
Binding of SARS-CoV Covalent Non-Covalent Inhibitors to the SARS-CoV-2 Papain-Like Protease and Ovarian Tumor Domain Deubiquitinases



Abstract
1. Introduction
2. Results and Discussion
2.1. Covalent OTUB2 Inhibitors
2.1.1. OTUB2 (5QIP) in Complex with PCM-0102153
2.1.2. OTUB2 (5QIQ) in Complex with PCM-0103050
2.1.3. OTUB2 (5QIR) in Complex with PCM-0102305
2.1.4. OTUB2 (5QIS) in Complex with PCM-0102500
2.1.5. OTUB2 (5QIT) in Complex with PCM-0102821
2.1.6. OTUB2 (5QIU) in Complex with PCM-0103011
2.1.7. OTUB2 (5QIV) in Complex with PCM-0102998
2.1.8. OTUB2 (5QIW) in Complex with PCM-0102660
2.1.9. OTUB2 (5QIX) in Complex with PCM-0103007
2.1.10. OTUB2 (5QIY) in Complex with PCM-0102954
2.1.11. OTUB2 (5QIZ) in Complex with PCM-0103080
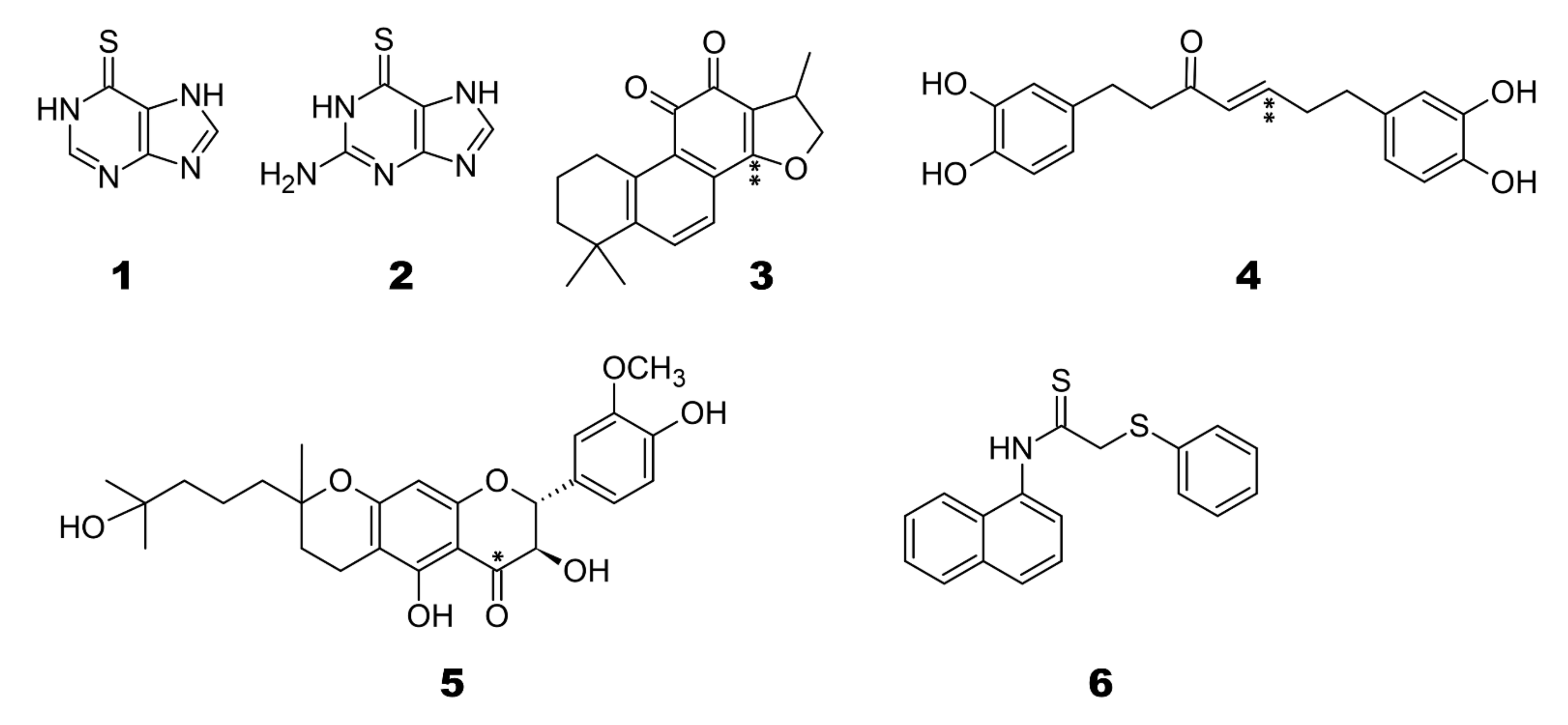
2.2. Binding of Known Covalent SARS-CoV PLpro Inhibitors to OTUB2/OTUB1/SARS-CoV-2 PLpro
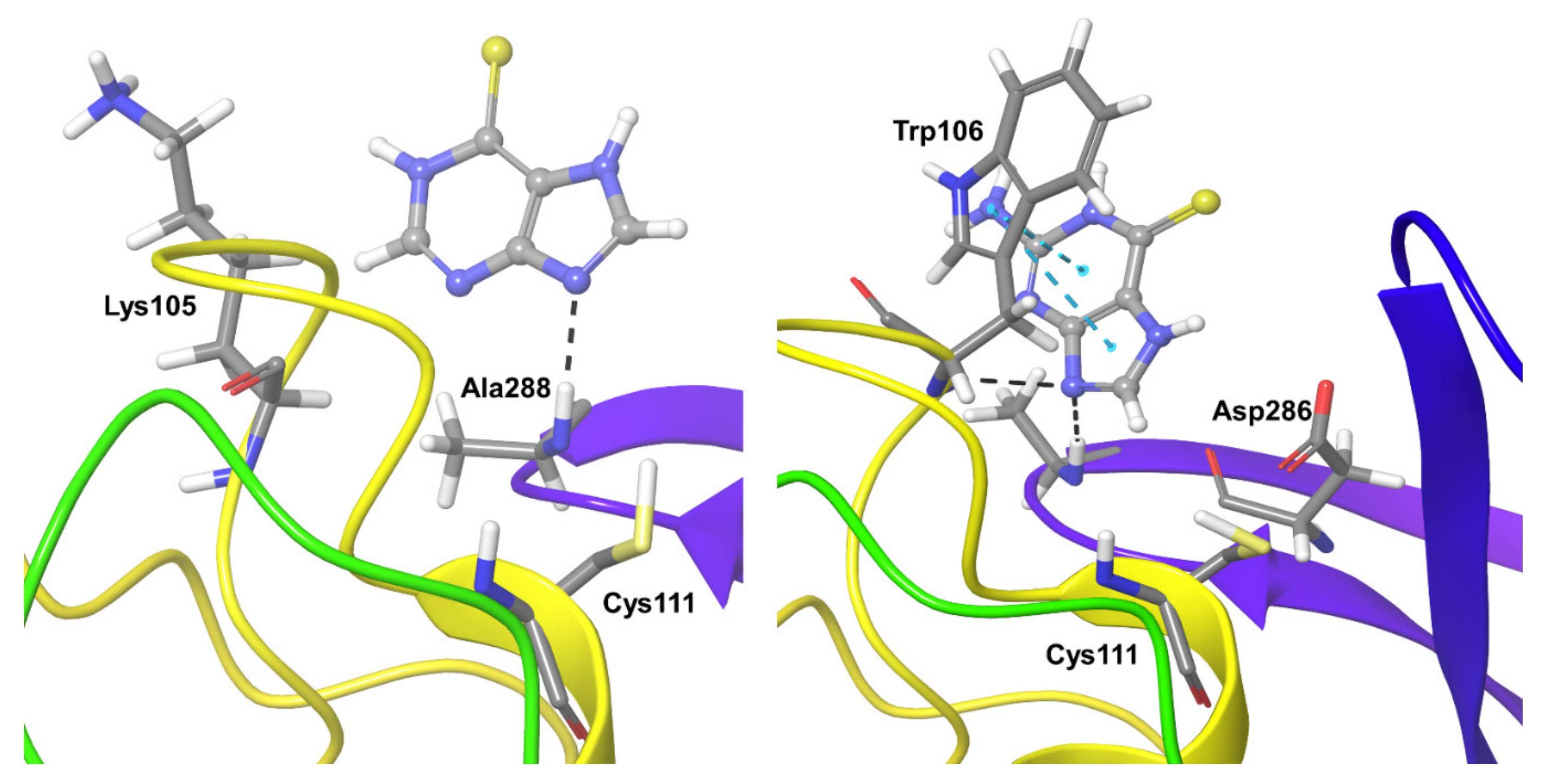
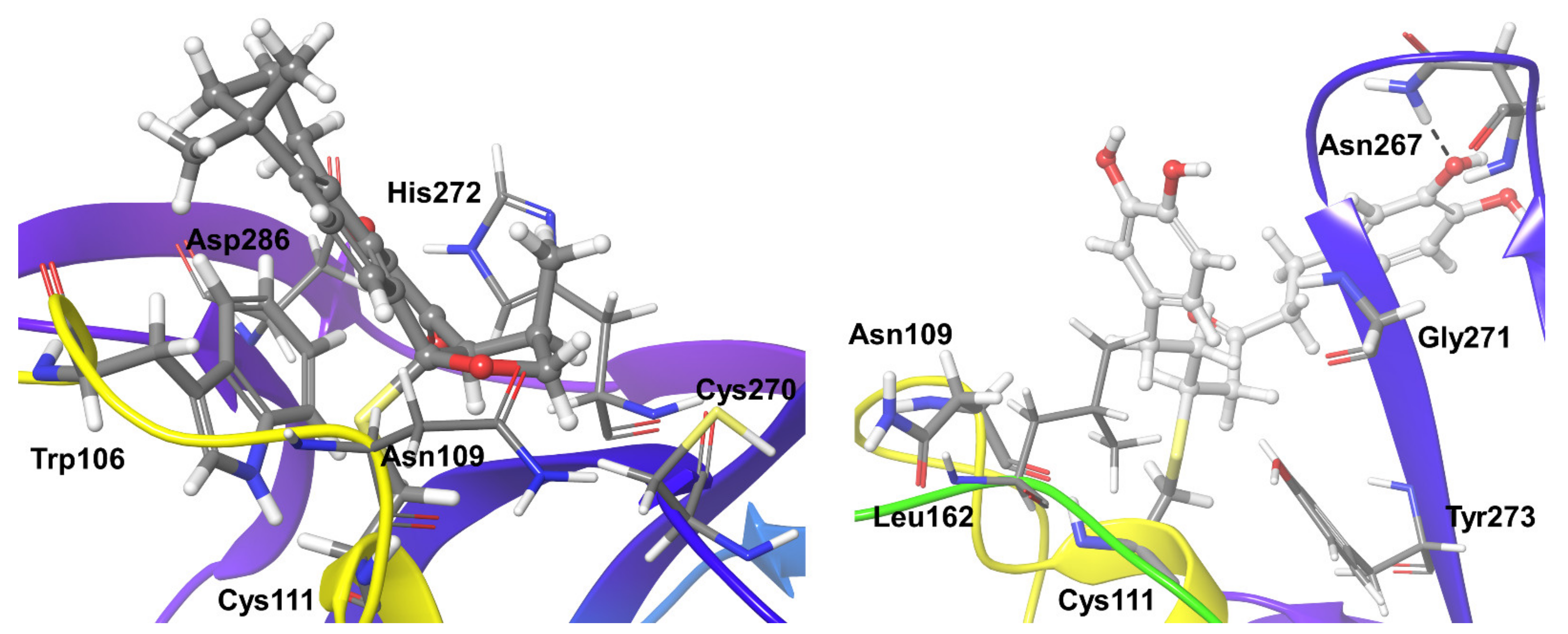
2.3. Molecular Dynamics Refinement of SARS-CoV-2 PLpro Inhibitors
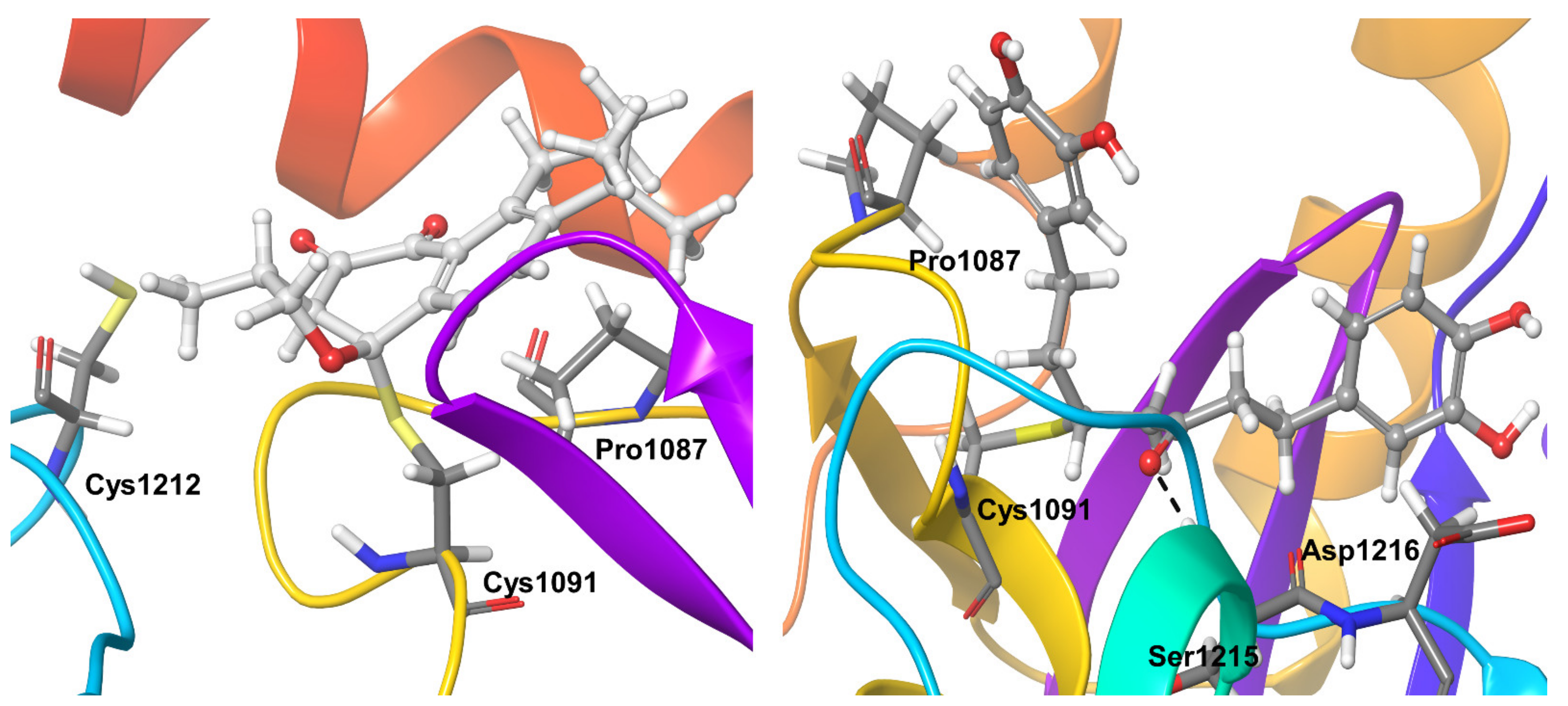
2.4. Molecular Dynamics Refinement of OTUB1-Bound Inhibitors
2.5. Molecular Dynamics Refinement of OTUB-2 Inhibitors
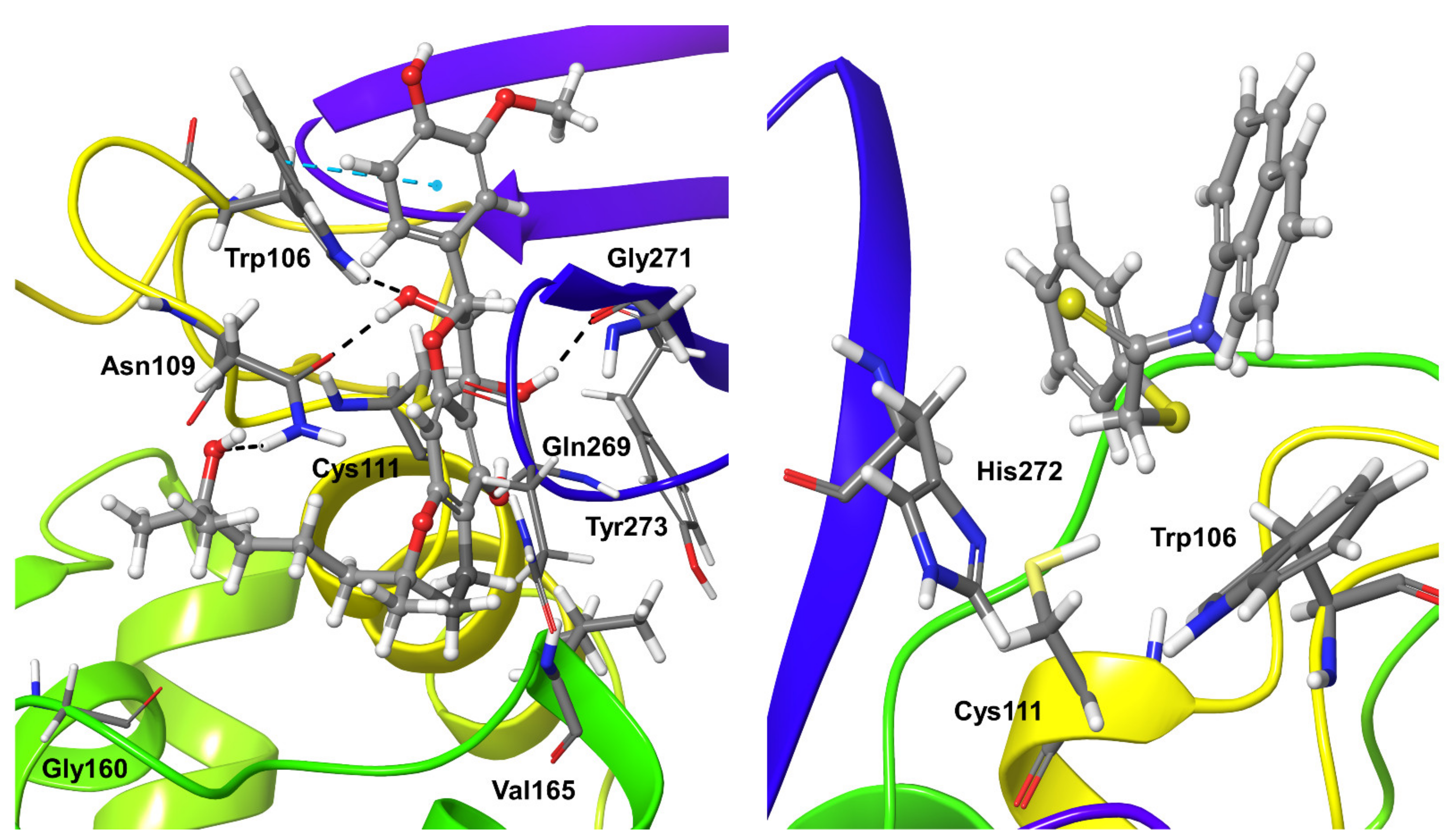
2.6. Molecular Dynamics Refinement of Covalent SARS-CoV-2 PLpro Inhibitors
3. Materials and Methods
3.1. Protein Structures and Preparation
3.2. Covalent and Non-Covalent Ligand Docking
3.3. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (Previously 2019-NCoV) Infection by a Highly Potent Pan-Coronavirus Fusion Inhibitor Targeting Its Spike Protein That Harbors a High Capacity to Mediate Membrane Fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Kneller, D.W.; Phillips, G.; O’Neill, H.M.; Jedrzejczak, R.; Stols, L.; Langan, P.; Joachimiak, A.; Coates, L.; Kovalevsky, A. Structural Plasticity of SARS-CoV-2 3CL M pro Active Site Cavity Revealed by Room Temperature X-Ray Crystallography. Nat. Commun. 2020, 11, 3202. [Google Scholar] [CrossRef] [PubMed]
- Kandeel, M.; Al-Nazawi, M. Virtual Screening and Re-purposing of FDA Approved Drugs against COVID-19 Main Protease. Life Sci. 2020, 251, 117627. [Google Scholar] [CrossRef]
- Ratia, K.; Pegan, S.; Takayama, J.; Sleeman, K.; Coughlin, M.; Baliji, S.; Chaudhuri, R.; Fu, W.; Prabhakar, B.S.; Johnson, M.E.; et al. A Noncovalent Class of Papain-like Protease/Deubiquitinase Inhibitors Blocks SARS Virus Replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16119–16124. [Google Scholar] [CrossRef]
- Van Kasteren, P.B.; Bailey-Elkin, B.A.; James, T.W.; Ninaber, D.K.; Beugeling, C.; Khajehpour, M.; Snijder, E.J.; Mark, B.L.; Kikkert, M. Deubiquitinase Function of Arterivirus Papain-like Protease 2 Suppresses the Innate Immune Response in Infected Host Cells. Proc. Natl. Acad. Sci. USA 2013, 110, E838–E847. [Google Scholar] [CrossRef]
- Ratia, K.; Kilianski, A.; Baez-Santos, Y.M.; Baker, S.C.; Mesecar, A. Structural Basis for the Ubiquitin-Linkage Specificity and DeISGylating Activity of SARS-CoV Papain-like Protease. PLoS Pathog. 2014, 10, e1004113. [Google Scholar] [CrossRef]
- Báez-Santos, Y.M.; St. John, S.E.; Mesecar, A.D. The SARS-Coronavirus Papain-like Protease: Structure, Function and Inhibition by Designed Antiviral Compounds. Antiviral Res. 2015, 115, 21–38. [Google Scholar] [CrossRef]
- Shagufta; Ahmad, I. The Race to Treat COVID-19: Potential Therapeutic Agents for the Prevention and Treatment of SARS-CoV-2. Eur. J. Med. Chem. 2021, 213, 113157. [Google Scholar] [CrossRef]
- Smith, M.; Smith, J.C. Repurposing Therapeutics for COVID-19: Supercomputer-Based Docking to the SARS-CoV-2 Viral Spike Protein and Viral Spike Protein-Human ACE2 Interface. J. Chem. Inf. Model. 2020, 60, 5832–5852. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of Therapeutic Targets for SARS-CoV-2 and Discovery of Potential Drugs by Computational Methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Arya, R.; Das, A.; Prashar, V.; Kumar, M. Potential Inhibitors against Papain-like Protease of Novel Coronavirus (SARS-CoV-2) from FDA Approved Drugs. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like Protease Regulates SARS-CoV-2 Viral Spread and Innate Immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and Inhibition of the Papain-like Protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, e106275. [Google Scholar] [CrossRef]
- Sivakumar, D.; Kumar, V.; Naumann, M.; Stein, M. Activation and Selectivity of OTUB-1 and OTUB-2 Deubiquitinylases. J. Biol. Chem. 2020, 295, 6972–6982. [Google Scholar] [CrossRef]
- Resnick, E.; Bradley, A.; Gan, J.; Douangamath, A.; Krojer, T.; Sethi, R.; Geurink, P.P.; Aimon, A.; Amitai, G.; Bellini, D.; et al. Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. J. Am. Chem. Soc. 2019, 141, 8951–8968. [Google Scholar] [CrossRef]
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking Covalent Inhibitors: A Parameter Free Approach to Pose Prediction and Scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef]
- Klein, P.; Barthels, F.; Johe, P.; Wagner, A.; Tenzer, S.; Distler, U.; Le, T.A.; Schmid, P.; Engel, V.; Engels, B.; et al. Naphthoquinones as Covalent Reversible Inhibitors of Cysteine Proteases—Studies on Inhibition Mechanism and Kinetics. Molecules 2020, 25, 2064. [Google Scholar] [CrossRef]
- Palmer, J.T.; Rasnick, D.; Klaus, J.L.; Brömme, D. Vinyl Sulfones as Mechanism-Based Cysteine Protease Inhibitors. J. Med. Chem. 1995, 38, 3193–3196. [Google Scholar] [CrossRef]
- Johansson, M.H. Reversible Michael Additions: Covalent Inhibitors and Prodrugs. Mini Rev. Med. Chem. 2012, 12, 1330–1344. [Google Scholar] [CrossRef]
- Ettari, R.; Micale, N.; Schirmeister, T.; Gelhaus, C.; Leippe, M.; Nizi, E.; Di Francesco, M.E.; Grasso, S.; Zappalà, M. Novel Peptidomimetics Containing a Vinyl Ester Moiety as Highly Potent and Selective Falcipain-2 Inhibitors. J. Med. Chem. 2009, 52, 2157–2160. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chou, C.-Y.; Chang, G.-G. Thiopurine Analogue Inhibitors of Severe Acute Respiratory Syndrome-Coronavirus Papain-like Protease, a Deubiquitinating and DeISGylating Enzyme. Antivir. Chem. Chemother. 2009, 19, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-Y.; Chien, C.-H.; Han, Y.-S.; Prebanda, M.T.; Hsieh, H.-P.; Turk, B.; Chang, G.-G.; Chen, X. Thiopurine Analogues Inhibit Papain-like Protease of Severe Acute Respiratory Syndrome Coronavirus. Biochem. Pharmacol. 2008, 75, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-Y.; Kim, J.H.; Kim, Y.M.; Jeong, H.J.; Kim, D.W.; Park, K.H.; Kwon, H.-J.; Park, S.-J.; Lee, W.S.; Ryu, Y.B. Tanshinones as Selective and Slow-Binding Inhibitors for SARS-CoV Cysteine Proteases. Bioorg. Med. Chem. 2012, 20, 5928–5935. [Google Scholar] [CrossRef]
- Park, J.-Y.; Jeong, H.J.; Kim, J.H.; Kim, Y.M.; Park, S.-J.; Kim, D.; Park, K.H.; Lee, W.S.; Ryu, Y.B. Diarylheptanoids from Alnus Japonica Inhibit Papain-like Protease of Severe Acute Respiratory Syndrome Coronavirus. Biol. Pharm. Bull. 2012, 35, 2036–2042. [Google Scholar] [CrossRef]
- Jk, C.; Mj, C.-L.; Kh, L.; Dw, K.; Hw, R.; Hj, Y.; Kh, P. Geranylated Flavonoids Displaying SARS-CoV Papain-like Protease Inhibition from the Fruits of Paulownia Tomentosa. Bioorg. Med. Chem. 2013, 21, 3051–3057. [Google Scholar] [CrossRef]
- Frieman, M.; Basu, D.; Matthews, K.; Taylor, J.; Jones, G.; Pickles, R.; Baric, R.; Engel, D.A. Yeast Based Small Molecule Screen for Inhibitors of SARS-CoV. PLoS ONE 2011, 6, e28479. [Google Scholar] [CrossRef]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; Oualid, F.E.; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity Profiling and Crystal Structures of Inhibitor-Bound SARS-CoV-2 Papain-like Protease: A Framework for Anti–COVID-19 Drug Design. Sci. Adv. 2020, 6, eabd4596. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the Comprehensive, Rapid, and Accurate Prediction of the Favorable Tautomeric States of Drug-like Molecules in Aqueous Solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A Software Program for PK(a) Prediction and Protonation State Generation for Drug-like Molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Schrödinger Release 2021-1: LigPrep; Schrödinger, LLC: New York, NY, USA, 2021.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Watts, K.S.; Dalal, P.; Murphy, R.B.; Sherman, W.; Friesner, R.A.; Shelley, J.C. ConfGen: A Conformational Search Method for Efficient Generation of Bioactive Conformers. J. Chem. Inf. Model. 2010, 50, 534–546. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing; Association for Computing Machinery: New York, NY, USA, 2006; p. 84. [Google Scholar]
- Ash, J.; Fourches, D. Characterizing the Chemical Space of ERK2 Kinase Inhibitors Using Descriptors Computed from Molecular Dynamics Trajectories. J. Chem. Inf. Model. 2017, 57, 1286–1299. [Google Scholar] [CrossRef]
- Kaczor, A.A.; Targowska-Duda, K.M.; Patel, J.Z.; Laitinen, T.; Parkkari, T.; Adams, Y.; Nevalainen, T.J.; Poso, A. Comparative Molecular Field Analysis and Molecular Dynamics Studies of α/β Hydrolase Domain Containing 6 (ABHD6) Inhibitors. J. Mol. Model. 2015, 21, 250. [Google Scholar] [CrossRef]
- Sivakumar, D.; Gorai, B.; Sivaraman, T. Screening Efficient BH3-Mimetics to HBcl-B by Means of Peptidodynmimetic Method. Mol. BioSyst. 2013, 9, 700–712. [Google Scholar] [CrossRef]
- Sivakumar, D.; Mudedla, S.; Jang, S.; Kim, H.; Park, H.; Choi, Y.; Oh, J.; Wu, S. Computational Study on Selective PDE9 Inhibitors on PDE9-Mg/Mg, PDE9-Zn/Mg, and PDE9-Zn/Zn Systems. Biomolecules 2021, 11, 709. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Target Protein | Hydrogen Bond Interactions | Hydrophobic Interactions * |
|---|---|---|---|
| 1 | SARS-CoV-2 PLpro | - | - |
| OTUB1 | Pro1263 | His1265 | |
| OTUB2 | Thr222, Asn226 | His224 | |
| 2 | SARS-CoV2 PLpro | Ala288 | - |
| OTUB1 | Glu1060, Pro1263 | - | |
| OTUB2 | Lys221, Thr222, Asn226 | His224 | |
| 3 | SARS-CoV2 PLpro | Cys111 | |
| OTUB1 | - | - | |
| OTUB2 | - | - | |
| 4 | SARS-CoV2 PLpro | Cys111, Gly163, Tyr268 | Tyr264, His272 |
| OTUB1 | Pro1087, Asp1216 | Tyr1261 | |
| OTUB2 | Thr45, Gly47 | His224 | |
| 5 | SARS-CoV2 PLpro | Cys111, Tyr112, Tyr264 | Tyr264, His272 |
| OTUB1 | Gly1264 | - | |
| OTUB2 | Arg49, Ser223 | - | |
| 6 | SARS-CoV2 PLpro | - | His272, Trp106 |
| OTUB1 | Glu1060 | - | |
| OTUB2 | Thr45 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sivakumar, D.; Stein, M. Binding of SARS-CoV Covalent Non-Covalent Inhibitors to the SARS-CoV-2 Papain-Like Protease and Ovarian Tumor Domain Deubiquitinases. Biomolecules 2021, 11, 802. https://doi.org/10.3390/biom11060802
Sivakumar D, Stein M. Binding of SARS-CoV Covalent Non-Covalent Inhibitors to the SARS-CoV-2 Papain-Like Protease and Ovarian Tumor Domain Deubiquitinases. Biomolecules. 2021; 11(6):802. https://doi.org/10.3390/biom11060802
Chicago/Turabian StyleSivakumar, Dakshinamurthy, and Matthias Stein. 2021. "Binding of SARS-CoV Covalent Non-Covalent Inhibitors to the SARS-CoV-2 Papain-Like Protease and Ovarian Tumor Domain Deubiquitinases" Biomolecules 11, no. 6: 802. https://doi.org/10.3390/biom11060802
APA StyleSivakumar, D., & Stein, M. (2021). Binding of SARS-CoV Covalent Non-Covalent Inhibitors to the SARS-CoV-2 Papain-Like Protease and Ovarian Tumor Domain Deubiquitinases. Biomolecules, 11(6), 802. https://doi.org/10.3390/biom11060802