
Identification of Hub Genes and Key Pathways Associated with Anti-VEGF Resistant Glioblastoma Using Gene Expression Data Analysis

Abstract
1. Introduction
2. Materials and Methods
2.1. Microarray Data Collection and Processing
2.2. Differential Expression Analysis and Identification of DEGs
2.3. GO and Pathway Functional Enrichment Analyses
2.4. PPI Network Construction and Module Functional Analysis
2.5. Identification of Hub Genes
2.6. Expression and Survival Analyses of Hub Genes
3. Results
3.1. Identification of DEGs
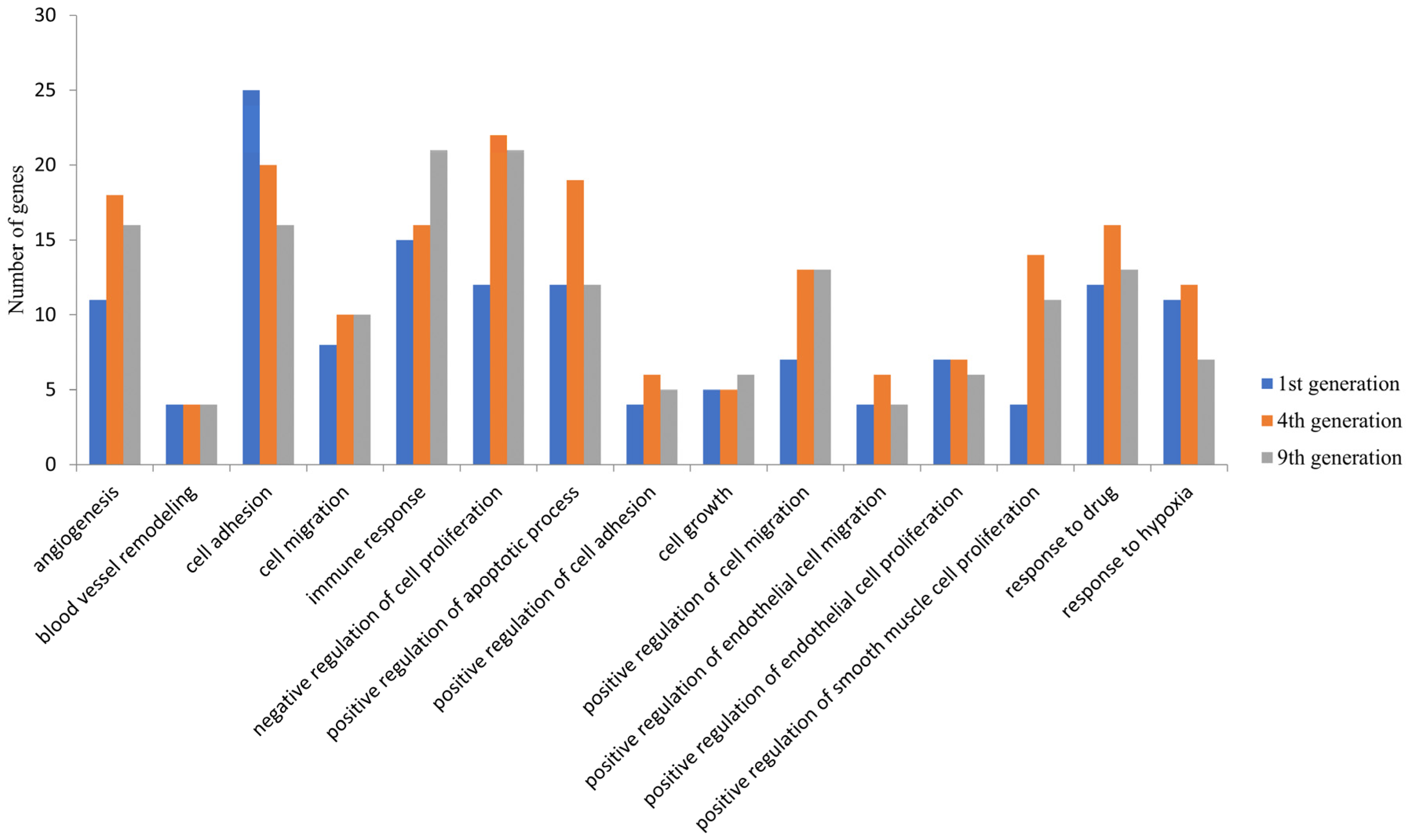
3.2. GO Function and KEGG Pathway Enrichment Analysis
3.3. Classification of Differentially Expressed Genes
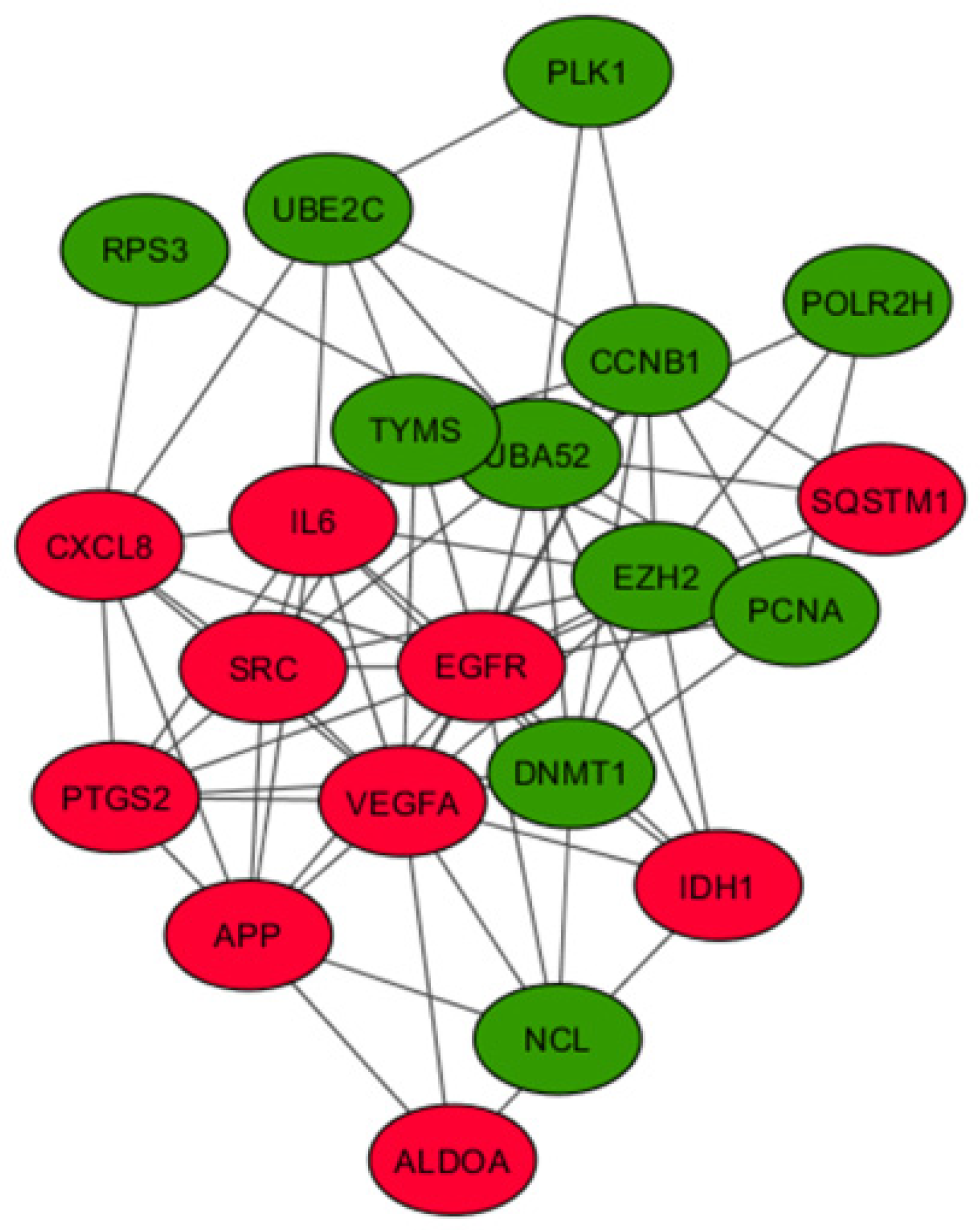
3.4. PPI Network Construction, Module Analysis, and Identification of Hub Genes
3.5. VEGF Pathway Association
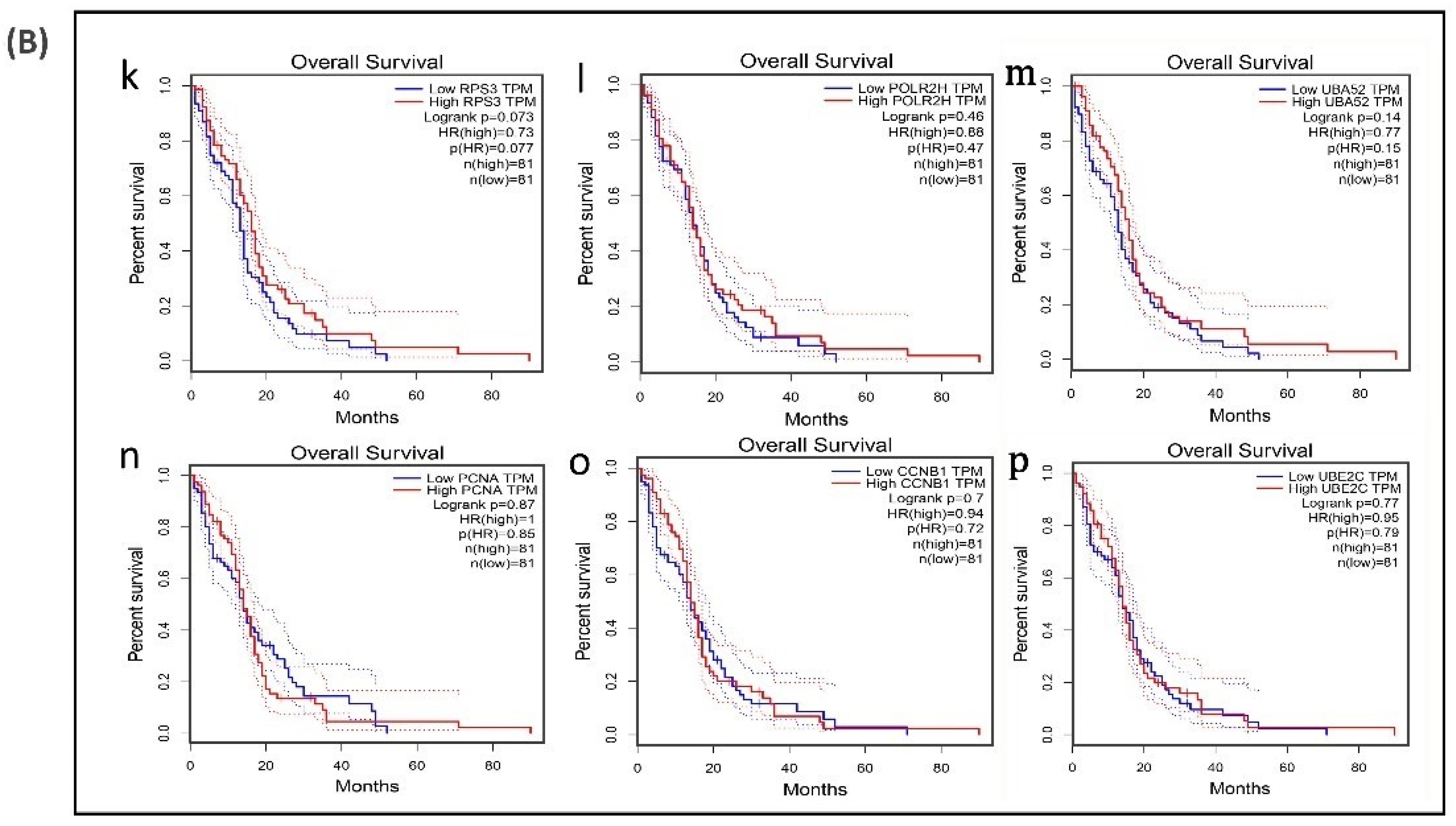
3.6. Survival Analysis of Hub Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Folkman, J. Anti-angiogenesis: New concept for therapy of solid tumors. Ann. Surg. 1972, 175, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I. The vasculature and limb development. Cell Differ. 1985, 16, 1–11. [Google Scholar] [CrossRef]
- Shibuya, M. VEGF-VEGFR Signals in Health and Disease. Biomol. Ther. 2014, 22, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Karamysheva, A.F. Mechanisms of angiogenesis. Biochem. Mosc. 2008, 73, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Lohela, M.; Bry, M.; Tammela, T.; Alitalo, K. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr. Opin. Cell Biol. 2009, 21, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Abhinand, C.S.; Raju, R.; Soumya, S.J.; Arya, P.S.; Sudhakaran, P.R. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J. Cell Commun. Signal. 2016, 10, 347–354. [Google Scholar] [CrossRef]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef]
- Wang, Y.; Fei, D.; Vanderlaan, M.; Song, A. Biological activity of bevacizumab, a humanized anti-VEGF antibody in vitro. Angiogenesis 2004, 7, 335–345. [Google Scholar] [CrossRef]
- Ebos, J.M.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated Metastasis after Short-Term Treatment with a Potent Inhibitor of Tumor Angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic Therapy Elicits Malignant Progression of Tumors to Increased Local Invasion and Distant Metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef]
- Piao, Y.; Liang, J.; Holmes, L.; Henry, V.; Sulman, E.; de Groot, J.F. Acquired Resistance to Anti-VEGF Therapy in Glioblastoma Is Associated with a Mesenchymal Transition. Clin. Cancer Res. 2013, 19, 4392–4403. [Google Scholar] [CrossRef]
- Taylor, O.G.; Brzozowski, J.S.; Skelding, K.A. Glioblastoma Multiforme: An Overview of Emerging Therapeutic Targets. Front. Oncol. 2019, 9, 963. [Google Scholar] [CrossRef]
- Patrizii, M.; Bartucci, M.; Pine, S.R.; Sabaawy, H.E. Utility of Glioblastoma Patient-Derived Orthotopic Xenografts in Drug Discovery and Personalized Therapy. Front. Oncol. 2018, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44. [Google Scholar] [CrossRef]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2012, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.M.; Segal, E.; Koller, D.; Kim, S.K. A Gene-Coexpression Network for Global Discovery of Conserved Genetic Modules. Science 2003, 302, 249–255. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2–27. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Caja, L.; Tzavlaki, K.; Dadras, M.S.; Tan, E.-J.; Hatem, G.; Maturi, N.P.; Morén, A.; Wik, L.; Watanabe, Y.; Savary, K.; et al. Snail regulates BMP and TGFβ pathways to control the differentiation status of glioma-initiating cells. Oncogene 2018, 37, 2515–2531. [Google Scholar] [CrossRef]
- Piao, Y.; Liang, J.; Holmes, L.; Zurita, A.J.; Henry, V.; Heymach, J.V.; de Groot, J.F. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro Oncol. 2012, 14, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Frattini, V.; Bansal, M.; Castano, A.M.; Sherman, D.; Hutchinson, K.; Bruce, J.N.; Califano, A.; Liu, G.; Cardozo, T.; et al. An ID2-dependent mechanism for VHL inactivation in cancer. Nature 2016, 529, 172–177. [Google Scholar] [CrossRef]
- Prayson, R.A.; Castilla, E.A.; Vogelbaum, M.A.; Barnett, G.H. Cyclooxygenase-2 (COX-2) expression by immunohistochemistry in glioblastoma multiforme. Ann. Diagn. Pathol. 2002, 6, 148–153. [Google Scholar] [CrossRef]
- New, P. Cyclooxygenase in the treatment of glioma: Its complex role in signal transduction. Cancer Control 2004, 11, 152–164. [Google Scholar] [CrossRef]
- Qiu, J.; Shi, Z.; Jiang, J. Cyclooxygenase-2 in glioblastoma multiforme. Drug Discov. Today 2017, 22, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Liu, L.; Liu, X.; Qi, L.; Wang, W.; Zhao, G.; Wang, L.; Dai, Y. Activation of PTGS2 /NF-κB signaling pathway enhances radiation resistance of glioma. Cancer Med. 2019, 8, 1175–1185. [Google Scholar] [CrossRef]
- Perez, J.G.; Tran, N.L.; Rosenblum, M.G.; Schneider, C.S.; Connolly, N.P.; Kim, A.J.; Woodworth, G.F.; Winkles, J.A. The TWEAK receptor Fn14 is a potential cell surface portal for targeted delivery of glioblastoma therapeutics. Oncogene 2016, 35, 2145–2155. [Google Scholar] [CrossRef]
- Hersh, D.S.; Harder, B.G.; Roos, A.; Peng, S.; E Heath, J.; Legesse, T.; Kim, A.J.; Woodworth, G.F.; Tran, N.L.; Winkles, J.A. The TNF receptor family member Fn14 is highly expressed in recurrent glioblastoma and in GBM patient-derived xenografts with acquired temozolomide resistance. Neuro Oncol. 2018, 20, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.; Hebda, J.K.; Le Guelte, A.; Galan-Moya, E.-M.; Smith, S.S.; Azzi, S.; Bidère, N.; Gavard, J. Glioblastoma Cell-Secreted Interleukin-8 Induces Brain Endothelial Cell Permeability via CXCR2. PLoS ONE 2012, 7, e45562. [Google Scholar] [CrossRef] [PubMed]
- Infanger, D.W.; Cho, Y.; Lopez, B.S.; Mohanan, S.; Liu, S.C.; Gursel, D.; Boockvar, J.A.; Fischbach, C. Glioblastoma Stem Cells Are Regulated by Interleukin-8 Signaling in a Tumoral Perivascular Niche. Cancer Res. 2013, 73, 7079–7089. [Google Scholar] [CrossRef]
- Luo, X.; Xu, S.; Zhong, Y.; Tu, T.; Xu, Y.; Li, X.; Wang, B.; Yang, F. High gene expression levels of VEGFA and CXCL8 in the peritumoral brain zone are associated with the recurrence of glioblastoma: A bioinformatics analysis. Oncol. Lett. 2019, 18, 6171–6179. [Google Scholar] [CrossRef] [PubMed]
- Nigim, F.; Cavanaugh, J.; Patel, A.P.; Curry, W.T.; Esaki, S.-I.; Kasper, E.M.; Chi, A.S.; Louis, D.N.; Martuza, R.L.; Rabkin, S.D.; et al. Targeting Hypoxia-Inducible Factor 1α in a New Orthotopic Model of Glioblastoma Recapitulating the Hypoxic Tumor Microenvironment. J. Neuropathol. Exp. Neurol. 2015, 74, 710–722. [Google Scholar] [CrossRef]
- Tang, H.; Zhao, J.; Zhang, L.; Zhao, J.; Zhuang, Y.; Liang, P. SRPX2 Enhances the Epithelial—Mesenchymal Transition and Temozolomide Resistance in Glioblastoma Cells. Cell. Mol. Neurobiol. 2016, 36, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Kohsaka, S.; Hinohara, K.; Wang, L.; Nishimura, T.; Urushido, M.; Yachi, K.; Tsuda, M.; Tanino, M.; Kimura, T.; Nishihara, H.; et al. Epiregulin enhances tumorigenicity by activating the ERK/MAPK pathway in glioblastoma. Neuro Oncol. 2014, 16, 960–970. [Google Scholar] [CrossRef]
- Xu, Y.; Kang, J.; Yuan, Z.; Li, H.; Su, J.; Li, Y.; Kong, X.; Zhang, H.; Wang, W.; Sun, L. Suppression of CLIC4/mtCLIC enhances hydrogen peroxide-induced apoptosis in C6 glioma cells. Oncol. Rep. 2013, 29, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Loilome, W.; Joshi, A.D.; Ap Rhys, C.M.J.; Piccirillo, S.G.M.; Angelo, V.L.; Gallia, G.L.; Riggins, G.J. Glioblastoma cell growth is suppressed by disruption of fibroblast growth factor pathway signaling. J. Neuro Oncol. 2009, 94, 359–366. [Google Scholar] [CrossRef]
- Otani, Y.; Ichikawa, T.; Kurozumi, K.; Inoue, S.; Ishida, J.; Oka, T.; Shimizu, T.; Tomita, Y.; Hattori, Y.; Uneda, A.; et al. Fibroblast growth factor 13 regulates glioma cell invasion and is important for bevacizumab-induced glioma invasion. Oncogene 2017, 37, 777–786. [Google Scholar] [CrossRef]
- Jimenez-Pascual, A.; Mitchell, K.; Siebzehnrubl, F.A.; Lathia, J.D. FGF2: A novel druggable target for glioblastoma? Expert Opin. Ther. Targets 2020, 24, 311–318. [Google Scholar] [CrossRef]
- Lewis-Tuffin, L.J.; Feathers, R.; Hari, P.; Durand, N.; Li, Z.; Rodriguez, F.J.; Bakken, K.; Carlson, B.L.; Schroeder, M.; Sarkaria, J.N.; et al. Src family kinases differentially influence glioma growth and motility. Mol. Oncol. 2015, 9, 1783–1798. [Google Scholar] [CrossRef] [PubMed]
- Comba, A.; Kadiyala, P.; E Argento, A.; Patel, P.; Nunez, F.J.; Saxena, M.; Castro, M.G.; Lowenstein, P.R. CSIG-39. Fyn, an oncogene that reduces glioblastoma survival yet sensitizes to chemo-radiotherapy. Neuro Oncol. 2017, 19, vi58. [Google Scholar] [CrossRef]
- Comba, A.; Dunn, P.J.; E Argento, A.; Kadiyala, P.; Ventosa, M.; Patel, P.; Zamler, D.B.; Núñez, F.J.; Zhao, L.; Castro, M.G.; et al. Fyn tyrosine kinase, a downstream target of receptor tyrosine kinases, modulates antiglioma immune responses. Neuro Oncol. 2020, 22, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Wang, Y.; Sun, Y.; Zhou, X. Transcription factor 7 functions as an unfavorable prognostic marker of glioblastoma multiforme by promoting proliferation by upregulating c-Myc. Neuroreport 2018, 29, 745–752. [Google Scholar] [CrossRef]
- Wallmann, T.; Zhang, X.-M.; Wallerius, M.; Bolin, S.; Joly, A.-L.; Sobocki, C.; Leiss, L.; Jiang, Y.; Bergh, J.; Holland, E.C.; et al. Microglia Induce PDGFRB Expression in Glioma Cells to Enhance Their Migratory Capacity. IScience 2018, 9, 71–83. [Google Scholar] [CrossRef]
- Song, K.; Yuan, Y.; Lin, Y.; Wang, Y.-X.; Zhou, J.; Gai, Q.-J.; Zhang, L.; Mao, M.; Yao, X.-X.; Qin, Y.; et al. ERBB3, IGF1R, and TGFBR2 expression correlate with PDGFR expression in glioblastoma and participate in PDGFR inhibitor resistance of glioblastoma cells. Am. J. Cancer Res. 2018, 8, 792–809. [Google Scholar]
- Xu, L.; Chen, Y.; Dutra-Clarke, M.; Mayakonda, A.; Hazawa, M.; Savinoff, S.E.; Doan, N.; Said, J.W.; Yong, W.H.; Watkins, A.; et al. BCL6 promotes glioma and serves as a therapeutic target. Proc. Natl. Acad. Sci. USA 2017, 114, 3981–3986. [Google Scholar] [CrossRef] [PubMed]
- Fabre, M.-S.; Stanton, N.M.; Slatter, T.L.; Lee, S.; Senanayake, D.; Gordon, R.M.A.; Castro, M.L.; Rowe, M.R.; Taha, A.; Royds, J.A.; et al. The oncogene BCL6 is up-regulated in glioblastoma in response to DNA damage, and drives survival after therapy. PLoS ONE 2020, 15, e0231470. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, D.A. Phosphoinositide 3-kinase signalling pathways. J. Cell Sci. 2001, 114, 1439–1445. [Google Scholar] [PubMed]
- Ashcroft, M.; Ludwig, R.L.; Woods, D.B.; Copeland, T.D.; Weber, H.O.; Macrae, E.J.; Vousden, K.H. Phosphorylation of HDM2 by Akt. Oncogene 2002, 21, 1955–1962. [Google Scholar] [CrossRef]
- Zell, J.A.; Cinar, P.; Mobasher, M.; Ziogas, A.; Meyskens, F.L., Jr.; Anton-Culver, H. Survival for Patients with Invasive Cutaneous Melanoma Among Ethnic Groups: The Effects of Socioeconomic Status and Treatment. J. Clin. Oncol. 2008, 26, 66–75. [Google Scholar] [CrossRef]
- Bae, E.; Sato, M.; Kim, R.-J.; Kwak, M.-K.; Naka, K.; Gim, J.; Kadota, M.; Tang, B.; Flanders, K.C.; Kim, T.-A.; et al. Definition of Smad3 Phosphorylation Events That Affect Malignant and Metastatic Behaviors in Breast Cancer Cells. Cancer Res. 2014, 74, 6139–6149. [Google Scholar] [CrossRef]
- Keller, S.; Schmidt, M.H.H. EGFR and EGFRvIII Promote Angiogenesis and Cell Invasion in Glioblastoma: Combination Therapies for an Effective Treatment. Int. J. Mol. Sci. 2017, 18, 1295. [Google Scholar] [CrossRef]
- Wang, X.; Deng, J.; Yuan, J.; Tang, X.; Wang, Y.; Chen, H.; Liu, Y.; Zhou, L. Curcumin exerts its tumor suppressive function via inhibition of NEDD4 oncoprotein in glioma cancer cells. Int. J. Oncol. 2017, 51, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Pandey, V.; Bhaskara, V.K.; Babu, P.P. Implications of mitogen-activated protein kinase signaling in glioma. J. Neurosci. Res. 2015, 94, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Croci, D.O.; Cerliani, J.P.; Dalotto-Moreno, T.; Méndez-Huergo, S.P.; Mascanfroni, I.D.; Dergan-Dylon, S.; Toscano, M.A.; Caramelo, J.J.; García-Vallejo, J.J.; Ouyang, J.; et al. Glycosylation-Dependent Lectin-Receptor Interactions Preserve Angiogenesis in Anti-VEGF Refractory Tumors. Cell 2014, 156, 744–758. [Google Scholar] [CrossRef]
- Croci, D.O.; Cerliani, J.P.; Pinto, N.A.; Morosi, L.G.; Rabinovich, G.A. Regulatory role of glycans in the control of hypoxia-driven angiogenesis and sensitivity to anti-angiogenic treatment. Glycobiology 2014, 24, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef]
- Ahmad, F.; Patrick, S.; Sheikh, T.; Sharma, V.; Pathak, P.; Malgulwar, P.B.; Kumar, A.; Joshi, S.D.; Sarkar, C.; Sen, E. Telomerase reverse transcriptase (TERT)—Enhancer of zeste homolog 2 (EZH2) network regulates lipid metabolism and DNA damage responses in glioblastoma. J. Neurochem. 2017, 143, 671–683. [Google Scholar] [CrossRef]
- Yan, K.; Wu, Q.; Yan, D.H.; Lee, C.H.; Rahim, N.; Tritschler, I.; DeVecchio, J.; Kalady, M.F.; Hjelmeland, A.B.; Rich, J.N. Glioma cancer stem cells secrete Gremlin1 to promote their maintenance within the tumor hierarchy. Genes Dev. 2014, 28, 1085–1100. [Google Scholar] [CrossRef]
- Wu, D.; Lin, J.; Zhu, Y.; Zhang, H.; Zhong, Y. Expression of Testis-Specific Gene Antigen 10 (TSGA10) is Associated with Apoptosis and Cell Migration in Bladder Cancer Cells and Tumor Stage and Overall Survival in Patients with Bladder Cancer. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 5289–5298. [Google Scholar] [CrossRef]
- Bao, L.; You, B.; Shi, S.; Shan, Y.; Zhang, Q.; Yue, H.; Zhang, J.; Zhang, W.; Shi, Y.; Liu, Y.; et al. Metastasis-associated miR-23a from nasopharyngeal carcinoma-derived exosomes mediates angiogenesis by repressing a novel target gene TSGA10. Oncogene 2018, 37, 2873–2889. [Google Scholar] [CrossRef]
- Jaiprasart, P.; Dogra, S.; Neelakantan, D.; Devapatla, B.; Woo, S. Identification of signature genes associated with therapeutic resistance to anti-VEGF therapy. Oncotarget 2020, 11, 99–114. [Google Scholar] [CrossRef]
- Mooney, K.L.; Choy, W.; Sidhu, S.; Pelargos, P.; Bui, T.T.; Voth, B.; Barnette, N.; Yang, I. The role of CD44 in glioblastoma multiforme. J. Clin. Neurosci. 2016, 34, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Toole, B.P. Hyaluronan-CD44 interactions in cancer: Paradoxes and possibilities. Clin. Cancer Res. 2009, 15, 7462–7468. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, S.C.; Guerrero, P.A.; Hirota, S.; Chen, Z.; Morales, J.E.; Aghi, M.; Mccarty, J.H. Neuropilin-1 modulates TGFβ signaling to drive glioblastoma growth and recurrence after anti-angiogenic therapy. PLoS ONE 2017, 12, e0185065. [Google Scholar] [CrossRef] [PubMed]
- Winkler, F.; Osswald, M.; Wick, W. Anti-Angiogenics: Their Role in the Treatment of Glioblastoma. Oncol. Res. Treat. 2017, 41, 181–186. [Google Scholar] [CrossRef]
- Sunitha, P.; Raju, R.; Sajil, C.K.; Abhinand, C.S.; Nair, A.S.; Oommen, O.V.; Sugunan, V.S.; Sudhakaran, P.R. Temporal VEGFA responsive genes in HUVECs: Gene signatures and potential ligands/receptors fine-tuning angiogenesis. J. Cell Commun. Signal. 2019, 13, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Abhinand, C.S.; Athira, P.A.; Soumya, S.J.; Sudhakaran, P.R. Multiple targets directed multiple ligands: An in silico and in vitro approach to evaluating the effect of triphala on angiogenesis. Biomolecules 2020, 10, 177. [Google Scholar] [CrossRef] [PubMed]
- Lamano, J.B.; Lamano, J.B.; Li, Y.D.; DiDomenico, J.D.; Choy, W.; Veliceasa, D.; Oyon, D.E.; Fakurnejad, S.; Ampie, L.; Kesavabhotla, K.; et al. Glioblastoma-Derived IL6 Induces Immunosuppressive Peripheral Myeloid Cell PD-L1 and Promotes Tumor Growth. Clin. Cancer Res. 2019, 25, 3643–3657. [Google Scholar] [CrossRef]
- Li, J.; Liang, R.; Song, C.; Xiang, Y.; Liu, Y. Prognostic significance of epidermal growth factor receptor expression in glioma patients. OncoTargets Ther. 2018, 11, 731–742. [Google Scholar] [CrossRef]
- Chang, C.-Y.; Li, M.-C.; Liao, S.-L.; Huang, Y.-L.; Shen, C.-C.; Pan, H.-C. Prognostic and clinical implication of IL-6 expression in glioblastoma multiforme. J. Clin. Neurosci. 2005, 12, 930–933. [Google Scholar] [CrossRef]
- Kim, W.; Youn, H.; Lee, S.; Kim, E.; Kim, D.; Lee, J.S.; Lee, J.-M.; Youn, B. RNF138-mediated ubiquitination of rpS3 is required for resistance of glioblastoma cells to radiation-induced apoptosis. Exp. Mol. Med. 2018, 50, e434. [Google Scholar] [CrossRef] [PubMed]
- Sharma, I.; Singh, A.; Sharma, K.C.; Saxena, S. Gene Expression Profiling of Chemokines and Their Receptors in Low and High Grade Astrocytoma. Asian Pac. J. Cancer Prev. 2017, 18, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Treps, L.; Perret, R.; Edmond, S.; Ricard, D.; Gavard, J. Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J. Extracell. Vesicles 2017, 6, 1359479. [Google Scholar] [CrossRef]
- Polivka, J.; Rohan, V.; Pesta, M.; Repik, T.; Pitule, P.; Topolčan, O. Isocitrate Dehydrogenase-1 Mutations as Prognostic Biomarker in Glioblastoma Multiforme Patients in West Bohemia. BioMed Res. Int. 2014, 2014, 735659. [Google Scholar] [CrossRef] [PubMed]
- Niyazi, M.; Harter, P.N.; Hattingen, E.; Rottler, M.; Von Baumgarten, L.; Proescholdt, M.; Belka, C.; Lauber, K.; Mittelbronn, M. Bevacizumab and radiotherapy for the treatment of glioblastoma: Brothers in arms or unholy alliance? Oncotarget 2015, 7, 2313–2328. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DEGs | Gene Symbol |
|---|---|
| Upregulated | ACSL3, ACTA2, ADGRG1, AKR1B1, ASAP2, ASS1, ATP2B4, BAALC, BHLHE40, CALD1, CCDC92, CLIC4, CSF3, CXCL8, FAM65A, FGF2, HIST2H2BE, HLA-B, HLA-DPA1, HLA-DRA, IDH1, IER3, IGFBP7, IL1B, IL6, INSIG1, KDELR3, LRRN3, MLLT11, MSMO1, MTSS1, NAMPT, NME4, NOTCH3, PAMR1, PDGFRB, PHKB, PPP1R3C, PTGS2, RCAN1, RFTN2, RHBDF1, RND3, RRAD, RSPO3, SEZ6L2, SH3BGRL, SMARCA1, SNX7, SPP1, SRPX2, SYNC, SYNDIG1, TAP1, TFPI2, TGM2, TMCO3, TMEM171, TMEM98, TOX2, TSPAN13, VAT1L |
| Downregulated | EIF6, NTSR1, IMP4, RPPH1, TOP3A, LYAR, THOC5, CXADR, FOXR2, DHRS11, DBNDD2, DDX27, DNAAF3, ALKBH2, IPO4, DUS3, MRPL12, PRPF19, INTS1, DDX18, FERMT3, EMG1, SNU13, SERPINB7, RPS19BP1, UBL7, RPS21, CCL26, SLC43A3, IL13RA2, NHP2, SNAPC4, GRWD1, PHB2, MRTO4, PDSS1, SPOUT1, DNLZ, GPATCH4, CCL20, EIF2B2, ZNF593, NOP16, NUDT14, MCM10, SLIRP, RRS1, KLRG1, ATF5, CDK4, COQ3, GEMIN4, GEMIN6, FTSJ3, PAK1IP1, MRPS26, NTS, TOMM22, EXOSC5, ATAD3A, GAR1, RPF2, TEAD4, PPAN, UBE2G2, C9orf142, PRPF4, PPIH, RPL36, TTLL12, WDR46, PSMG3, PRIM1, CSTF2, TOMM6, AHSA1, TFB2M, SLC5A6, NOP56, TIMM10, C10orf2, CLPTM1L, DPH2, C19orf48, FEN1, UBA52, FARSA, ELOF1, S100A2, NOL6, STOML2, ADSL, CHCHD10, POLR2H, PA2G4, E2F2, PRKAR1B, LBHD1, PRELID1, MRPL21, EXOSC4, TRIML2, NPM3, SNORD104, EBNA1BP2, WDR18, EIF3K, STC2, POLD1, SFXN4, SNORA67, METTL17, ATIC, EIF3G, SDCCAG3, MAGEB2, PNP, HCLS1, NDUFB9, DHRS2, CDC25A, ESM1 |
| Both up- and downregulated | CRABP2, EDNRA, GJB2, IGFBP5, MATN2, MFAP4, NREP, OLFML2A, PCOLCE, PDGFRA, RGS2, SERPINB2, TGFB3, TIMP2, TRIM9 |
| DEGs Classification | Gene Symbol |
|---|---|
| Growth factors | HBEGF, VEGFA, EGFR, PDGFRB, FGF13, FGF2, TFPI, TFPI2, TGFA, TGIF1, WNT5A, BDNF, NRG1, TIMP1, PDGFRA, TGFB3 |
| Cytokines | CSF3, NAMPT, CCL3, IL6, CXCL2, CXCL8, GREM1, IL11, IL36RN, LIF, CLCF1, CCL3L3, IL36B, IL1B, IL1A, SPP1, IL1R1, NRG1, EREG |
| BMPs | BMP6, GREM1, NBL1, TWSG1 |
| Proto-oncogenes | FYN, MLLT11, PDGFRB, BCL6, SRC, CBLB, CRKL |
| Genes involved in GBM | ESM1, PDGFRA, TGFB3 |
| Receptor–Ligand pairs | CD44-SPP1, CD44-HBEGF, CD44-FGF2, F3-IL6, F3-TFPI, IL6ST-CLCF1, IL6ST-IL6, IL6ST-LIF, IL6ST-IL11, ITGB1-SPP1, ITGB1-THBS1, ITGB1-VEGFA, NRP2-VEGFA, PLAUR -SERPINE1, EGFR-EREG, EGFR-FGF13, EGFR-HBEGF, EGFR-TGFA, EGFR-VEGFA |
| Enzymes (Glycosyl transferases) | GBE1, PLOD2, HAS1, ST3GAL1, EDEM1 |
| Term | Description | Count | p-Value | Gene Symbol |
|---|---|---|---|---|
| Module 3 | ||||
| hsa04110 | Cell cycle | 9 | 6.28 × 10−7 | E2F2, CDKN1A, SKP2, MCM2, MCM3, CDK4, CDC25A, CDC25B, MCM6 |
| hsa04060 | Cytokine–cytokine receptor interaction | 7 | 0.003 | CXCL1, CCL3, CCL20, CXCL2, IL1B, IL1A, IL11 |
| hsa05206 | MicroRNAs in cancer | 7 | 0.007 | E2F2, CDKN1A, PTGS2, HMOX1, THBS1, CDC25A, CDC25B |
| hsa04668 | TNF signaling pathway | 6 | 4.46 × 10−4 | CXCL1, PTGS2, CCL20, CXCL2, IL1B, MMP3 |
| hsa05200 | Pathways in cancer | 6 | 0.049 | E2F2, CDKN1A, HIF1A, PTGS2, SKP2, CDK4 |
| hsa05219 | Bladder cancer | 5 | 1.03 × 10−4 | E2F2, CDKN1A, THBS1, CDK4, SRC |
| hsa04066 | HIF1 signaling pathway | 4 | 0.020 | CDKN1A, HIF1A, HMOX1, SERPINE1 |
| hsa04621 | NOD-like receptor signaling pathway | 3 | 0.045 | CXCL1, CXCL2, IL1B |
| hsa05214 | Glioma | 3 | 0.049 | E2F2, CDKN1A, CDK4 |
| Module 4 | ||||
| hsa05200 | Pathways in cancer | 6 | 0.002 | VEGFA, TGFA, PDGFRB, JAK1, FGF13, FGF2 |
| hsa04151 | PI3K-Akt signaling pathway | 5 | 0.008 | VEGFA, PDGFRB, JAK1, FGF13, FGF2 |
| hsa05212 | Pancreatic cancer | 3 | 0.011 | VEGFA, TGFA, JAK1 |
| hsa05205 | Proteoglycans in cancer | 4 | 0.012 | VEGFA, HBEGF, SDC4, FGF2 |
| hsa05218 | Melanoma | 3 | 0.013 | PDGFRB, FGF13, FGF2 |
| hsa04015 | Rap1 signaling pathway | 4 | 0.014 | VEGFA, PDGFRB, FGF13, FGF2 |
| hsa04014 | Ras signaling pathway | 4 | 0.017 | VEGFA, PDGFRB, FGF13, FGF2 |
| hsa04010 | MAPK signaling pathway | 4 | 0.023 | DUSP1, PDGFRB, FGF13, FGF2 |
| hsa05219 | Bladder cancer | 2 | 0.038 | VEGFA, HBEGF |
| DEGs | Gene Symbol |
|---|---|
| Upregulated | IL6, VEGFA, SRC, APP, CXCL8, IDH1, SQSTM1, EGFR, PTGS2, ALDOA |
| Downregulated | NCL, RPS3, UBA52, DNMT1, CCNB1, EZH2, PLK1, POLR2H, UBE2C, TYMS, PCNA |
| Table | Description | Count | p-Value | Gene Symbol |
|---|---|---|---|---|
| hsa05219 | Bladder cancer | 4 | 0.0002 | CXCL8, SRC, EGFR, VEGFA |
| hsa04068 | FoxO signaling pathway | 4 | 0.006 | IL6, CCNB1, PLK1, EGFR |
| hsa05206 | MicroRNAs in cancer | 5 | 0.008 | DNMT1, PTGS2, EGFR, EZH2, VEGFA |
| hsa04370 | VEGF signaling pathway | 3 | 0.013 | SRC, PTGS2, VEGFA |
| hsa05120 | Epithelial cell signaling in Helicobacter pylori infection | 3 | 0.016 | CXCL8, SRC, EGFR |
| hsa05200 | Pathways in cancer | 5 | 0.024 | IL6, CXCL8, PTGS2, EGFR, VEGFA |
| hsa04066 | HIF1 signaling pathway | 3 | 0.031 | IL6, EGFR, VEGFA |
| hsa04110 | Cell cycle | 3 | 0.049 | CCNB1, PCNA, PLK1 |
| Upregulated Hub Genes | Survival Rate (in Months) | Log-Rank p | p(HR) | Downregulated Hub Genes | Survival Rate (in Months) | Log-Rank p | p (HR) |
|---|---|---|---|---|---|---|---|
| IL6 | 49 | 0.07 | 0.074 | PCNA | 52 | 0.87 | 0.85 |
| VEGFA | 49 | 0.05 | 0.046 | POLR2H | 52 | 0.46 | 0.47 |
| SRC | 52 | 0.24 | 0.25 | RPS3 | 52 | 0.07 | 0.077 |
| CXCL8 | 49 | 0.04 | 0.05 | UBA52 | 52 | 0.14 | 0.15 |
| IDH1 | 49 | 0.03 | 0.029 | CCNB1 | 71 | 0.7 | 0.72 |
| PTGS2 | 52 | 0.54 | 0.54 | UBE2C | 71 | 0.77 | 0.79 |
| EGFR | 49 | 0.93 | 0.9 | ||||
| APP | 71 | 0.46 | 0.47 | ||||
| ALDOA | 71 | 0.11 | 0.11 | ||||
| SQSTM1 | 71 | 0.13 | 0.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arya, K.R.; Bharath Chand, R.P.; Abhinand, C.S.; Nair, A.S.; Oommen, O.V.; Sudhakaran, P.R. Identification of Hub Genes and Key Pathways Associated with Anti-VEGF Resistant Glioblastoma Using Gene Expression Data Analysis. Biomolecules 2021, 11, 403. https://doi.org/10.3390/biom11030403
Arya KR, Bharath Chand RP, Abhinand CS, Nair AS, Oommen OV, Sudhakaran PR. Identification of Hub Genes and Key Pathways Associated with Anti-VEGF Resistant Glioblastoma Using Gene Expression Data Analysis. Biomolecules. 2021; 11(3):403. https://doi.org/10.3390/biom11030403
Chicago/Turabian StyleArya, Kesavan R., Ramachandran P. Bharath Chand, Chandran S. Abhinand, Achuthsankar S. Nair, Oommen V. Oommen, and Perumana R. Sudhakaran. 2021. "Identification of Hub Genes and Key Pathways Associated with Anti-VEGF Resistant Glioblastoma Using Gene Expression Data Analysis" Biomolecules 11, no. 3: 403. https://doi.org/10.3390/biom11030403
APA StyleArya, K. R., Bharath Chand, R. P., Abhinand, C. S., Nair, A. S., Oommen, O. V., & Sudhakaran, P. R. (2021). Identification of Hub Genes and Key Pathways Associated with Anti-VEGF Resistant Glioblastoma Using Gene Expression Data Analysis. Biomolecules, 11(3), 403. https://doi.org/10.3390/biom11030403