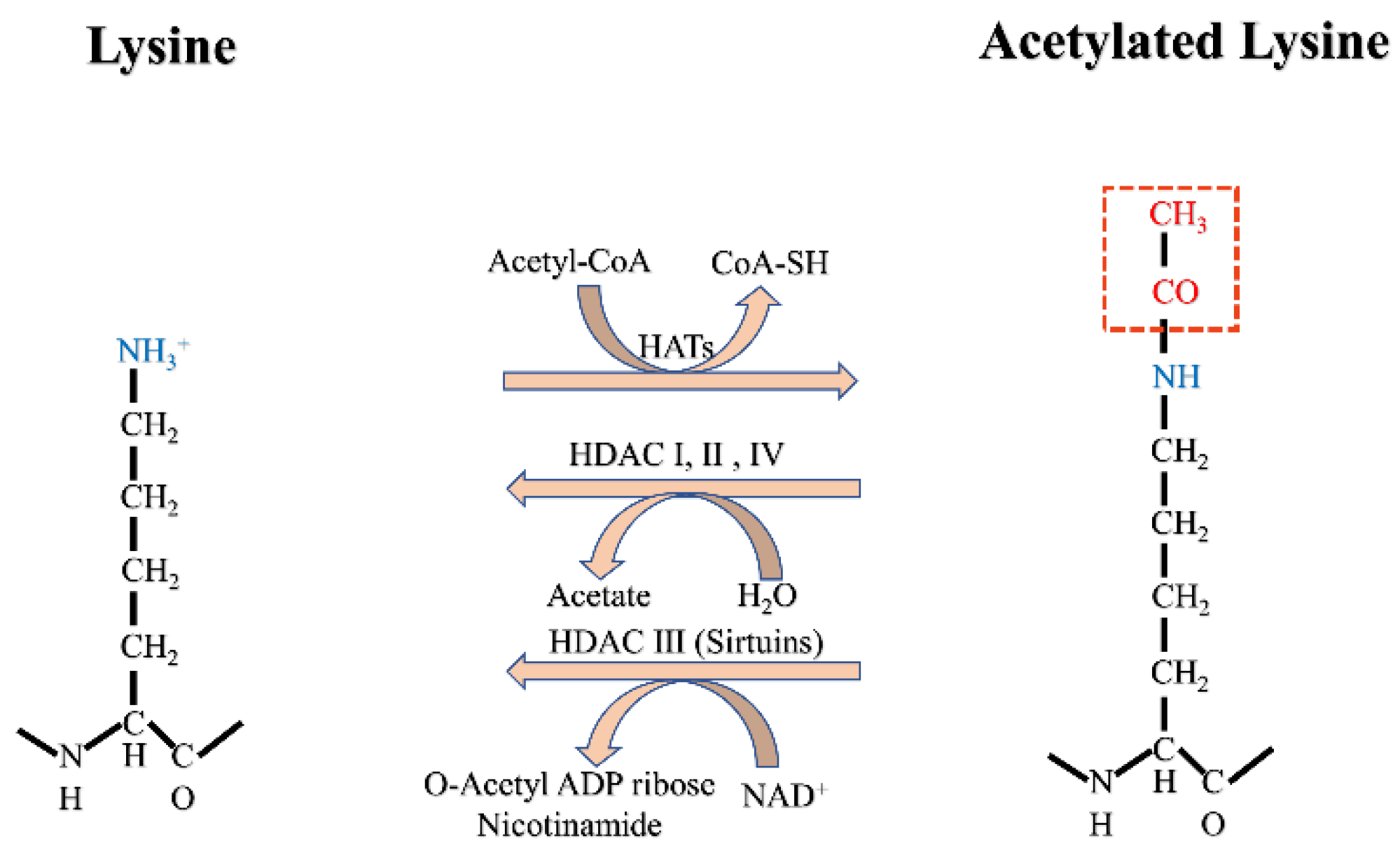
2. Acetylation
2.1. Acetylation of Histones and Non-Histone Proteins
2.2. Acetylation in the DDR
2.2.1. Histone Acetylation in the DDR
2.2.2. Non-Histone Acetylation in the DDR
2.2.3. Roles of HATs and HDACs/SIRTs in DDR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modification | Writers | Erasers | Readers | Substrates in the DDR | References |
|---|---|---|---|---|---|
| Acetylation (histone) | P160, P300/CBP | HDAC1-11, | YAF9, ENL, AF9, Taf14, | H1K85 | |
| TAFII230, MYST, | SIRT1-7 | Sas5(Yeats), | H2AX K5, K36 | [44,45,46,49,50,51] | |
| GNAT, PCAF | PHDs | H3K4, K18, K56 H4K16 | |||
| Acetylation (non-histone) | GCN5, P300, MYST19, | HDAC1-11 | Tip60, APE1, | ||
| KAT1, TAT1, ESCO1-2 | SIRT1-7, LEF1, TCF1 | NA | PFKFB3 K472, OGG1, Cdc25A, P53K382/K120, WRN K1117/K1127, MLH1, RRM2 PARP1 K949, | [34,87,88,89,90,91] | |
| Succinylation | NA | KDAC: SIRT5, SIRT7 | NA | H3K12, P53K120, H4K77, | |
| ACOX1(acyl-CoA oxidase 1), FEN1 K200, NPM1 | [19,92,93,94,95,96] | ||||
| Palmitoylation | DHHC1-23 | APT1-2 | NA | Rap1-interacting factor 1(Rif1) C466/C473 | [97] |
| N-myristoylation | NMT1-2 | SIRT1-3, SIRT6 | NA | Finkel-Biskis-Reilly (FBR) v-fos | [98] |
| Crotonylation | P300/CBP, MOF | HDAC1-3, SIRT1-3 | YEATS, PHD | RPA1 | [21] |
2.3. Acetylation in Cancer
2.3.1. HATs and Cancer
2.3.2. HDACs and Cancer
2.3.3. Summary
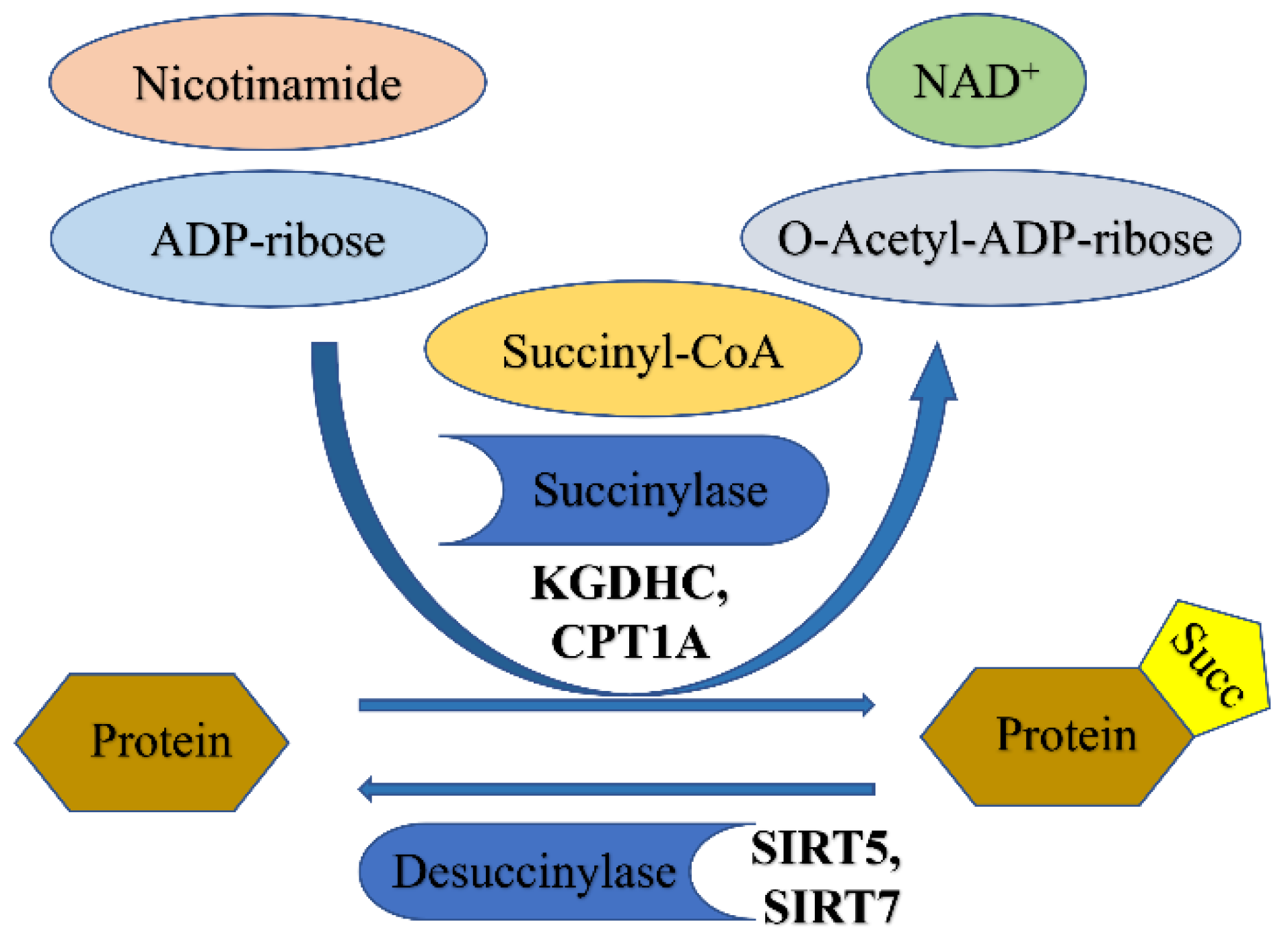
3. Succinylation
3.1. Succinylation
3.1.1. Enzymatic Regulation of Succinylation
3.1.2. Non-Enzymatic Succinylation
3.1.3. De-Succinylation
3.1.4. Lysine-Succinyl Readers
3.2. Succinylation in the DDR
3.3. Succinylation in Cancer
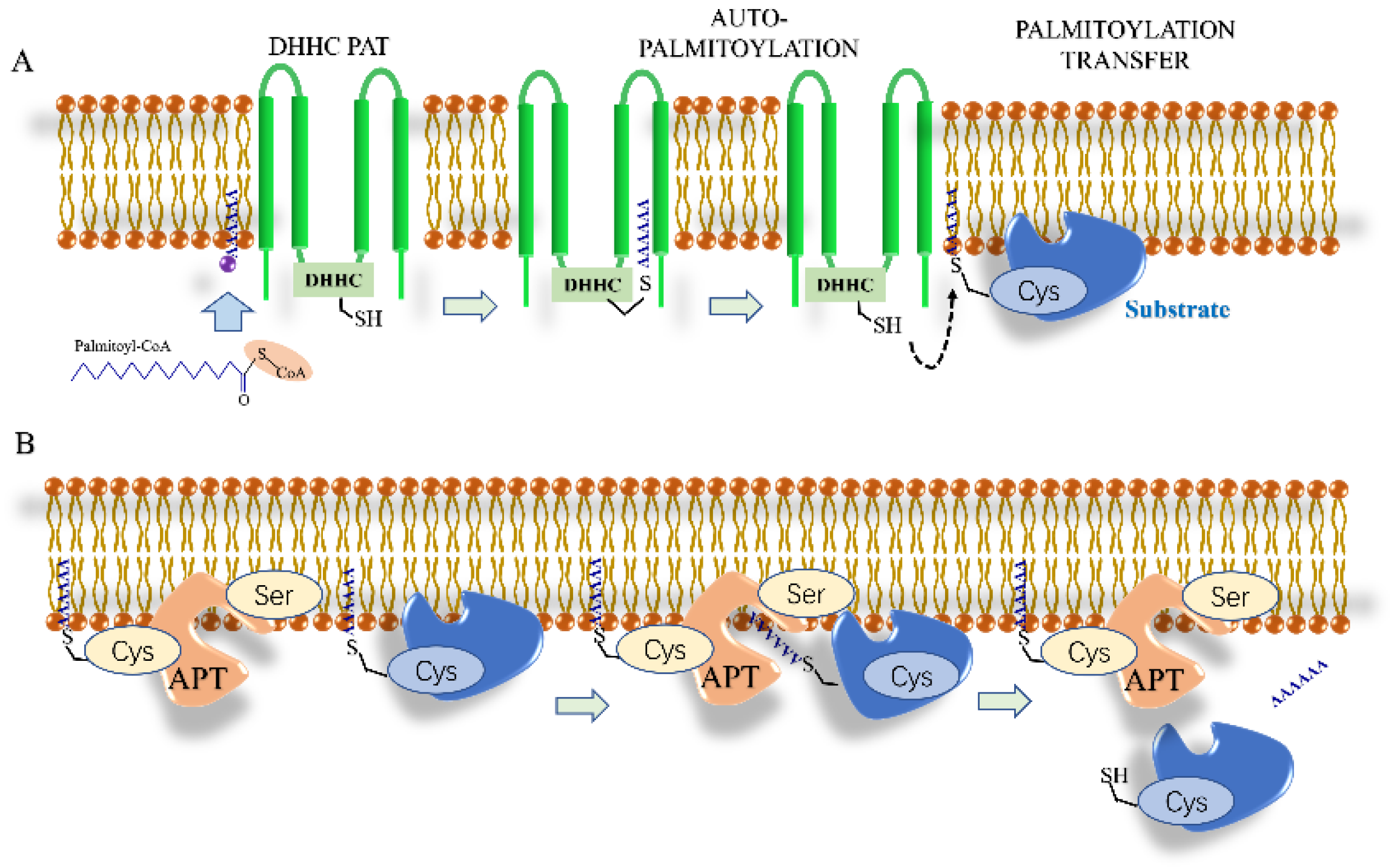
4. Palmitoylation
4.1. S-Palmitoylation
Depalmitoylation
4.2. Palmitoylation in the DDR
4.3. Palmitoylation in Cancer
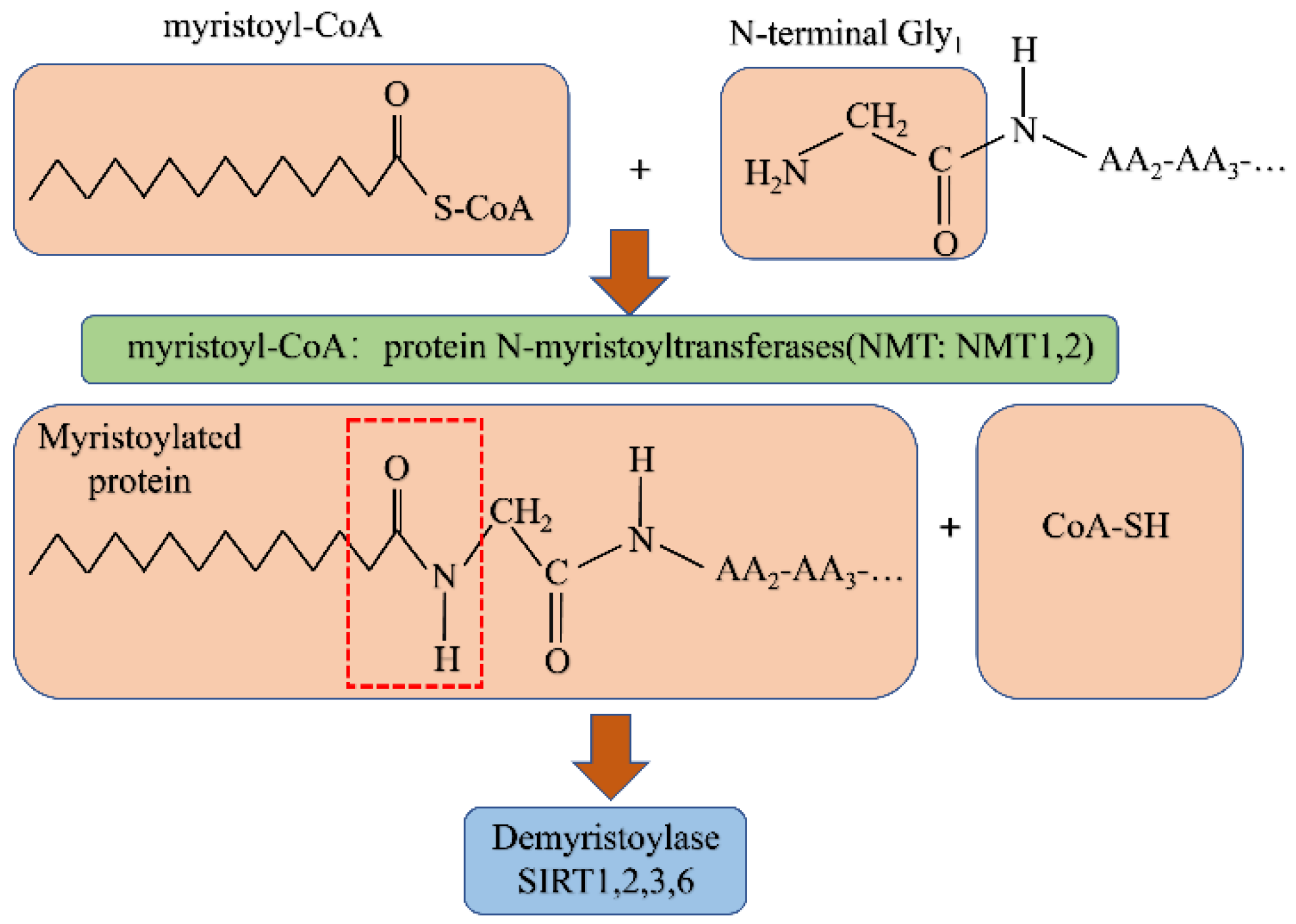
5. N-myristoylation
5.1. N-myristoylation
Demyristoylation
5.2. N-Myristoylation in the DDR
5.3. N-myristoylation in Cancer
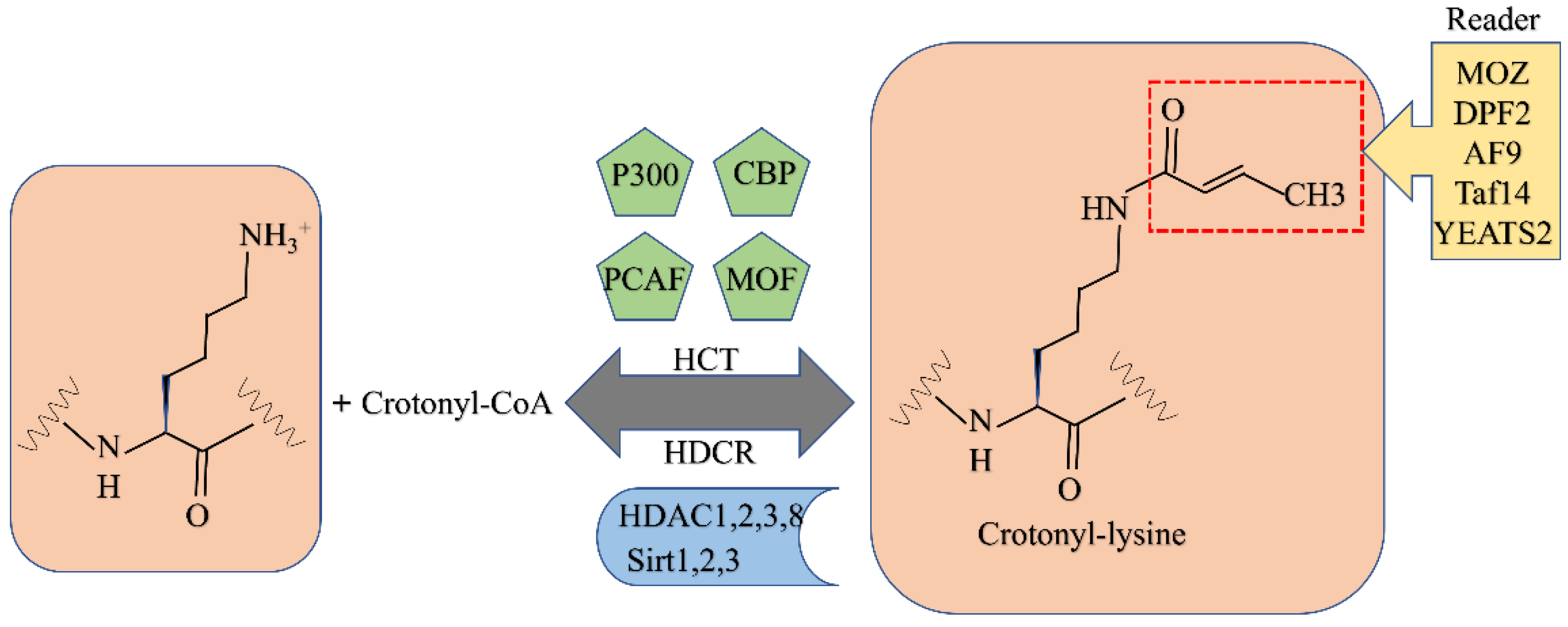
6. Crotonylation
6.1. Crotonylation
6.1.1. Writers for Crotonylation
6.1.2. Erasers for Crotonylation
6.1.3. Readers for Crotonylation
6.2. Crotonylation in the DDR
6.3. Crotonylation in Cancer
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kim, S.I.; Pfeifer, G.P. The epigenetic DNA modification 5-carboxylcytosine promotes high levels of cyclobutane pyrimidine dimer formation upon UVB irradiation. Genome Instab. Dis. 2021, 2, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P. Mechanisms of UV-induced mutations and skin cancer. Genome Instab. Dis. 2020, 1, 99–113. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA damage response pathway: Biomarker and therapeutic strategy for cancer immunotherapy. Acta Pharm. Sin. B 2021, 11, 2983–2994. [Google Scholar] [CrossRef]
- Rahnamay, F.P.; Danesh, P.R.; Asemi, Z.; Yousefi, B. DNA damage response and repair in pancreatic cancer development and therapy. DNA Repair 2021, 103, 103116. [Google Scholar] [CrossRef]
- Wang, M.; Chen, S.; Ao, D. Targeting DNA repair pathway in cancer: Mechanisms and clinical application. Med. Comm. 2021, 2, 654–691. [Google Scholar] [CrossRef]
- Bateman, A.C. DNA mismatch repair protein immunohistochemistry—An illustrated guide. Histopathology 2021, 79, 128–138. [Google Scholar] [CrossRef]
- Putnam, C.D. Strand discrimination in DNA mismatch repair. DNA Repair 2021, 105, 103161. [Google Scholar] [CrossRef]
- Olivares-Hernández, A.; Del, B.M.E.; Parra, P.C.; Miramontes-González, J.P.; Figuero-Pérez, L.; Martín-Gómez, T.; Escala-Cornejo, R.; Bellido, H.L.; González, S.R.; Cruz-Hernández, J.J.; et al. Influence of DNA Mismatch Repair (MMR) System in Survival and Response to Immune Checkpoint Inhibitors (ICIs) in Non-Small Cell Lung Cancer (NSCLC): Retrospective Analysis. Biomedicines 2022, 10, 360. [Google Scholar] [CrossRef]
- Demin, A.A.; Hirota, K.; Tsuda, M.; Adamowicz, M.; Hailstone, R.; Brazina, J.; Gittens, W.; Kalasova, I.; Shao, Z.; Zha, S.; et al. XRCC1 prevents toxic PARP1 trapping during DNA base excision repair. Mol. Cell 2021, 81, 3018–3030. [Google Scholar] [CrossRef]
- Malfatti, M.C.; Antoniali, G.; Codrich, M.; Tell, G. Coping with RNA damage with a focus on APE1, a BER enzyme at the crossroad between DNA damage repair and RNA processing/decay. DNA Repair 2021, 104, 103133. [Google Scholar] [CrossRef]
- Anabtawi, N.; Cvammen, W.; Kemp, M.G. Pharmacological inhibition of cryptochrome and REV-ERB promotes DNA repair and cell cycle arrest in cisplatin-treated human cells. Sci. Rep. 2021, 11, 17997. [Google Scholar] [CrossRef]
- Kraithong, T.; Hartley, S.; Jeruzalmi, D.; Pakotiprapha, D. A Peek Inside the Machines of Bacterial Nucleotide Excision Repair. Int. J. Mol. Sci. 2021, 22, 952. [Google Scholar] [CrossRef]
- Wang, R.; Wang, G. Protein Modification and Autophagy Activation. Adv. Exp. Med. Biol. 2019, 1206, 237–259. [Google Scholar] [CrossRef]
- Sabari, B.R.; Zhang, D.; Allis, C.D.; Zhao, Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell. Biol. 2017, 18, 90–101. [Google Scholar] [CrossRef]
- Chen, X.F.; Chen, X.; Tang, X. Short-chain fatty acid, acylation and cardiovascular diseases. Clin. Sci. 2020, 134, 657–676. [Google Scholar] [CrossRef]
- Huang, G.; Zheng, Y.; Wu, Y.Q.; Han, G.S.; Yu, Z.G. An Information Entropy-Based Approach for Computationally Identifying Histone Lysine Butyrylation. Front. Genet. 2019, 10, 1325. [Google Scholar] [CrossRef]
- Liu, J.; Shangguan, Y.; Tang, D.; Dai, Y. Histone succinylation and its function on the nucleosome. J. Cell. Mol. Med. 2021, 25, 7101–7109. [Google Scholar] [CrossRef]
- Lin, H. Protein cysteine palmitoylation in immunity and inflammation. FEBS J. 2021, 288, 7043–7059. [Google Scholar] [CrossRef]
- Jiang, G.; Li, C.; Lu, M.; Lu, K.; Li, H. Protein lysine crotonylation: Past, present, perspective. Cell. Death Dis. 2021, 12, 703. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Dai, T.; Sun, W.; Wei, Y.; Ren, J.; Zhang, L.; Zhang, M.; Zhou, F. Protein N-myristoylation: Functions and mechanisms in control of innate immunity. Cell. Mol. Immunol. 2021, 18, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Komath, S.S.; Fujita, M.; Hart, G.W.; Ferguson, M.; Kinoshita, T. Glycosylphosphatidylinositol Anchors. In Essentials of Glycobiology; National Library of Medicine: Bethesda, MD, USA, 2022; Chapter 12; pp. 141–154. [Google Scholar] [CrossRef]
- Yuan, Y.; Yuan, H.F.; Geng, Y.; Zhao, L.N.; Yun, H.L.; Wang, Y.F.; Yang, G.; Zhang, X.D. Aspirin modulates 2-hydroxyisobutyrylation of ENO1K281 to attenuate the glycolysis and proliferation of hepatoma cells. Biochem. Biophys. Res. Commun. 2021, 560, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Koronowski, K.B.; Greco, C.M.; Huang, H.; Kim, J.K.; Fribourgh, J.L.; Crosby, P.; Mathur, L.; Ren, X.; Partch, C.L.; Jang, C.; et al. Ketogenesis impact on liver metabolism revealed by proteomics of lysineβ-hydroxybutyrylation. Cell Rep. 2021, 36, 109487. [Google Scholar] [CrossRef] [PubMed]
- Mcdonnell, E.; Crown, S.B.; Fox, D.B.; Kitir, B.; Ilkayeva, O.R.; Olsen, C.A.; Grimsrud, P.A.; Hirschey, M.D. Lipids Reprogram Metabolism to Become a Major Carbon Source for Histone Acetylation. Cell Rep. 2016, 17, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Trefely, S.; Lovell, C.D.; Snyder, N.W.; Wellen, K.E. Compartmentalised acyl-CoA metabolism and roles in chromatin regulation. Mol. Metab. 2020, 38, 100941. [Google Scholar] [CrossRef]
- Hou, J.Y.; Zhou, L.; Li, J.L.; Wang, D.P.; Cao, J.M. Emerging roles of non-histone protein crotonylation in biomedicine. Cell Biosci. 2021, 11, 101. [Google Scholar] [CrossRef]
- Slaughter, M.J.; Shanle, E.K.; Khan, A.; Chua, K.F.; Hong, T.; Boxer, L.D.; Allis, C.D.; Josefowicz, S.Z.; Garcia, B.A.; Rothbart, S.B.; et al. HDAC inhibition results in widespread alteration of the histone acetylation landscape and BRD4 targeting to gene bodies. Cell Rep. 2021, 34, 108638. [Google Scholar] [CrossRef]
- Luu, J.; Carabetta, V.J. Contribution of N(ε)-lysine Acetylation towards Regulation of Bacterial Pathogenesis. mSystems 2021, 6, e42221. [Google Scholar] [CrossRef]
- Sebastián, C.; Mostoslavsky, R. The Various Metabolic Sources of Histone Acetylation. Trends Endocrinol. Metab. 2017, 28, 85–87. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of Histone Modification. Adv. Exp. Med. Biol. 2021, 1283, 1–16. [Google Scholar] [CrossRef]
- Gu, W.; Cheng, Y.; Wang, S.; Sun, T.; Li, Z. PHD Finger Protein 19 Promotes Cardiac Hypertrophy via Epigenetically Regulating SIRT2. Cardiovasc. Toxicol. 2021, 21, 451–461. [Google Scholar] [CrossRef]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef]
- Li, L.; Jayabal, S.; Ghorbani, M.; Legault, L.M.; Mcgraw, S.; Watt, A.J.; Yang, X.J. ATAT1 regulates forebrain development and stress-induced tubulin hyperacetylation. Cell. Mol. Life Sci. 2019, 76, 3621–3640. [Google Scholar] [CrossRef]
- Sivalingam, K.; Doke, M.; Khan, M.A.; Samikkannu, T. Influence of psychostimulants and opioids on epigenetic modification of class III histone deacetylase (HDAC)-sirtuins in glial cells. Sci. Rep. 2021, 11, 21335. [Google Scholar] [CrossRef]
- Min, Z.; Gao, J.; Yu, Y. The Roles of Mitochondrial SIRT4 in Cellular Metabolism. Front. Endocrinol. 2018, 9, 783. [Google Scholar] [CrossRef]
- Kumar, S.; Lombard, D.B. Functions of the sirtuin deacylase SIRT5 in normal physiology and pathobiology. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 311–334. [Google Scholar] [CrossRef]
- Xing, S.; Li, F.; Zeng, Z.; Zhao, Y.; Yu, S.; Shan, Q.; Li, Y.; Phillips, F.C.; Maina, P.K.; Qi, H.H.; et al. Tcf1 and Lef1 transcription factors establish CD8(+) T cell identity through intrinsic HDAC activity. Nat. Immunol. 2016, 17, 695–703. [Google Scholar] [CrossRef]
- Li, F.; Zhao, X.; Zhang, Y.; Shao, P.; Ma, X.; Paradee, W.J.; Liu, C.; Wang, J.; Xue, H.H. T(FH) cells depend on Tcf1-intrinsic HDAC activity to suppress CTLA4 and guard B-cell help function. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Kim, C.; Jin, J.; Weyand, C.M.; Goronzy, J.J. The Transcription Factor TCF1 in T Cell Differentiation and Aging. Int. J. Mol. Sci. 2020, 21, 6497. [Google Scholar] [CrossRef]
- Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, E. The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications. Cell Metab. 2018, 27, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Sivanand, S.; Rhoades, S.; Jiang, Q.; Lee, J.V.; Benci, J.; Zhang, J.; Yuan, S.; Viney, I.; Zhao, S.; Carrer, A.; et al. Nuclear Acetyl-CoA Production by ACLY Promotes Homologous Recombination. Mol. Cell 2017, 67, 252–265. [Google Scholar] [CrossRef]
- Li, Y.; Li, Z.; Dong, L.; Tang, M.; Zhang, P.; Zhang, C.; Cao, Z.; Zhu, Q.; Chen, Y.; Wang, H.; et al. Histone H1 acetylation at lysine 85 regulates chromatin condensation and genome stability upon DNA damage. Nucleic Acids Res. 2018, 46, 7716–7730. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Xu, Y.; Price, B.D. Acetylation of H2AX on lysine 36 plays a key role in the DNA double-strand break repair pathway. FEBS Lett. 2010, 584, 2926–2930. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xie, L.; Ramachandran, S.; Lee, Y.; Yan, Z.; Zhou, L.; Krajewski, K.; Liu, F.; Zhu, C.; Chen, D.J.; et al. Non-canonical Bromodomain within DNA-PKcs Promotes DNA Damage Response and Radioresistance through Recognizing an IR-Induced Acetyl-Lysine on H2AX. Chem. Biol. 2015, 22, 849–861. [Google Scholar] [CrossRef][Green Version]
- Hu, R.; Wang, E.; Peng, G.; Dai, H.; Lin, S.Y. Zinc finger protein 668 interacts with Tip60 to promote H2AX acetylation after DNA damage. Cell Cycle 2013, 12, 2033–2041. [Google Scholar] [CrossRef][Green Version]
- Van, H.T.; Santos, M.A. Histone modifications and the DNA double-strand break response. Cell Cycle 2018, 17, 2399–2410. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Z.; Guo, X.; Li, F.; Wei, Q.; Chen, X.; Gong, D.; Xu, Y.; Chen, W.; Liu, Y.; et al. TRIM66 reads unmodified H3R2K4 and H3K56ac to respond to DNA damage in embryonic stem cells. Nat. Commun. 2019, 10, 4273. [Google Scholar] [CrossRef]
- Zhang, A.L.; Chen, L.; Ma, L.; Ding, X.J.; Tang, S.F.; Zhang, A.H.; Li, J. Role of H3K18ac-regulated nucleotide excision repair-related genes in arsenic-induced DNA damage and repair of HaCaT cells. Hum. Exp. Toxicol. 2020, 39, 1168–1177. [Google Scholar] [CrossRef]
- García-González, R.; Morejón-García, P.; Campillo-Marcos, I.; Salzano, M.; Lazo, P.A. VRK1 Phosphorylates Tip60/KAT5 and Is Required for H4K16 Acetylation in Response to DNA Damage. Cancers 2020, 12, 2986. [Google Scholar] [CrossRef]
- Su, J.; Wang, F.; Cai, Y.; Jin, J. The Functional Analysis of Histone Acetyltransferase MOF in Tumorigenesis. Int. J. Mol. Sci. 2016, 17, 99. [Google Scholar] [CrossRef]
- Ge, Z.; Nair, D.; Guan, X.; Rastogi, N.; Freitas, M.A.; Parthun, M.R. Sites of acetylation on newly synthesized histone H4 are required for chromatin assembly and DNA damage response signaling. Mol. Cell. Biol. 2013, 33, 3286–3298. [Google Scholar] [CrossRef]
- Shahar, O.D.; Gabizon, R.; Feine, O.; Alhadeff, R.; Ganoth, A.; Argaman, L.; Shimshoni, E.; Friedler, A.; Goldberg, M. Acetylation of lysine 382 and phosphorylation of serine 392 in p53 modulate the interaction between p53 and MDC1 in vitro. PLoS ONE 2013, 8, e78472. [Google Scholar] [CrossRef]
- Liu, X.; Tan, Y.; Zhang, C.; Zhang, Y.; Zhang, L.; Ren, P.; Deng, H.; Luo, J.; Ke, Y.; Du, X. NAT10 regulates p53 activation through acetylating p53 at K120 and ubiquitinating Mdm2. EMBO Rep. 2016, 17, 349–366. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef]
- Ghosh, D.; Bohr, V.A.; Karmakar, P. Acetylation of Werner protein at K1127 and K1117 is important for nuclear trafficking and DNA repair. DNA Repair 2019, 79, 22–31. [Google Scholar] [CrossRef]
- Yamamori, T.; Dericco, J.; Naqvi, A.; Hoffman, T.A.; Mattagajasingh, I.; Kasuno, K.; Jung, S.B.; Kim, C.S.; Irani, K. SIRT1 deacetylates APE1 and regulates cellular base excision repair. Nucleic Acids Res. 2010, 38, 832–845. [Google Scholar] [CrossRef]
- Dai, C.; Shi, D.; Gu, W. Negative regulation of the acetyltransferase TIP60-p53 interplay by UHRF1 (ubiquitin-like with PHD and RING finger domains 1). J. Biol. Chem. 2013, 288, 19581–19592. [Google Scholar] [CrossRef]
- Li, X.; Corsa, C.A.; Pan, P.W.; Wu, L.; Ferguson, D.; Yu, X.; Min, J.; Dou, Y. MOF and H4 K16 acetylation play important roles in DNA damage repair by modulating recruitment of DNA damage repair protein Mdc1. Mol. Cell. Biol. 2010, 30, 5335–5347. [Google Scholar] [CrossRef]
- Liu, R.; Gou, D.; Xiang, J.; Pan, X.; Gao, Q.; Zhou, P.; Liu, Y.; Hu, J.; Wang, K.; Tang, N. O-GlcNAc modified-TIP60/KAT5 is required for PCK1 deficiency-induced HCC metastasis. Oncogene 2021, 40, 6707–6719. [Google Scholar] [CrossRef]
- Squatrito, M.; Gorrini, C.; Amati, B. Tip60 in DNA damage response and growth control: Many tricks in one HAT. Trends Cell Biol. 2006, 16, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Bao, H.; Zhang, X.P.; Liu, F.; Wang, W. Regulation of Tip60-dependent p53 acetylation in cell fate decision. FEBS Lett. 2019, 593, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Sun, Y.; Chen, S.; Roy, K.; Price, B.D. The FATC domains of PIKK proteins are functionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J. Biol. Chem. 2006, 281, 15741–15746. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, K.; Fradet-Turcotte, A.; Avvakumov, N.; Lambert, J.P.; Roques, C.; Pandita, R.K.; Paquet, E.; Herst, P.; Gingras, A.C.; Pandita, T.K.; et al. The TIP60 Complex Regulates Bivalent Chromatin Recognition by 53BP1 through Direct H4K20me Binding and H2AK15 Acetylation. Mol. Cell 2016, 62, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Ikura, T.; Tashiro, S.; Kakino, A.; Shima, H.; Jacob, N.; Amunugama, R.; Yoder, K.; Izumi, S.; Kuraoka, I.; Tanaka, K.; et al. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol. Cell. Biol. 2007, 27, 7028–7040. [Google Scholar] [CrossRef]
- Murr, R.; Loizou, J.I.; Yang, Y.G.; Cuenin, C.; Li, H.; Wang, Z.Q.; Herceg, Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 91–99. [Google Scholar] [CrossRef]
- Schiltz, R.L.; Mizzen, C.A.; Vassilev, A.; Cook, R.G.; Allis, C.D.; Nakatani, Y. Overlapping but distinct patterns of histone acetylation by the human coactivators p300 and PCAF within nucleosomal substrates. J. Biol. Chem. 1999, 274, 1189–1192. [Google Scholar] [CrossRef]
- Liu, C.; Yang, Q.; Zhu, Q.; Lu, X.; Li, M.; Hou, T.; Li, Z.; Tang, M.; Li, Y.; Wang, H.; et al. CBP mediated DOT1L acetylation confers DOT1L stability and promotes cancer metastasis. Theranostics 2020, 10, 1758–1776. [Google Scholar] [CrossRef]
- Marmorstein, R.; Roth, S.Y. Histone acetyltransferases: Function, structure, and catalysis. Curr. Opin. Genet. Dev. 2001, 11, 155–161. [Google Scholar] [CrossRef]
- Tjeertes, J.V.; Miller, K.M.; Jackson, S.P. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 2009, 28, 1878–1889. [Google Scholar] [CrossRef]
- Grant, P.A.; Eberharter, A.; John, S.; Cook, R.G.; Turner, B.M.; Workman, J.L. Expanded lysine acetylation specificity of Gcn5 in native complexes. J. Biol. Chem. 1999, 274, 5895–5900. [Google Scholar] [CrossRef]
- Guo, R.; Chen, J.; Mitchell, D.L.; Johnson, D.G. GCN5 and E2F1 stimulate nucleotide excision repair by promoting H3K9 acetylation at sites of damage. Nucleic Acids Res. 2011, 39, 1390–1397. [Google Scholar] [CrossRef]
- Aricthota, S.; Rana, P.P.; Haldar, D. Histone acetylation dynamics in repair of DNA double-strand breaks. Front. Genet. 2022, 13, 926577. [Google Scholar] [CrossRef]
- Vaquero, A. The conserved role of sirtuins in chromatin regulation. Int. J. Dev. Biol. 2009, 53, 303–322. [Google Scholar] [CrossRef]
- Jing, H.; Lin, H. Sirtuins in epigenetic regulation. Chem. Rev. 2015, 115, 2350–2375. [Google Scholar] [CrossRef]
- Yuan, J.; Pu, M.; Zhang, Z.; Lou, Z. Histone H3-K56 acetylation is important for genomic stability in mammals. Cell Cycle 2009, 8, 1747–1753. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a18713. [Google Scholar] [CrossRef]
- Scher, M.B.; Vaquero, A.; Reinberg, D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007, 21, 920–928. [Google Scholar] [CrossRef]
- Vaquero, A.; Sternglanz, R.; Reinberg, D. NAD+-dependent deacetylation of H4 lysine 16 by class III HDACs. Oncogene 2007, 26, 5505–5520. [Google Scholar] [CrossRef]
- Michishita, E.; Mccord, R.A.; Berber, E.; Kioi, M.; Padilla-Nash, H.; Damian, M.; Cheung, P.; Kusumoto, R.; Kawahara, T.L.; Barrett, J.C.; et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 2008, 452, 492–496. [Google Scholar] [CrossRef]
- Barber, M.F.; Michishita-Kioi, E.; Xi, Y.; Tasselli, L.; Kioi, M.; Moqtaderi, Z.; Tennen, R.I.; Paredes, S.; Young, N.L.; Chen, K.; et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 2012, 487, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 2010, 17, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Battu, A.; Ray, A.; Wani, G.; Qian, J.; He, J.; Wang, Q.E.; Wani, A.A. Damaged DNA-binding protein down-regulates epigenetic mark H3K56Ac through histone deacetylase 1 and 2. Mutat. Res. 2015, 776, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.P.; Spitz, G.S.; Tharkar, S.; Quayle, S.N.; Shearstone, J.R.; Jones, S.; Mcdowell, M.E.; Wellman, H.; Tyler, J.K.; Cairns, B.R.; et al. HDAC1,2 inhibition impairs EZH2- and BBAP-mediated DNA repair to overcome chemoresistance in EZH2 gain-of-function mutant diffuse large B-cell lymphoma. Oncotarget 2015, 6, 4863–4887. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, C.; Li, T.; Yu, S.; Gan, T.; Hu, J.; Cui, J.; Zheng, X. The Deubiquitinase USP38 Promotes NHEJ Repair through Regulation of HDAC1 Activity and Regulates Cancer Cell Response to Genotoxic Insults. Cancer Res. 2020, 80, 719–731. [Google Scholar] [CrossRef]
- Li, F.L.; Liu, J.P.; Bao, R.X.; Yan, G.; Feng, X.; Xu, Y.P.; Sun, Y.P.; Yan, W.; Ling, Z.Q.; Xiong, Y.; et al. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat. Commun. 2018, 9, 508. [Google Scholar] [CrossRef]
- Lozada, E.M.; Andrysik, Z.; Yin, M.; Redilla, N.; Rice, K.; Stambrook, P.J. Acetylation and deacetylation of Cdc25A constitutes a novel mechanism for modulating Cdc25A functions with implications for cancer. Oncotarget 2016, 7, 20425–20439. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, C.; Moses, N.; Haakenson, J.; Xiang, S.; Quan, D.; Fang, B.; Yang, Z.; Bai, W.; Bepler, G.; et al. HDAC6 regulates DNA damage response via deacetylating MLH1. J. Biol. Chem. 2019, 294, 5813–5826. [Google Scholar] [CrossRef]
- Zhang, L.; Li, D.Q. MORC2 regulates DNA damage response through a PARP1-dependent pathway. Nucleic Acids Res. 2019, 47, 8502–8520. [Google Scholar] [CrossRef]
- Chen, G.; Luo, Y.; Warncke, K.; Sun, Y.; Yu, D.S.; Fu, H.; Behera, M.; Ramalingam, S.S.; Doetsch, P.W.; Duong, D.M.; et al. Acetylation regulates ribonucleotide reductase activity and cancer cell growth. Nat. Commun. 2019, 10, 3213. [Google Scholar] [CrossRef]
- Liu, X.; Rong, F.; Tang, J.; Zhu, C.; Chen, X.; Jia, S.; Wang, Z.; Sun, X.; Deng, H.; Zha, H.; et al. Repression of p53 function by SIRT5-mediated desuccinylation at Lysine 120 in response to DNA damage. Cell Death Differ. 2022, 29, 722–736. [Google Scholar] [CrossRef]
- Jing, Y.; Ding, D.; Tian, G.; Kwan, K.; Liu, Z.; Ishibashi, T.; Li, X.D. Semisynthesis of site-specifically succinylated histone reveals that succinylation regulates nucleosome unwrapping rate and DNA accessibility. Nucleic Acids Res. 2020, 48, 9538–9549. [Google Scholar] [CrossRef]
- Chen, X.F.; Tian, M.X.; Sun, R.Q.; Zhang, M.L.; Zhou, L.S.; Jin, L.; Chen, L.L.; Zhou, W.J.; Duan, K.L.; Chen, Y.J.; et al. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018, 19, e45124. [Google Scholar] [CrossRef]
- Shi, R.; Wang, Y.; Gao, Y.; Xu, X.; Mao, S.; Xiao, Y.; Song, S.; Wang, L.; Tian, B.; Zhao, Y.; et al. Succinylation at a key residue of FEN1 is involved in the DNA damage response to maintain genome stability. Am. J. Physiol. Cell Physiol. 2020, 319, C657–C666. [Google Scholar] [CrossRef]
- Gao, X.; Bao, H.; Liu, L.; Zhu, W.; Zhang, L.; Yue, L. Systematic analysis of lysine acetylome and succinylome reveals the correlation between modification of H2A.X complexes and DNA damage response in breast cancer. Oncol. Rep. 2020, 43, 1819–1830. [Google Scholar] [CrossRef]
- Fontana, G.A.; Hess, D.; Reinert, J.K.; Mattarocci, S.; Falquet, B.; Klein, D.; Shore, D.; Thomä, N.H.; Rass, U. Rif1 S-acylation mediates DNA double-strand break repair at the inner nuclear membrane. Nat. Commun. 2019, 10, 2535. [Google Scholar] [CrossRef]
- Abbott, D.W.; Holt, J.T. Finkel-Biskis-Reilly mouse osteosarcoma virus v-fos inhibits the cellular response to ionizing radiation in a myristoylation-dependent manner. J. Biol. Chem. 1997, 272, 14005–14008. [Google Scholar] [CrossRef][Green Version]
- Pelka, P.; Ablack, J.N.; Torchia, J.; Turnell, A.S.; Grand, R.J.; Mymryk, J.S. Transcriptional control by adenovirus E1A conserved region 3 via p300/CBP. Nucleic Acids Res. 2009, 37, 1095–1106. [Google Scholar] [CrossRef]
- Zhao, L.J.; Loewenstein, P.M.; Green, M. The adenoviral E1A N-terminal domain represses MYC transcription in human cancer cells by targeting both p300 and TRRAP and inhibiting MYC promoter acetylation of H3K18 and H4K16. Genes Cancer 2016, 7, 98–109. [Google Scholar] [CrossRef]
- Peng, Y.; Wang, Y.; Tang, N.; Sun, D.; Lan, Y.; Yu, Z.; Zhao, X.; Feng, L.; Zhang, B.; Jin, L.; et al. Andrographolide inhibits breast cancer through suppressing COX-2 expression and angiogenesis via inactivation of p300 signaling and VEGF pathway. J. Exp. Clin. Cancer Res. 2018, 37, 248. [Google Scholar] [CrossRef]
- Welti, J.; Sharp, A.; Brooks, N.; Yuan, W.; Mcnair, C.; Chand, S.N.; Pal, A.; Figueiredo, I.; Riisnaes, R.; Gurel, B.; et al. Targeting the p300/CBP Axis in Lethal Prostate Cancer. Cancer Discov. 2021, 11, 1118–1137. [Google Scholar] [CrossRef]
- Gruber, M.; Ferrone, L.; Puhr, M.; Santer, F.R.; Furlan, T.; Eder, I.E.; Sampson, N.; Schäfer, G.; Handle, F.; Culig, Z. p300 is upregulated by docetaxel and is a target in chemoresistant prostate cancer. Endocr. Relat. Cancer 2020, 27, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.N.; Avery, V.M.; Carrasco-Pozo, C. Metabolic Roles of Androgen Receptor and Tip60 in Androgen-Dependent Prostate Cancer. Int. J. Mol. Sci. 2020, 21, 6622. [Google Scholar] [CrossRef]
- Ravichandran, P.; Davis, S.A.; Vashishtha, H.; Gucwa, A.L.; Ginsburg, D.S. Nuclear Localization Is Not Required for Tip60 Tumor Suppressor Activity in Breast and Lung Cancer Cells. DNA Cell Biol. 2020, 39, 2077–2084. [Google Scholar] [CrossRef]
- Judes, G.; Dubois, L.; Rifaï, K.; Idrissou, M.; Mishellany, F.; Pajon, A.; Besse, S.; Daures, M.; Degoul, F.; Bignon, Y.J.; et al. TIP60: An actor in acetylation of H3K4 and tumor development in breast cancer. Epigenomics 2018, 10, 1415–1430. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, X.; Price, B.D. Tip60: Connecting chromatin to DNA damage signaling. Cell Cycle 2010, 9, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Avvakumov, N.; Côté, J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 2007, 26, 5395–5407. [Google Scholar] [CrossRef]
- Gorrini, C.; Squatrito, M.; Luise, C.; Syed, N.; Perna, D.; Wark, L.; Martinato, F.; Sardella, D.; Verrecchia, A.; Bennett, S.; et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 2007, 448, 1063–1067. [Google Scholar] [CrossRef]
- Fisher, J.B.; Kim, M.S.; Blinka, S.; Ge, Z.D.; Wan, T.; Duris, C.; Christian, D.; Twaroski, K.; North, P.; Auchampach, J.; et al. Stress-induced cell-cycle activation in Tip60 haploinsufficient adult cardiomyocytes. PLoS ONE 2012, 7, e31569. [Google Scholar] [CrossRef]
- Liu, L.; Qiu, S.; Liu, Y.; Liu, Z.; Zheng, Y.; Su, X.; Chen, B.; Chen, H. Chidamide and 5-flurouracil show a synergistic antitumor effect on human colon cancer xenografts in nude mice. Neoplasma 2016, 63, 193–200. [Google Scholar] [CrossRef][Green Version]
- Kiweler, N.; Schwarz, H.; Nguyen, A.; Matschos, S.; Mullins, C.; Piée-Staffa, A.; Brachetti, C.; Roos, W.P.; Schneider, G.; Linnebacher, M.; et al. The epigenetic modifier HDAC2 and the checkpoint kinase ATM determine the responses of microsatellite instable colorectal cancer cells to 5-fluorouracil. Cell Biol. Toxicol. 2022; advance online publication. [Google Scholar] [CrossRef]
- Spurling, C.C.; Godman, C.A.; Noonan, E.J.; Rasmussen, T.P.; Rosenberg, D.W.; Giardina, C. HDAC3 overexpression and colon cancer cell proliferation and differentiation. Mol. Carcinog. 2008, 47, 137–147. [Google Scholar] [CrossRef]
- Wu, G.; Yu, W.; Zhang, M.; Yin, R.; Wu, Y.; Liu, Q. MicroRNA-145-3p suppresses proliferation and promotes apotosis and autophagy of osteosarcoma cell by targeting HDAC4. Artif. Cells Nanomed. Biotechnol. 2018, 46, 579–586. [Google Scholar] [CrossRef]
- Lee, B.S.; Kim, Y.S.; Kim, H.J.; Kim, D.H.; Won, H.R.; Kim, Y.S.; Kim, C.H. HDAC4 degradation by combined TRAIL and valproic acid treatment induces apoptotic cell death of TRAIL-resistant head and neck cancer cells. Sci. Rep. 2018, 8, 12520. [Google Scholar] [CrossRef]
- Li, X.H.; Huang, M.L.; Wang, S.M.; Wang, Q. Selective inhibition of bicyclic tetrapeptide histone deacetylase inhibitor on HDAC4 and K562 leukemia cell. Asian Pac. J. Cancer Prev. 2013, 14, 7095–7100. [Google Scholar] [CrossRef][Green Version]
- Nie, X.; Jia, W.; Li, X.; Pan, X.; Yin, R.; Liu, N.; Su, Z. FBXW7 induces apoptosis in glioblastoma cells by regulating HDAC7. Cell Biol. Int. 2021, 45, 2150–2158. [Google Scholar] [CrossRef]
- Zhu, C.; Chen, Q.; Xie, Z.; Ai, J.; Tong, L.; Ding, J.; Geng, M. The role of histone deacetylase 7 (HDAC7) in cancer cell proliferation: Regulation on c-Myc. J. Mol. Med. 2011, 89, 279–289. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, S.H.; Choi, M.C.; Lee, J.; Oh, D.Y.; Im, S.A.; Bang, Y.J.; Kim, T.Y. Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors. Biochem. Biophys. Res. Commun. 2008, 368, 318–322. [Google Scholar] [CrossRef]
- Schmid, N.; Dietrich, K.G.; Forne, I.; Burges, A.; Szymanska, M.; Meidan, R.; Mayr, D.; Mayerhofer, A. Sirtuin 1 and Sirtuin 3 in Granulosa Cell Tumors. Int. J. Mol. Sci. 2021, 22, 2047. [Google Scholar] [CrossRef]
- Ceballos, M.P.; Angel, A.; Delprato, C.B.; Livore, V.I.; Ferretti, A.C.; Lucci, A.; Comanzo, C.G.; Alvarez, M.L.; Quiroga, A.D.; Mottino, A.D.; et al. Sirtuin 1 and 2 inhibitors enhance the inhibitory effect of sorafenib in hepatocellular carcinoma cells. Eur. J. Pharmacol. 2021, 892, 173736. [Google Scholar] [CrossRef]
- Tan, J.; Liu, Y.; Maimaiti, Y.; Wang, C.; Yan, Y.; Zhou, J.; Ruan, S.; Huang, T. Combination of SIRT1 and Src overexpression suggests poor prognosis in luminal breast cancer. Onco. Targets Ther. 2018, 11, 2051–2061. [Google Scholar] [CrossRef] [PubMed]
- Huffman, D.M.; Grizzle, W.E.; Bamman, M.M.; Kim, J.S.; Eltoum, I.A.; Elgavish, A.; Nagy, T.R. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007, 67, 6612–6618. [Google Scholar] [CrossRef] [PubMed]
- Saunders, L.R.; Verdin, E. Sirtuins: Critical regulators at the crossroads between cancer and aging. Oncogene 2007, 26, 5489–5504. [Google Scholar] [CrossRef] [PubMed]
- Marcu, M.G.; Jung, Y.J.; Lee, S.; Chung, E.J.; Lee, M.J.; Trepel, J.; Neckers, L. Curcumin is an inhibitor of p300 histone acetylatransferase. Med. Chem. 2006, 2, 169–174. [Google Scholar] [CrossRef]
- Kusio-Kobialka, M.; Dudka-Ruszkowska, W.; Ghizzoni, M.; Dekker, F.J.; Piwocka, K. Inhibition of PCAF by anacardic acid derivative leads to apoptosis and breaks resistance to DNA damage in BCR-ABL-expressing cells. Anticancer Agents Med. Chem. 2013, 13, 762–767. [Google Scholar] [CrossRef]
- Yang, H.; Pinello, C.E.; Luo, J.; Li, D.; Wang, Y.; Zhao, L.Y.; Jahn, S.C.; Saldanha, S.A.; Chase, P.; Planck, J.; et al. Small-molecule inhibitors of acetyltransferase p300 identified by high-throughput screening are potent anticancer agents. Mol. Cancer Ther. 2013, 12, 610–620. [Google Scholar] [CrossRef]
- Di Martile, M.; Del, B.D.; Trisciuoglio, D. The multifaceted role of lysine acetylation in cancer: Prognostic biomarker and therapeutic target. Oncotarget 2016, 7, 55789–55810. [Google Scholar] [CrossRef]
- Yang, G.; Yuan, Y.; Yuan, H.; Wang, J.; Yun, H.; Geng, Y.; Zhao, M.; Li, L.; Weng, Y.; Liu, Z.; et al. Histone acetyltransferase 1 is a succinyltransferase for histones and non-histones and promotes tumorigenesis. EMBO Rep. 2021, 22, e50967. [Google Scholar] [CrossRef]
- Yang, Y.; Gibson, G.E. Succinylation Links Metabolism to Protein Functions. Neurochem. Res. 2019, 44, 2346–2359. [Google Scholar] [CrossRef]
- Zhang, Z.; Tan, M.; Xie, Z.; Dai, L.; Chen, Y.; Zhao, Y. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 2011, 7, 58–63. [Google Scholar] [CrossRef]
- Sreedhar, A.; Wiese, E.K.; Hitosugi, T. Enzymatic and metabolic regulation of lysine succinylation. Genes Dis. 2020, 7, 166–171. [Google Scholar] [CrossRef]
- Qin, Y.P.; Yu, H.B.; Yuan, S.Y.; Yang, Z.; Ren, F.; Wang, Q.; Li, F.; Ren, J.H.; Cheng, S.T.; Zhou, Y.J.; et al. KAT2A Promotes Hepatitis B Virus Transcription and Replication Through Epigenetic Regulation of cccDNA Minichromosome. Front. Microbiol. 2021, 12, 795388. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, Y.R.; Liu, K.; Yin, Z.; Liu, R.; Xia, Y.; Tan, L.; Yang, P.; Lee, J.H.; Li, X.J.; et al. KAT2A coupled with the α-KGDH complex acts as a histone H3 succinyltransferase. Nature 2017, 552, 273–277. [Google Scholar] [CrossRef]
- Li, X.; Zhang, C.; Zhao, T.; Su, Z.; Li, M.; Hu, J.; Wen, J.; Shen, J.; Wang, C.; Pan, J.; et al. Lysine-222 succinylation reduces lysosomal degradation of lactate dehydrogenase a and is increased in gastric cancer. J. Exp. Clin. Cancer Res. 2020, 39, 172. [Google Scholar] [CrossRef] [PubMed]
- Kurmi, K.; Hitosugi, S.; Wiese, E.K.; Boakye-Agyeman, F.; Gonsalves, W.I.; Lou, Z.; Karnitz, L.M.; Goetz, M.P.; Hitosugi, T. Carnitine Palmitoyltransferase 1A Has a Lysine Succinyltransferase Activity. Cell Rep. 2018, 22, 1365–1373. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, C.; Li, X.; Shen, J.; Xu, Y.; Shi, H.; Mu, X.; Pan, J.; Zhao, T.; Li, M.; et al. CPT1A-mediated succinylation of S100A10 increases human gastric cancer invasion. J. Cell. Mol. Med. 2019, 23, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Gut, P.; Matilainen, S.; Meyer, J.G.; Pällijeff, P.; Richard, J.; Carroll, C.J.; Euro, L.; Jackson, C.B.; Isohanni, P.; Minassian, B.A.; et al. SUCLA2 mutations cause global protein succinylation contributing to the pathomechanism of a hereditary mitochondrial disease. Nat. Commun. 2020, 11, 5927. [Google Scholar] [CrossRef] [PubMed]
- Enos-Berlage, J.L.; Downs, D.M. Mutations in sdh (succinate dehydrogenase genes) alter the thiamine requirement of Salmonella typhimurium. J. Bacteriol. 1997, 179, 3989–3996. [Google Scholar] [CrossRef]
- Li, F.; He, X.; Ye, D.; Lin, Y.; Yu, H.; Yao, C.; Huang, L.; Zhang, J.; Wang, F.; Xu, S.; et al. NADP(+)-IDH Mutations Promote Hypersuccinylation that Impairs Mitochondria Respiration and Induces Apoptosis Resistance. Mol. Cell 2015, 60, 661–675. [Google Scholar] [CrossRef]
- Wang, F.; Wang, K.; Xu, W.; Zhao, S.; Ye, D.; Wang, Y.; Xu, Y.; Zhou, L.; Chu, Y.; Zhang, C.; et al. SIRT5 Desuccinylates and Activates Pyruvate Kinase M2 to Block Macrophage IL-1β Production and to Prevent DSS-Induced Colitis in Mice. Cell Rep. 2017, 19, 2331–2344. [Google Scholar] [CrossRef]
- Sadhukhan, S.; Liu, X.; Ryu, D.; Nelson, O.D.; Stupinski, J.A.; Li, Z.; Chen, W.; Zhang, S.; Weiss, R.S.; Locasale, J.W.; et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl. Acad. Sci. USA 2016, 113, 4320–4325. [Google Scholar] [CrossRef]
- Ogura, M.; Nakamura, Y.; Tanaka, D.; Zhuang, X.; Fujita, Y.; Obara, A.; Hamasaki, A.; Hosokawa, M.; Inagaki, N. Overexpression of SIRT5 confirms its involvement in deacetylation and activation of carbamoyl phosphate synthetase 1. Biochem. Biophys. Res. Commun. 2010, 393, 73–78. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, J.; Chung, M.; Feng, L.; Sun, H.; Hao, Q. Identification of the YEATS domain of GAS41 as a pH-dependent reader of histone succinylation. Proc. Natl. Acad. Sci. USA 2018, 115, 2365–2370. [Google Scholar] [CrossRef]
- Tang, M.; Li, Z.; Zhang, C.; Lu, X.; Tu, B.; Cao, Z.; Li, Y.; Chen, Y.; Jiang, L.; Wang, H.; et al. SIRT7-mediated ATM deacetylation is essential for its deactivation and DNA damage repair. Sci. Adv. 2019, 5, v1118. [Google Scholar] [CrossRef]
- Li, L.; Shi, L.; Yang, S.; Yan, R.; Zhang, D.; Yang, J.; He, L.; Li, W.; Yi, X.; Sun, L.; et al. SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat. Commun. 2016, 7, 12235. [Google Scholar] [CrossRef]
- Xiangyun, Y.; Xiaomin, N.; Linping, G.; Yunhua, X.; Ziming, L.; Yongfeng, Y.; Zhiwei, C.; Shun, L. Desuccinylation of pyruvate kinase M2 by SIRT5 contributes to antioxidant response and tumor growth. Oncotarget 2017, 8, 6984–6993. [Google Scholar] [CrossRef]
- Ko, P.J.; Dixon, S.J. Protein palmitoylation and cancer. EMBO Rep. 2018, 19, e46666. [Google Scholar] [CrossRef]
- Fhu, C.W.; Ali, A. Protein Lipidation by Palmitoylation and Myristoylation in Cancer. Front. Cell Dev. Biol. 2021, 9, 673647. [Google Scholar] [CrossRef]
- Putilina, T.; Wong, P.; Gentleman, S. The DHHC domain: A new highly conserved cysteine-rich motif. Mol. Cell. Biochem. 1999, 195, 219–226. [Google Scholar] [CrossRef]
- Liu, H.; Yan, P.; Ren, J.; Wu, C.; Yuan, W.; Rao, M.; Zhang, Z.; Kong, E. Identifying the Potential Substrates of the Depalmitoylation Enzyme Acyl-protein Thioesterase 1. Curr. Mol. Med. 2019, 19, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Won, S.J.; Davda, D.; Labby, K.J.; Hwang, S.Y.; Pricer, R.; Majmudar, J.D.; Armacost, K.A.; Rodriguez, L.A.; Rodriguez, C.L.; Chong, F.S.; et al. Molecular Mechanism for Isoform-Selective Inhibition of Acyl Protein Thioesterases 1 and 2 (APT1 and APT2). ACS Chem. Biol. 2016, 11, 3374–3382. [Google Scholar] [CrossRef]
- Lin, D.T.; Conibear, E. Enzymatic protein depalmitoylation by acyl protein thioesterases. Biochem. Soc. Trans. 2015, 43, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.A.; Gilman, A.G. A cytoplasmic acyl-protein thioesterase that removes palmitate from G protein alpha subunits and p21(RAS). J. Biol. Chem. 1998, 273, 15830–15837. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.T.; Conibear, E. ABHD17 proteins are novel protein depalmitoylases that regulate N-Ras palmitate turnover and subcellular localization. ELife 2015, 4, e11306. [Google Scholar] [CrossRef]
- Greaves, J.; Chamberlain, L.H. DHHC palmitoyl transferases: Substrate interactions and (patho)physiology. Trends Biochem. Sci. 2011, 36, 245–253. [Google Scholar] [CrossRef]
- Cao, N.; Li, J.K.; Rao, Y.Q.; Liu, H.; Wu, J.; Li, B.; Zhao, P.; Zeng, L.; Li, J. A potential role for protein palmitoylation and zDHHC16 in DNA damage response. BMC Mol. Biol. 2016, 17, 12. [Google Scholar] [CrossRef]
- Fan, X.; Sun, S.; Yang, H.; Ma, H.; Zhao, C.; Niu, W.; Fan, J.; Fang, Z.; Chen, X. SETD2 Palmitoylation Mediated by ZDHHC16 in Epidermal Growth Factor Receptor-Mutated Glioblastoma Promotes Ionizing Radiation-Induced DNA Damage. Int. J. Radiat. Oncol. Biol. Phys. 2022, 113, 648–660. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, Y.; Chen, J.; Zhu, H.; Whiteheart, S.W. Dynamic cycling of t-SNARE acylation regulates platelet exocytosis. J. Biol. Chem. 2018, 293, 3593–3606. [Google Scholar] [CrossRef]
- Chan, P.; Han, X.; Zheng, B.; Deran, M.; Yu, J.; Jarugumilli, G.K.; Deng, H.; Pan, D.; Luo, X.; Wu, X. Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat. Chem. Biol. 2016, 12, 282–289. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, F.; Fu, K.; Liu, X.; Lien, I.C.; Li, H. Potential Role of S-Palmitoylation in Cancer Stem Cells of Lung Adenocarcinoma. Front. Cell Dev. Biol. 2021, 9, 734897. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, C.; Xiao, M.; Han, Y.; Zhang, S.; Xu, B. Bioinformatics Analysis of the Prognostic and Biological Significance of ZDHHC-Protein Acyltransferases in Kidney Renal Clear Cell Carcinoma. Front. Oncol. 2020, 10, 565414. [Google Scholar] [CrossRef]
- Lu, F.; Shen, S.H.; Wu, S.; Zheng, P.; Lin, K.; Liao, J.; Jiang, X.; Zeng, G.; Wei, D. Hypomethylation-induced prognostic marker zinc finger DHHC-type palmitoyltransferase 12 contributes to glioblastoma progression. Ann. Transl. Med. 2022, 10, 334. [Google Scholar] [CrossRef]
- Planey, S.L.; Keay, S.K.; Zhang, C.O.; Zacharias, D.A. Palmitoylation of cytoskeleton associated protein 4 by DHHC2 regulates antiproliferative factor-mediated signaling. Mol. Biol. Cell 2009, 20, 1454–1463. [Google Scholar] [CrossRef]
- Shen, L.F.; Chen, Y.J.; Liu, K.M.; Haddad, A.; Song, I.W.; Roan, H.Y.; Chen, L.Y.; Yen, J.; Chen, Y.J.; Wu, J.Y.; et al. Role of S-Palmitoylation by ZDHHC13 in Mitochondrial function and Metabolism in Liver. Sci. Rep. 2017, 7, 2182. [Google Scholar] [CrossRef]
- Ducker, C.E.; Griffel, L.K.; Smith, R.A.; Keller, S.N.; Zhuang, Y.; Xia, Z.; Diller, J.D.; Smith, C.D. Discovery and characterization of inhibitors of human palmitoyl acyltransferases. Mol. Cancer Ther. 2006, 5, 1647–1659. [Google Scholar] [CrossRef]
- Yeste-Velasco, M.; Mao, X.; Grose, R.; Kudahetti, S.C.; Lin, D.; Marzec, J.; Vasiljević, N.; Chaplin, T.; Xue, L.; Xu, M.; et al. Identification of ZDHHC14 as a novel human tumour suppressor gene. J. Pathol. 2014, 232, 566–577. [Google Scholar] [CrossRef]
- Tian, H.; Lu, J.Y.; Shao, C.; Huffman, K.E.; Carstens, R.M.; Larsen, J.E.; Girard, L.; Liu, H.; Rodriguez-Canales, J.; Frenkel, E.P.; et al. Systematic siRNA Screen Unmasks NSCLC Growth Dependence by Palmitoyltransferase DHHC5. Mol. Cancer Res. 2015, 13, 784–794. [Google Scholar] [CrossRef]
- Chen, X.; Ma, H.; Wang, Z.; Zhang, S.; Yang, H.; Fang, Z. EZH2 Palmitoylation Mediated by ZDHHC5 in p53-Mutant Glioma Drives Malignant Development and Progression. Cancer Res. 2017, 77, 4998–5010. [Google Scholar] [CrossRef]
- Yuan, M.; Song, Z.H.; Ying, M.D.; Zhu, H.; He, Q.J.; Yang, B.; Cao, J. N-myristoylation: From cell biology to translational medicine. Acta Pharmacol. Sin. 2020, 41, 1005–1015. [Google Scholar] [CrossRef]
- Xie, L.; Zeng, J.; Luo, H.; Pan, W.; Xie, J. The roles of bacterial GCN5-related N-acetyltransferases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, J.T.; Habeeb, M.T.; Czub, M.P.; Majorek, K.A.; Arolli, X.; Variot, C.; Anonick, M.; Minor, W.; Ballicora, M.A.; Becker, D.P.; et al. Gcn5-Related N-Acetyltransferases (GNATs) With a Catalytic Serine Residue Can Play Ping-Pong Too. Front. Mol. Biosci. 2021, 8, 646046. [Google Scholar] [CrossRef] [PubMed]
- Albaugh, B.N.; Denu, J.M. Catalysis by protein acetyltransferase Gcn5. Biochim. Biophys. Acta Gene Regul. Mech. 2021, 1864, 194627. [Google Scholar] [CrossRef] [PubMed]
- Kosciuk, T.; Price, I.R.; Zhang, X.; Zhu, C.; Johnson, K.N.; Zhang, S.; Halaby, S.L.; Komaniecki, G.P.; Yang, M.; Dehart, C.J.; et al. NMT1 and NMT2 are lysine myristoyltransferases regulating the ARF6 GTPase cycle. Nat. Commun. 2020, 11, 1067. [Google Scholar] [CrossRef] [PubMed]
- Mackey, J.R.; Lai, J.; Chauhan, U.; Beauchamp, E.; Dong, W.F.; Glubrecht, D.; Sim, Y.W.; Ghosh, S.; Bigras, G.; Lai, R.; et al. N-myristoyltransferase proteins in breast cancer: Prognostic relevance and validation as a new drug target. Breast Cancer Res. Treat 2021, 186, 79–87. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, F.; Li, H.; Zhang, X.; Wu, Q.; Liu, Y.; Qian, M.; Guo, S.; Yang, Y.; Xue, X.; et al. N-Myristoylation by NMT1 Is POTEE-Dependent to Stimulate Liver Tumorigenesis via Differentially Regulating Ubiquitination of Targets. Front. Oncol. 2021, 11, 681366. [Google Scholar] [CrossRef]
- Kumar, S.; Sharma, R.K. N-terminal region of the catalytic domain of human N-myristoyltransferase 1 acts as an inhibitory module. PLoS ONE 2015, 10, e127661. [Google Scholar] [CrossRef]
- Spiegelman, N.A.; Hong, J.Y.; Hu, J.; Jing, H.; Wang, M.; Price, I.R.; Cao, J.; Yang, M.; Zhang, X.; Lin, H. A Small-Molecule SIRT2 Inhibitor That Promotes K-Ras4a Lysine Fatty-Acylation. ChemMedChem 2019, 14, 744–748. [Google Scholar] [CrossRef]
- Matsuno, H.; Tsuchimine, S.; Fukuzato, N.; O’Hashi, K.; Kunugi, H.; Sohya, K. Sirtuin 6 is a regulator of dendrite morphogenesis in rat hippocampal neurons. Neurochem. Int. 2021, 145, 104959. [Google Scholar] [CrossRef]
- Burnaevskiy, N.; Fox, T.G.; Plymire, D.A.; Ertelt, J.M.; Weigele, B.A.; Selyunin, A.S.; Way, S.S.; Patrie, S.M.; Alto, N.M. Proteolytic elimination of N-myristoyl modifications by the Shigella virulence factor IpaJ. Nature 2013, 496, 106–109. [Google Scholar] [CrossRef]
- Schreiber, M.; Baumann, B.; Cotten, M.; Angel, P.; Wagner, E.F. Fos is an essential component of the mammalian UV response. EMBO J. 1995, 14, 5338–5349. [Google Scholar] [CrossRef]
- Jotte, R.M.; Holt, J.T. Myristylation of FBR v-fos dictates the differentiation pathways in malignant osteosarcoma. J. Cell Biol. 1996, 135, 457–467. [Google Scholar] [CrossRef]
- Kim, S.; Alsaidan, O.A.; Goodwin, O.; Li, Q.; Sulejmani, E.; Han, Z.; Bai, A.; Albers, T.; Beharry, Z.; Zheng, Y.G.; et al. Blocking Myristoylation of Src Inhibits Its Kinase Activity and Suppresses Prostate Cancer Progression. Cancer Res. 2017, 77, 6950–6962. [Google Scholar] [CrossRef]
- Bielawska, A.; Bielawski, J.; Szulc, Z.M.; Mayroo, N.; Liu, X.; Bai, A.; Elojeimy, S.; Rembiesa, B.; Pierce, J.; Norris, J.S.; et al. Novel analogs of D-e-MAPP and B13. Part 2: Signature effects on bioactive sphingolipids. Bioorg. Med. Chem. 2008, 16, 1032–1045. [Google Scholar] [CrossRef]
- Díaz, B.; Ostapoff, K.T.; Toombs, J.E.; Lo, J.; Bonner, M.Y.; Curatolo, A.; Adsay, V.; Brekken, R.A.; Arbiser, J.L. Tris DBA palladium is highly effective against growth and metastasis of pancreatic cancer in an orthotopic model. Oncotarget 2016, 7, 51569–51580. [Google Scholar] [CrossRef]
- Elsey, J.; Bubley, J.A.; Zhu, L.; Rao, S.; Sasaki, M.; Pollack, B.P.; Yang, L.; Arbiser, J.L. Palladium based nanoparticles for the treatment of advanced melanoma. Sci. Rep. 2019, 9, 3255. [Google Scholar] [CrossRef]
- Bhandarkar, S.S.; Bromberg, J.; Carrillo, C.; Selvakumar, P.; Sharma, R.K.; Perry, B.N.; Govindarajan, B.; Fried, L.; Sohn, A.; Reddy, K.; et al. Tris (dibenzylideneacetone) dipalladium, a N-myristoyltransferase-1 inhibitor, is effective against melanoma growth in vitro and in vivo. Clin. Cancer Res. 2008, 14, 5743–5748. [Google Scholar] [CrossRef]
- Kolluri, S.K.; Balduf, C.; Hofmann, M.; Göttlicher, M. Novel target genes of the Ah (dioxin) receptor: Transcriptional induction of N-myristoyltransferase 2. Cancer Res. 2001, 61, 8534–8539. [Google Scholar]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef]
- Wang, S.; Mu, G.; Qiu, B.; Wang, M.; Yu, Z.; Wang, W.; Wang, J.; Yang, Y. The Function and related Diseases of Protein Crotonylation. Int. J. Biol. Sci. 2021, 17, 3441–3455. [Google Scholar] [CrossRef]
- Wan, J.; Liu, H.; Ming, L. Lysine crotonylation is involved in hepatocellular carcinoma progression. Biomed. Pharmacother. 2019, 111, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, Z. Histone crotonylation-centric gene regulation. Epigenetics Chromatin. 2021, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Hundertmark, T.; Gärtner, S.; Rathke, C.; Renkawitz-Pohl, R. Nejire/dCBP-mediated histone H3 acetylation during spermatogenesis is essential for male fertility in Drosophila melanogaster. PLoS ONE 2018, 13, e203622. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wei, W.; Liu, Y.; Yang, X.; Wu, J.; Zhang, Y.; Zhang, Q.; Shi, T.; Du, J.X.; Zhao, Y.; et al. MOF as an evolutionarily conserved histone crotonyltransferase and transcriptional activation by histone acetyltransferase-deficient and crotonyltransferase-competent CBP/p300. Cell Discov. 2017, 3, 17016. [Google Scholar] [CrossRef] [PubMed]
- Kollenstart, L.; de Groot, A.; Janssen, G.; Cheng, X.; Vreeken, K.; Martino, F.; Côté, J.; van Veelen, P.A.; van Attikum, H. Gcn5 and Esa1 function as histone crotonyltransferases to regulate crotonylation-dependent transcription. J. Biol. Chem. 2019, 294, 20122–20134. [Google Scholar] [CrossRef]
- Bao, X.; Wang, Y.; Li, X.; Li, X.M.; Liu, Z.; Yang, T.; Wong, C.F.; Zhang, J.; Hao, Q.; Li, X.D. Identification of ‘erasers’ for lysine crotonylated histone marks using a chemical proteomics approach. ELife 2014, 3, e02999. [Google Scholar] [CrossRef]
- Wei, W.; Liu, X.; Chen, J.; Gao, S.; Lu, L.; Zhang, H.; Ding, G.; Wang, Z.; Chen, Z.; Shi, T.; et al. Class I histone deacetylases are major histone decrotonylases: Evidence for critical and broad function of histone crotonylation in transcription. Cell Res. 2017, 27, 898–915. [Google Scholar] [CrossRef]
- Zhao, D.; Li, Y.; Xiong, X.; Chen, Z.; Li, H. YEATS Domain-A Histone Acylation Reader in Health and Disease. J. Mol. Biol. 2017, 429, 1994–2002. [Google Scholar] [CrossRef]
- Schulze, J.M.; Kane, C.M.; Ruiz-Manzano, A. The YEATS domain of Taf14 in Saccharomyces cerevisiae has a negative impact on cell growth. Mol. Genet. Genom. 2010, 283, 365–380. [Google Scholar] [CrossRef]
- Zhao, D.; Guan, H.; Zhao, S.; Mi, W.; Wen, H.; Li, Y.; Zhao, Y.; Allis, C.D.; Shi, X.; Li, H. YEATS2 is a selective histone crotonylation reader. Cell Res. 2016, 26, 629–632. [Google Scholar] [CrossRef]
- Li, Y.; Sabari, B.R.; Panchenko, T.; Wen, H.; Zhao, D.; Guan, H.; Wan, L.; Huang, H.; Tang, Z.; Zhao, Y.; et al. Molecular Coupling of Histone Crotonylation and Active Transcription by AF9 YEATS Domain. Mol. Cell 2016, 62, 181–193. [Google Scholar] [CrossRef]
- Xiong, X.; Panchenko, T.; Yang, S.; Zhao, S.; Yan, P.; Zhang, W.; Xie, W.; Li, Y.; Zhao, Y.; Allis, C.D.; et al. Selective recognition of histone crotonylation by double PHD fingers of MOZ and DPF2. Nat. Chem. Biol. 2016, 12, 1111–1118. [Google Scholar] [CrossRef]
- Abu-Zhayia, E.R.; Machour, F.E.; Ayoub, N. HDAC-dependent decrease in histone crotonylation during DNA damage. J. Mol. Cell Biol. 2019, 11, 804–806. [Google Scholar] [CrossRef]
- Yu, H.; Bu, C.; Liu, Y.; Gong, T.; Liu, X.; Liu, S.; Peng, X.; Zhang, W.; Peng, Y.; Yang, J.; et al. Global crotonylome reveals CDYL-regulated RPA1 crotonylation in homologous recombination-mediated DNA repair. Sci. Adv. 2020, 6, y4697. [Google Scholar] [CrossRef]
- Wan, J.; Liu, H.; Chu, J.; Zhang, H. Functions and mechanisms of lysine crotonylation. J. Cell. Mol. Med. 2019, 23, 7163–7169. [Google Scholar] [CrossRef]
- Han, X.; Xiang, X.; Yang, H.; Zhang, H.; Liang, S.; Wei, J.; Yu, J. p300-Catalyzed Lysine Crotonylation Promotes the Proliferation, Invasion, and Migration of HeLa Cells via Heterogeneous Nuclear Ribonucleoprotein A1. Anal Cell Pathol. 2020, 2020, 5632342. [Google Scholar] [CrossRef]
- Kebede, A.F.; Nieborak, A.; Shahidian, L.Z.; Le Gras, S.; Richter, F.; Gómez, D.A.; Baltissen, M.P.; Meszaros, G.; Magliarelli, H.F.; Taudt, A.; et al. Histone propionylation is a mark of active chromatin. Nat. Struct. Mol. Biol. 2017, 24, 1048–1056. [Google Scholar] [CrossRef]
- Chen, Y.; Sprung, R.; Tang, Y.; Ball, H.; Sangras, B.; Kim, S.C.; Falck, J.R.; Peng, J.; Gu, W.; Zhao, Y. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell. Proteomics 2007, 6, 812–819. [Google Scholar] [CrossRef]
- Sabari, B.R.; Tang, Z.; Huang, H.; Yong-Gonzalez, V.; Molina, H.; Kong, H.E.; Dai, L.; Shimada, M.; Cross, J.R.; Zhao, Y.; et al. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol. Cell 2015, 58, 203–215. [Google Scholar] [CrossRef]
- Kaczmarska, Z.; Ortega, E.; Goudarzi, A.; Huang, H.; Kim, S.; Márquez, J.A.; Zhao, Y.; Khochbin, S.; Panne, D. Structure of p300 in complex with acyl-CoA variants. Nat. Chem. Biol. 2017, 13, 21–29. [Google Scholar] [CrossRef]
- Montgomery, D.C.; Sorum, A.W.; Meier, J.L. Chemoproteomic profiling of lysine acetyltransferases highlights an expanded landscape of catalytic acetylation. J. Am. Chem. Soc. 2014, 136, 8669–8676. [Google Scholar] [CrossRef] [PubMed]
- Dyda, F.; Klein, D.C.; Hickman, A.B. GCN5-related N-acetyltransferases: A structural overview. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 81–103. [Google Scholar] [CrossRef]
- Feldman, J.L.; Baeza, J.; Denu, J.M. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J. Biol. Chem. 2013, 288, 31350–31356. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Andres, O.; Sanchez-Niño, M.D.; Cannata-Ortiz, P.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; Sanz, A.B. Histone lysine crotonylation during acute kidney injury in mice. Dis. Model. Mech. 2016, 9, 633–645. [Google Scholar] [CrossRef]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Vollmuth, F.; Geyer, M. Interaction of propionylated and butyrylated histone H3 lysine marks with Brd4 bromodomains. Angew. Chem. Int. Ed. Engl. 2010, 49, 6768–6772. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, G.; Zheng, X.; Wang, Z.; Xu, X. Post-Translational Modifications by Lipid Metabolites during the DNA Damage Response and Their Role in Cancer. Biomolecules 2022, 12, 1655. https://doi.org/10.3390/biom12111655
Zhu G, Zheng X, Wang Z, Xu X. Post-Translational Modifications by Lipid Metabolites during the DNA Damage Response and Their Role in Cancer. Biomolecules. 2022; 12(11):1655. https://doi.org/10.3390/biom12111655
Chicago/Turabian StyleZhu, Guangrong, Xiangyang Zheng, Zhifeng Wang, and Xingzhi Xu. 2022. "Post-Translational Modifications by Lipid Metabolites during the DNA Damage Response and Their Role in Cancer" Biomolecules 12, no. 11: 1655. https://doi.org/10.3390/biom12111655
APA StyleZhu, G., Zheng, X., Wang, Z., & Xu, X. (2022). Post-Translational Modifications by Lipid Metabolites during the DNA Damage Response and Their Role in Cancer. Biomolecules, 12(11), 1655. https://doi.org/10.3390/biom12111655