


Cyanide Molecular Laser-Induced Breakdown Spectroscopy with Current Databases


Abstract
1. Introduction
2. Materials and Methods
2.1. Traditional Simulation of Diatomic Molecular Spectra
2.2. Line Positions and Strengths of Diatomic Spectra
2.3. Wigner–Witmer Diatomic Eigenfunction
2.4. Diatomic Line Position Fitting Algorithm
2.5. Measured Air Wavelength vs. Vacuum Wavenumbers
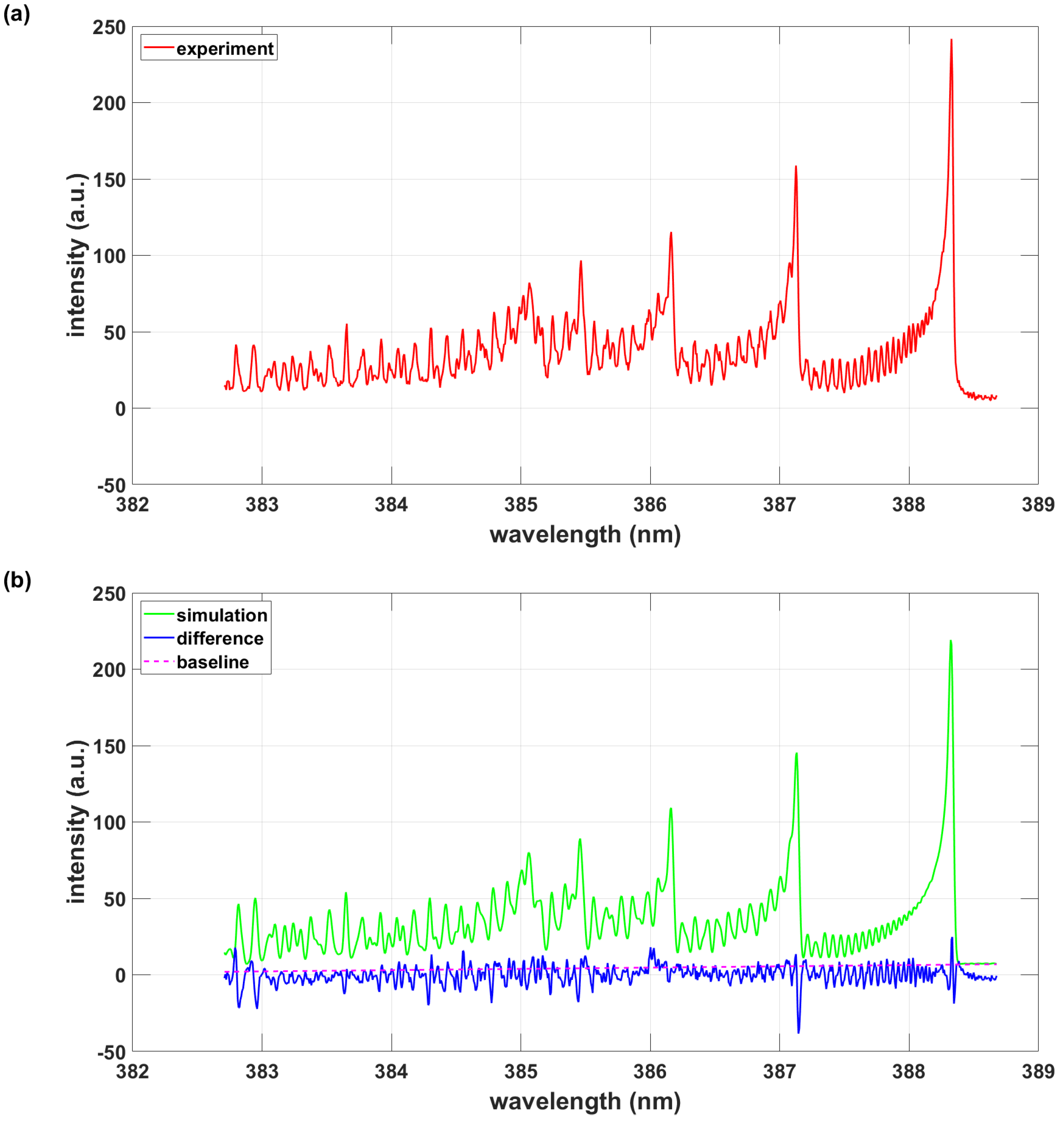
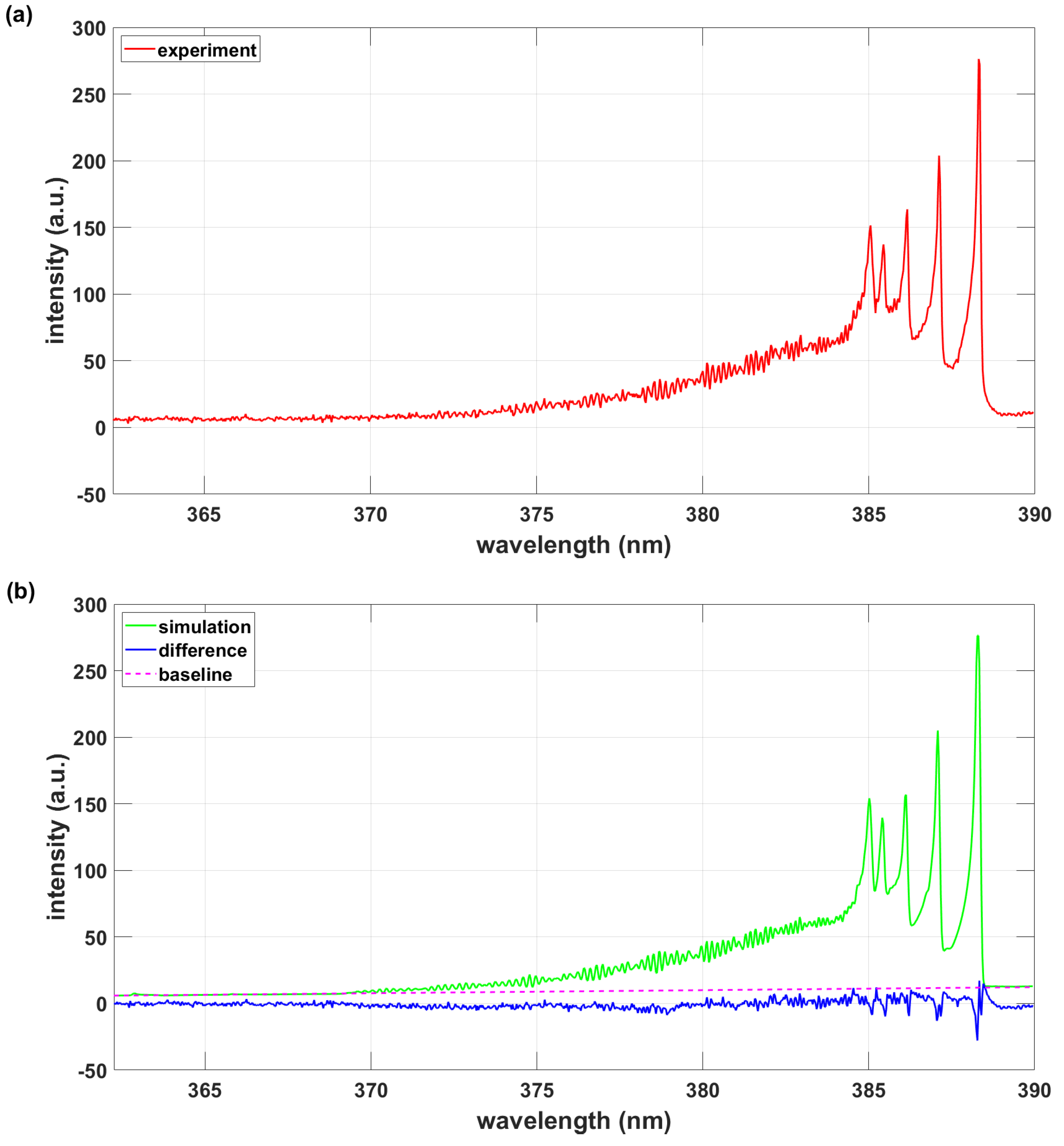
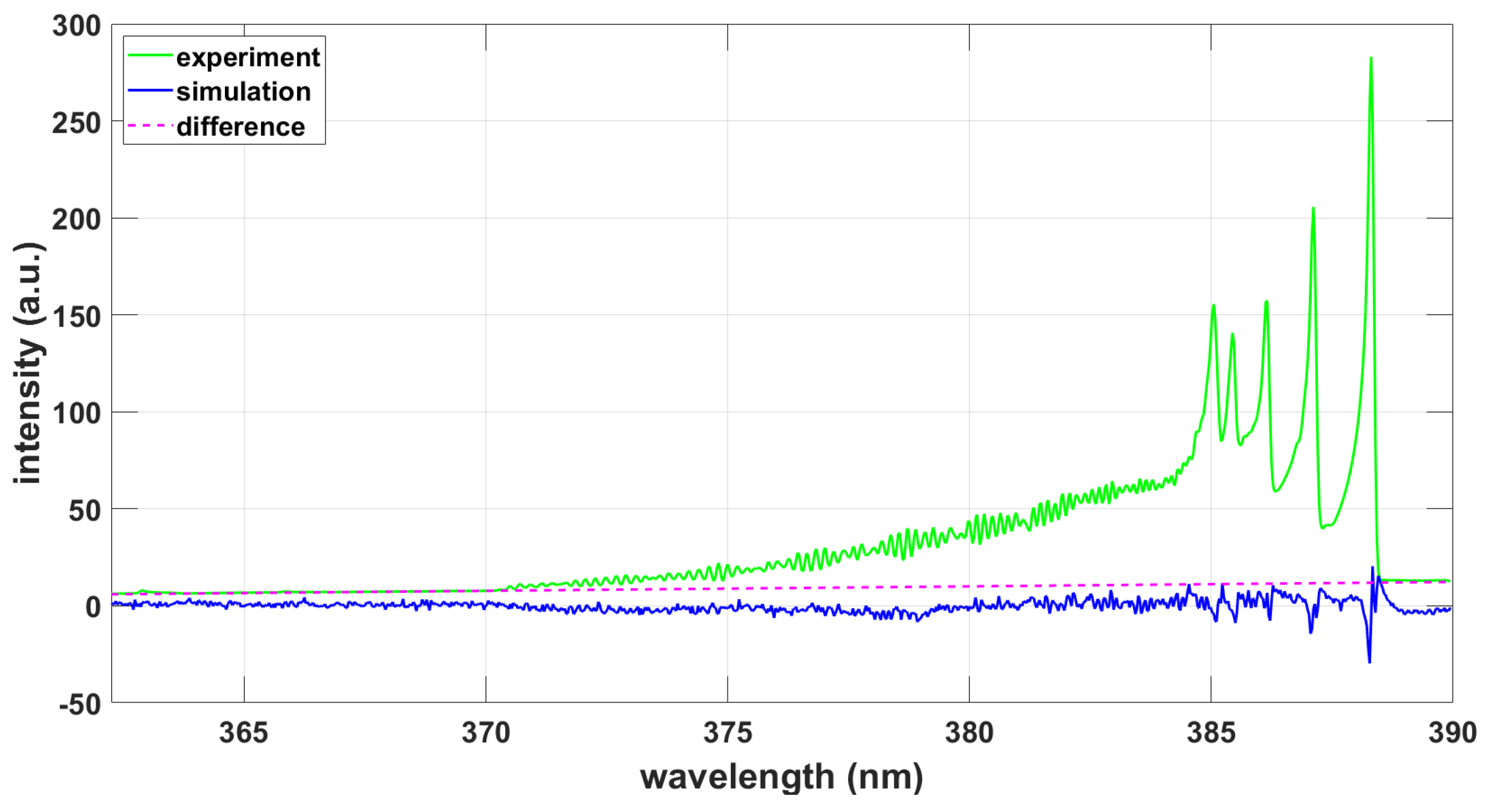
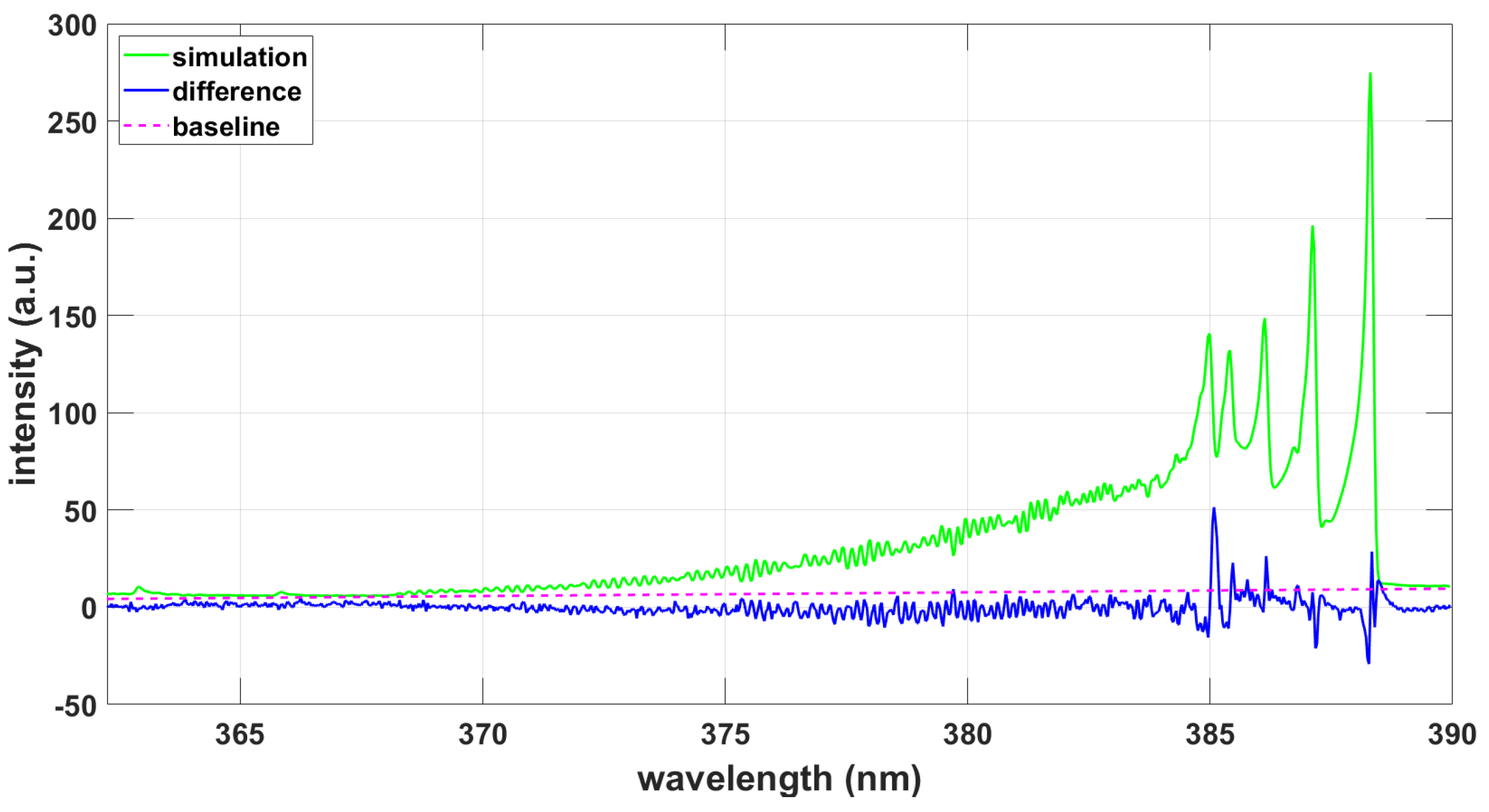
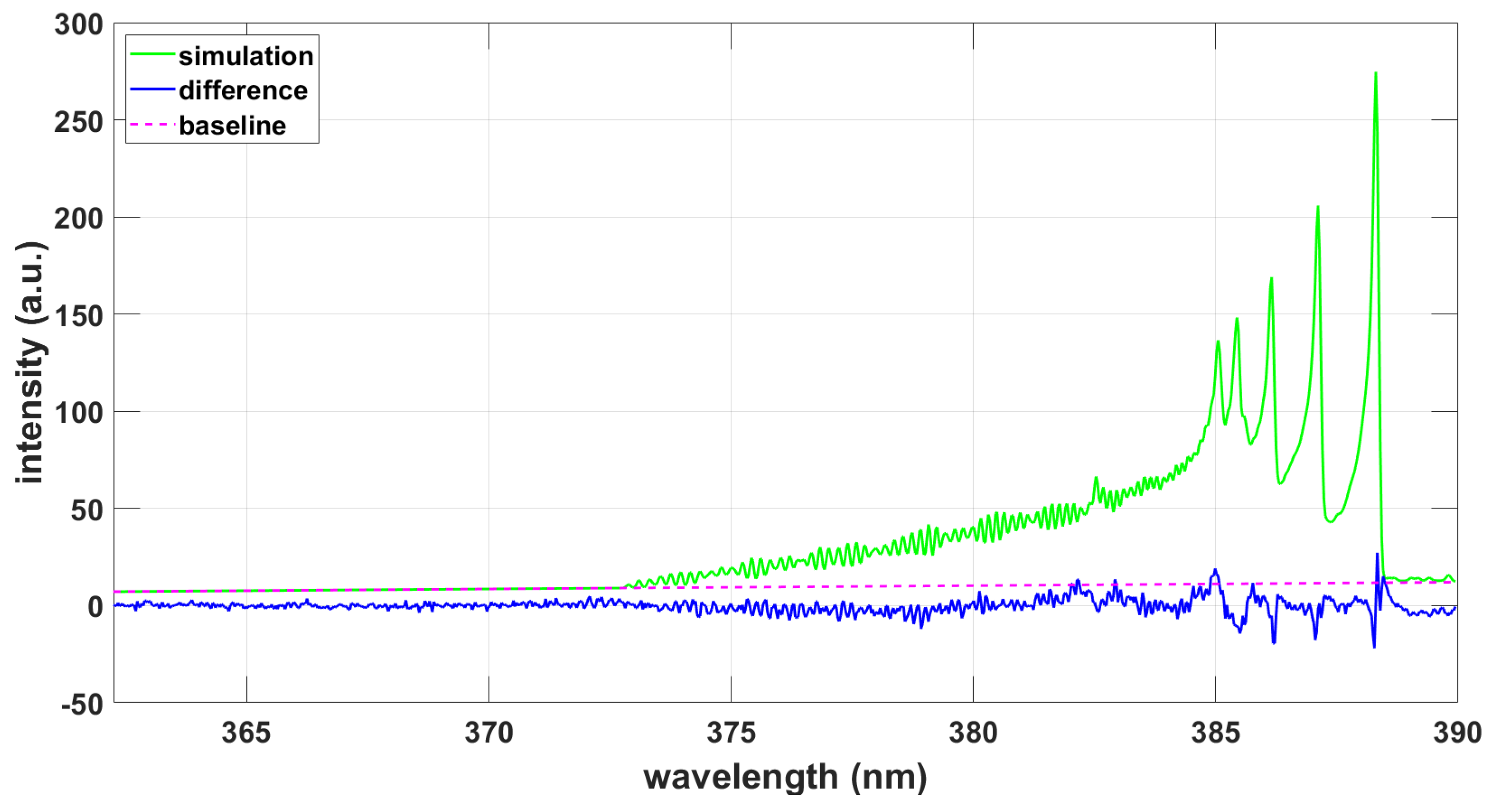
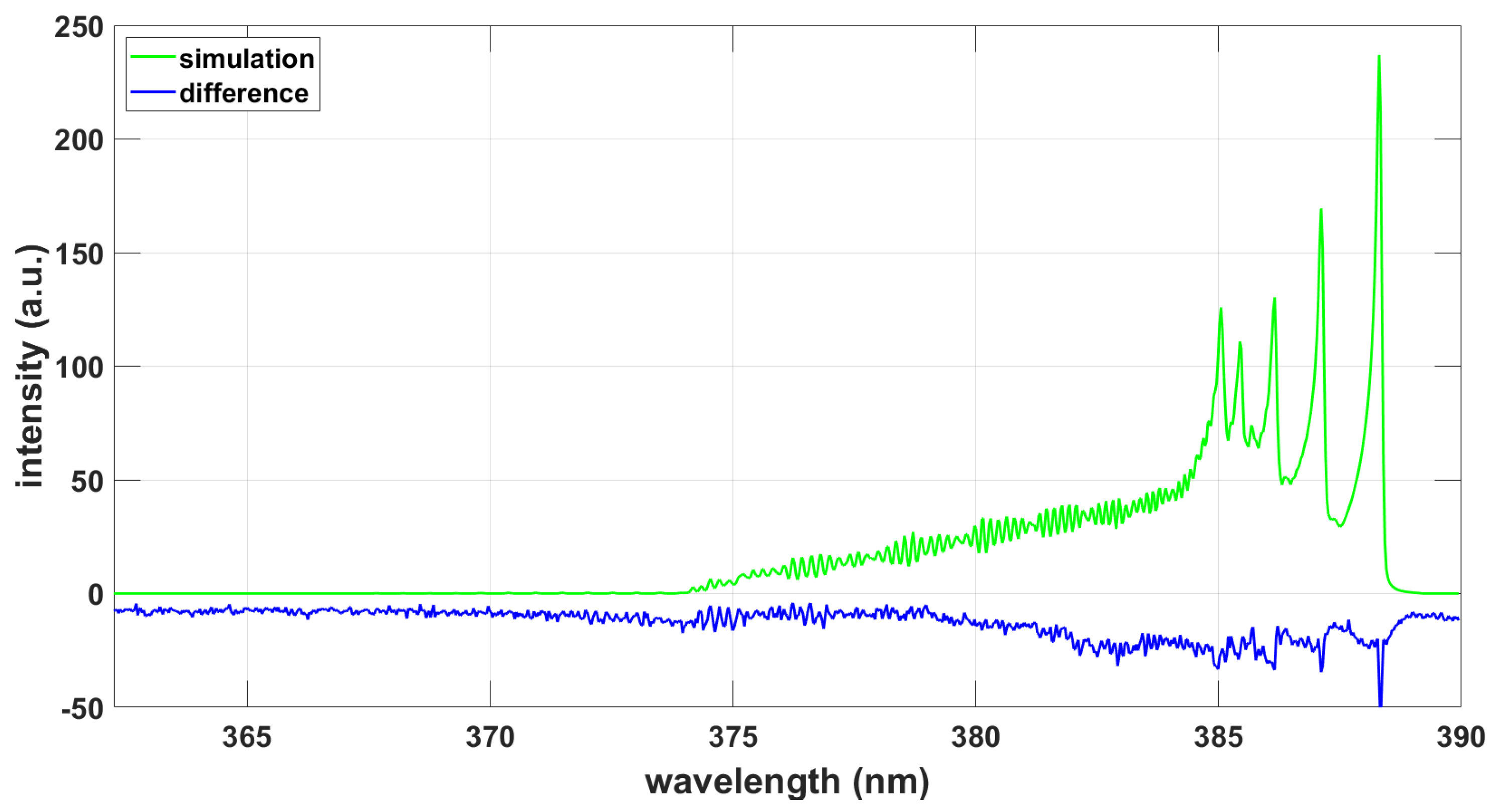
3. Results
- The PGOPHER program using PGOPHER data,
- The NMT program using PGOPHER- and ExoMol line strengths,
- The NMT program using CNv-lsf data, comparisons,
- The LIFBASE program.
3.1. Analysis of the 0.033 nm Spectral Resolution Data
3.1.1. PGOPHER Program Using PGOPHER Data
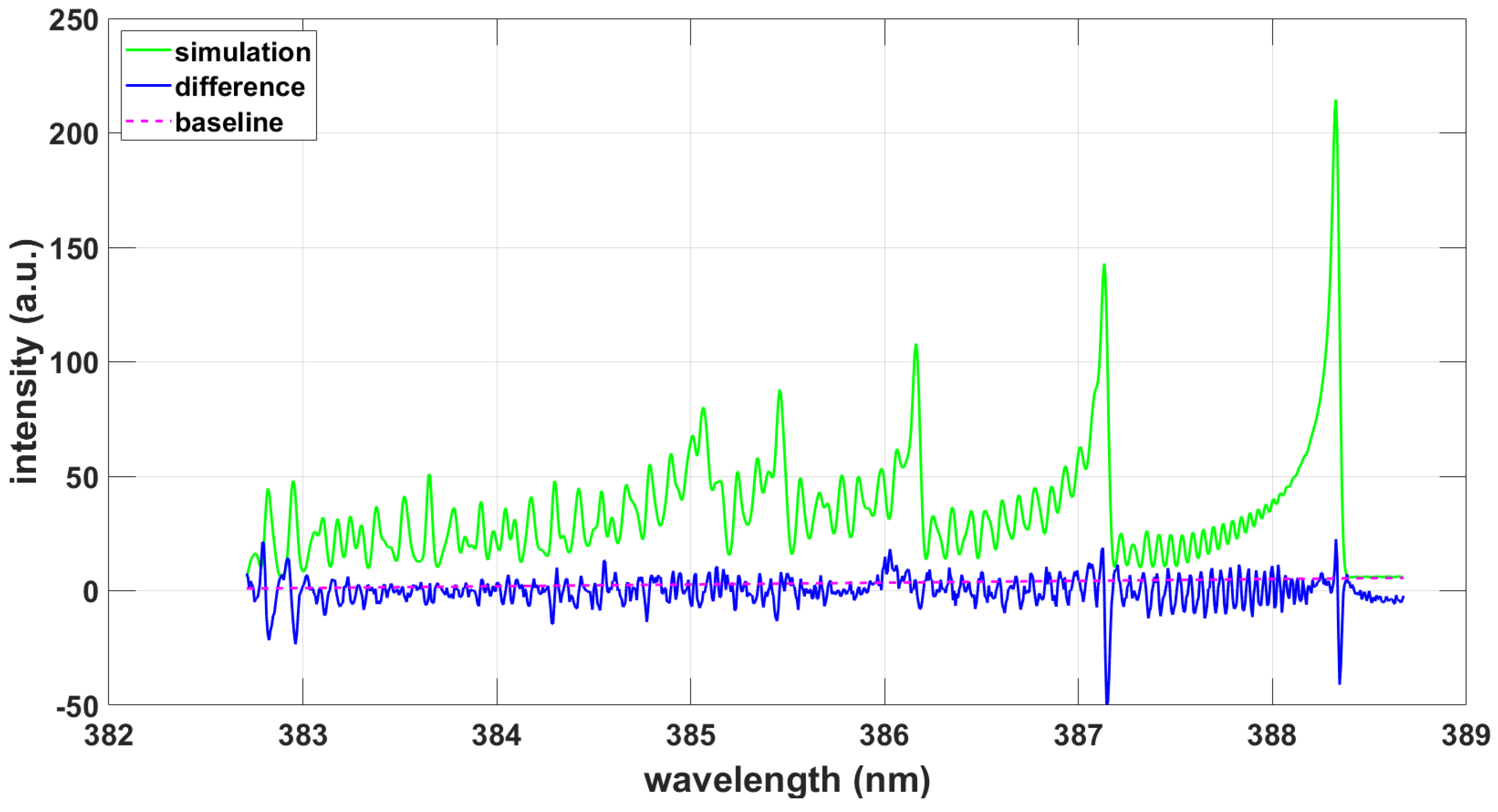
3.1.2. NMT Program Using PGOPHER and ExoMol Line Strengths
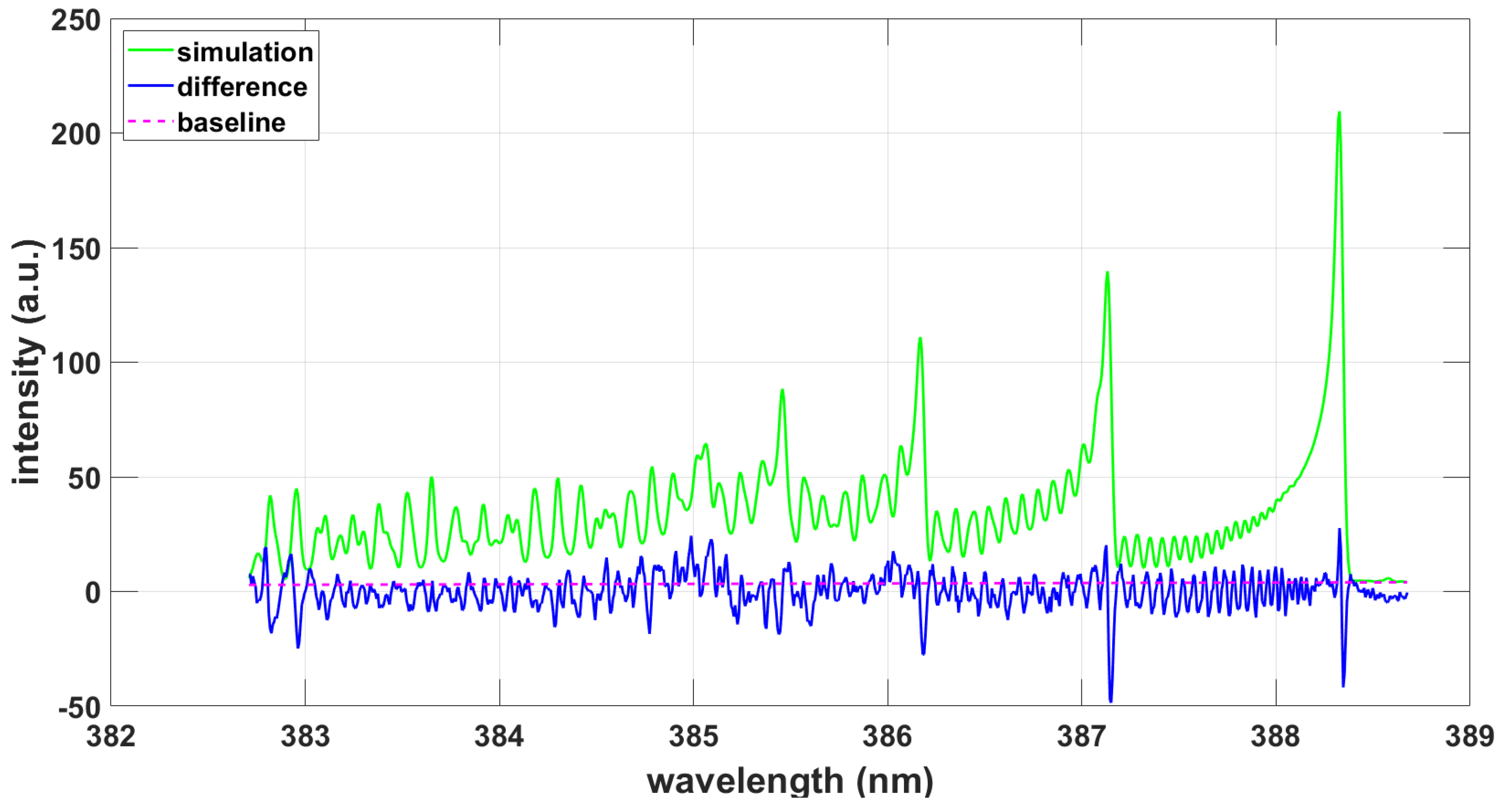
3.1.3. NMT Program Using CNv-lsf Data, Comparisons
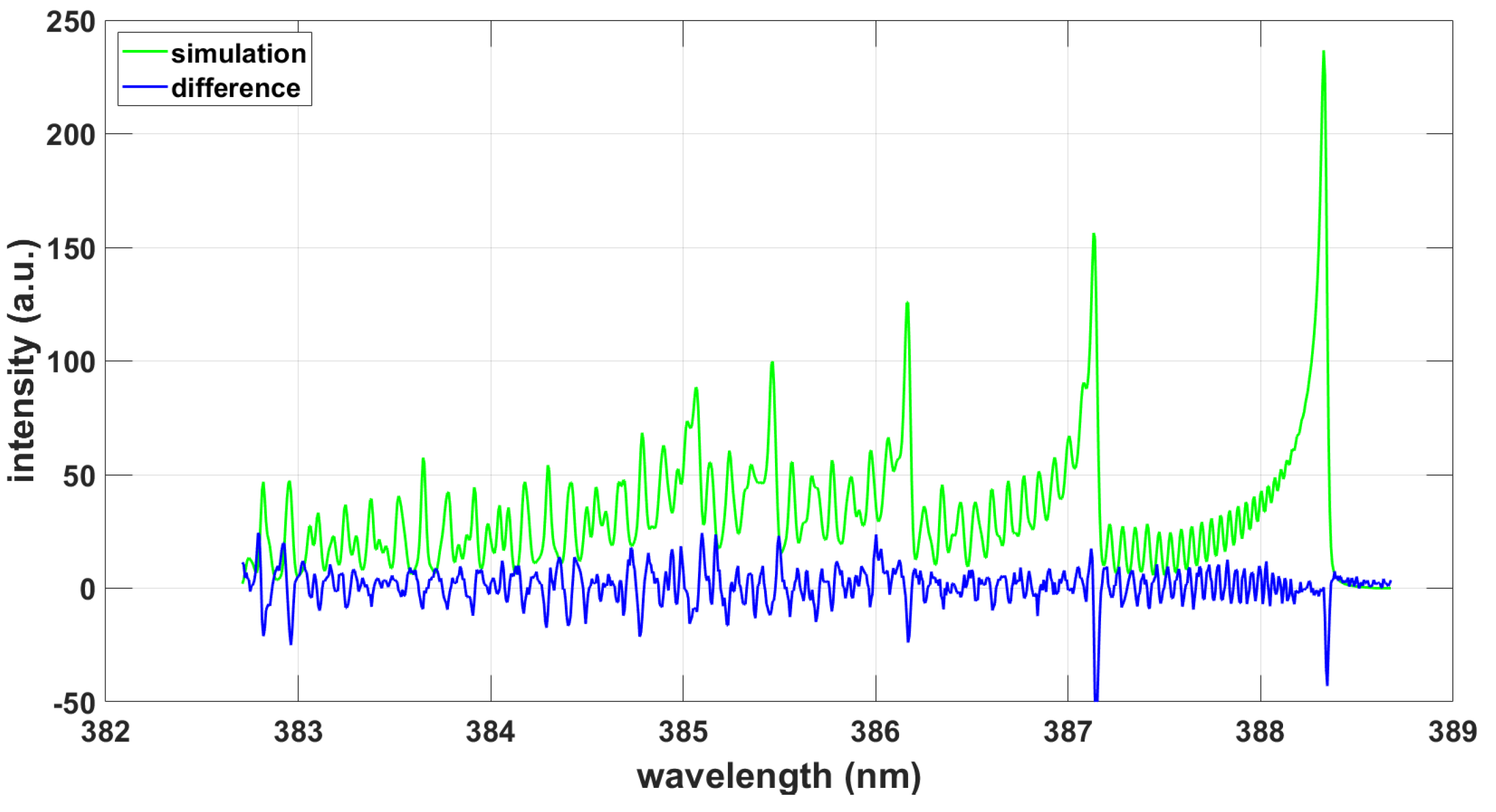
3.1.4. LIFBASE Program
3.2. Analysis of the 0.11 nm Spectral Resolution Data
3.2.1. PGOPHER Program Using PGOPHER Data
3.2.2. NMT Program Using PGOPHER and ExoMol Line Strengths
3.2.3. NMT Program Using CNv-lsf Data, Comparisons
3.2.4. LIFBASE Program
4. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BESP | Boltzmann equilibrium spectral program |
| CNr-lsf | Cyanide red system line-strength data |
| CNv-lsf | Cyanide violet system line-strength data |
| ExoMol | Molecular line lists for exoplanet and other hot atmospheres |
| FWHM | Full width at half maximum |
| HITEMP | High-temperature molecular spectroscopic database |
| LIFBASE | Program for simulating selected diatomic molecules |
| LSF | Line strength file |
| NEQAIR | Non-equilibrium and equilibrium radiative transport and spectra program |
| NIST | National Institute of Standards and Technology |
| NMT | Nelder–Mead temperature |
| OMA | Optical multichannel analyzer |
| PGOPHER | Program for simulating rotational, vibrational, and electronic spectra |
| SATP | Standard ambient temperature and pressure |
| SPECAIR | Program for calculating emission or absorption spectra of air plasma radiation |
References
- Roth, K.C.; Meyer, D.M.; Hawkins, I. Interstellar cyanogen and the temperature of the cosmic microwave background radiation. Astrophys. J. 1993, 413, L67–L71. [Google Scholar] [CrossRef]
- Leach, S. CN spectroscopy and cosmic background radiation measurements. Can. J. Chem. 2004, 82, 730–739. [Google Scholar] [CrossRef]
- Ram, R.S.; Davis, S.P.; Wallace, L.; Englman, R.; Appadoo, D.R.T.; Bernath, P.F. Fourier transform emission spectroscopy of the B2Σ+-X2Σ+ system of CN. J. Mol. Spectrosc. 2006, 37, 225–231. [Google Scholar] [CrossRef]
- Brooke, J.S.A.; Ram, R.S.; Western, C.M.; Li, G.; Schwenke, D.W.; Bernath, P.F. Einstein A Coefficients and Oscillator Strengths for the A2Π-X2Σ+ (Red) and B2Σ+-X2Σ+ (Violet) Systems and Rovibrational Transitions in the X2Σ+ State of CN. Astrophys. J. Suppl. Ser. 2014, 210, 1–15. [Google Scholar] [CrossRef]
- Davis, S.P. Laboratory spectrsoscopy of astrophysically interesting molecules. Publ. Astron. Soc. Pac. 1987, 99, 1105–1114. [Google Scholar] [CrossRef]
- Kunze, H.-J. Introduction to Plasma Spectroscopy; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Fujimoto, T. Plasma Spectroscopy; Clarendon Press: Oxford, UK, 2004. [Google Scholar]
- Ochkin, V.N. Spectroscopy of Low Temperature Plasma; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Demtröder, W. Laser Spectroscopy 1: Basic Principles, 5th ed.; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Demtröder, W. Laser Spectroscopy 2: Experimental Techniques, 5th ed.; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Miziolek, A.W.; Palleschi, V.; Schechter, I. (Eds.) Laser Induced Breakdown Spectroscopy (LIBS): Fundamentals and Applications; Cambridge Univ. Press: New York, NY, USA, 2006. [Google Scholar]
- Singh, J.P.; Thakur, S.N. (Eds.) Laser-Induced Breakdown Spectroscopy, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Tennyson, J.; Yurchenko, S.N.; Al-Refaie, A.F.; Clark, V.H.J.; Chubb, K.L.; Conway, E.K.; Dewan, A.; Gorman, M.N.; Hill, C.; Lynas-Gray, A.E.; et al. The 2020 release of the ExoMol database: Molecular line lists for exoplanet and other hot atmospheres. J. Quant. Spectrosc. Radiat. Transf. 2020, 255, 107228. [Google Scholar] [CrossRef]
- Luque, J.; Crosley, D.R. LIFBASE: Database and Spectral Simulation for Diatomic Molecules. 2021. Available online: https://www.sri.com/platform/lifbase-spectroscopy-tool (accessed on 25 November 2019).
- Western, C.M. PGOPHER, A Program for Simulating Rotational, Vibrational and Electronic Spectra. J. Quant. Spectrosc. Radiat. Transf. 2017, 186, 221–242. [Google Scholar] [CrossRef]
- McKemmish, L.K. Molecular diatomic spectroscopy data. WIREs Comput. Mol. Sci. 2021, 11, e1520. [Google Scholar] [CrossRef]
- Rothman, L.S.; Gordon, I.E.; Barber, R.J.; Dothe, H.; Gamache, R.R.; Goldman, A.; Perevalov, V.I.; Tashkun, S.A.; Tennyson, J. HITEMP, the high-temperature molecular spectroscopic database. J. Quant. Spectrosc. Radiat. Transf. 2010, 111, 2139–2150. [Google Scholar] [CrossRef]
- MATLAB Release R2022a Update 5; The MathWorks, Inc.: Natick, MA, USA, 2022.
- Parigger, C.G.; Hornkohl, J.O. Quantum Mechanics of the Diatomic Molecule with Applications; IOP Publishing: Bristol, UK, 2020. [Google Scholar]
- Parigger, C.G. Diatomic line strengths for fitting selected molecular transitions of ALO, C2, CN, OH, N2+, NO, and TiO, spectra. Foundations 2023, 3, 1–15. [Google Scholar] [CrossRef]
- Hornkohl, J.O.; Parigger, C.G.; Lewis, J.W.L. Temperature Measurement from CN spectra in a laser-induced plasma. J. Quant. Spectrosc. Radiat. Transf. 1991, 46, 405–411. [Google Scholar] [CrossRef]
- Dunham, J.L. The Energy Levels of a Rotating Vibrator. Phys. Rev. 1932, 41, 721–731. [Google Scholar] [CrossRef]
- National Institute of Standards and Technology (NIST) Chemistry WebBook, SRD 69, for the Cyano Radical, Constants of Diatomic Molecules. 2021. Available online: https://webbook.nist.gov (accessed on 20 February 2023).
- Whiting, E.E.; (Space Administration, Reacting Flow Environments Branch, Ames Research Center, CA, USA). Personal communication, 1995.
- Whiting, E.E.; Park, C.; Liu, Y.; Arnold, J.; Paterson, J. NEQAIR96, Nonequilibrium and Equilibrium Radiative Transport and Spectra Program: User’s Manual; Technical Report NASA RP-1389; NASA Ames Research Center: Moffet Field, CA, USA, 1996. [Google Scholar]
- Boulous, P.M.I.; Pfender, E. Thermal Plasmas-Fundamentals and Applications; Plenum Press: New York, NY, USA, 1994. [Google Scholar]
- McBride, B.; Gordan, S. Interim Revision NASA Report RP-1311, Part I; NASA Lewis Research Center: Cleveland, OH, USA, 1994. [Google Scholar]
- McBride, B.; Gordan, S. Interim Revision NASA Report RP-1311, Part II; NASA Lewis Research Center: Cleveland, OH, USA, 1996. [Google Scholar]
- Laux, C.O. Radiation and Nonequilibrium Collisional-Radiative Models, von Karman Institute Lecture Series 2002-07, Physico-Chemical Modeling of High Enthalpy and Plasma Flows; Fletcher, D., Charbonnier, J.-M., Sarma, G.S.R., Magin, T., Eds.; Rhode-Saint-Genèse: Flanders, Belgium, 2002. [Google Scholar]
- Syme, A.-M.; McKemmish, L.K. Experimental energy levels of 12C14N through MARVEL analysis. Mon. Not. R. Astron. Soc. 2020, 499, 25–39. [Google Scholar] [CrossRef]
- Syme, A.M.; McKemmish, L.K. Full spectroscopic model and trihybrid experimental-perturbative-variational line list for CN. Mon. Not. R. Astron. Soc. 2021, 505, 4383–4395. [Google Scholar] [CrossRef]
- Western, C.M.; (University of Bristol, Bristol, UK). Personal communication, 2019.
- Condon, E.U.; Shortley, G.H. The Theory of Atomic Spectra; Cambridge Univ Press: Cambridge, UK, 1964. [Google Scholar]
- Hilborn, R.C. Einstein coefficients, cross sections, f values, dipole moments, and all that. Am. J. Phys. 1982, 50, 982–986. [Google Scholar] [CrossRef]
- Thorne, A.P. Spectrophysics, 2nd ed.; Chapman and Hall: New York, NY, USA, 1988. [Google Scholar]
- Wigner, E.; Witmer, E.E. Über die Struktur der zweiatomigen Molekelspektren nach der Quantenmechanik. Z. Phys. 1928, 51, 859–886. [Google Scholar] [CrossRef]
- Wigner, E.; Witmer, E.E. On the structure of the spectra of two-atomic molecules according to quantum mechanics. In Quantum Chemistry: Classic Scientific Papers; Hettema, H., Ed.; World Scientific: Singapore, 2000; pp. 287–311. [Google Scholar]
- Barrell, H.; Sears, J.E. The Refraction and Dispersion of Air for the Visible Spectrum. Philos. Trans. R. Soc. 1939, 238, 1–64. [Google Scholar]
- Ciddor, P.E. Refractive index of air: New equations for the visible and near infrared. Appl. Opt. 1996, 35, 1567–1573. [Google Scholar] [CrossRef]
- Whiting, E.E. An empirical approximation to the Voigt profile. J. Quant. Spectrosc. Radiat. Transf. 1968, 8, 1379–1384. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Constant | X () | A () | B () | |
|---|---|---|---|---|
| 0.0 | 9245.28 | 25,752.0 | ||
| 2068.59 | 1812.5 | 2163.9 | ||
| 13.087 | 12.60 | 20.2 | ||
| −0.0118 | – | |||
| 1.8997 | 1.7151 | 1.973 | ||
| 0.01736 | 0.01708 | 0.023 | ||
| – | ||||
| – |
| Parameter | Value |
|---|---|
| 272.643 | |
| 1.2288 (m) | |
| 0.03555 (m) |
| Parameter | Value (m) |
|---|---|
| (k0) | 238.0185 |
| (k1) | 5,792,105 |
| (k2) | 57.362 |
| (k3) | 167,917 |
| Parameter | Value | Standard Deviation |
|---|---|---|
| Temperature (K) | 8103 | 187 |
| Gaussian width (cm) | 2.2 | 0.03 |
| Scale (a.u.) | 43,960 | 790 |
| T (K) | 8108 | 146 |
| Database | 0.033 nm Range CN B-X | 0.033 nm Range CN B-X and A-X |
|---|---|---|
| PGOPHER | 3598 | 5631 |
| ExoMol | 4302 | 17,181 |
| CNv-lsf | 2461 | 2461 |
| Database | 0.033 nm Range CN B-X | 0.033 nm Range CN B-X and A-X |
|---|---|---|
| PGOPHER | 3205 | 4532 |
| ExoMol | 3625 | 8270 |
| CNv-lsf | 2461 | 2461 |
| Database | < 0.05 | < 0.2 | < 1.0 | < 2.0 |
|---|---|---|---|---|
| PGOPHER | 1200 | 1422 | 1823 | 2012 |
| ExoMol | 158 | 463 | 1266 | 1935 |
| Parameter | Value | Standard Deviation |
|---|---|---|
| Temperature (K) | 8340 | 43 |
| Gaussian width (cm) | 7.1 | 0.07 |
| Scale (a.u.) | 107,300 | 850 |
| Database | 0.11 nm Range CN B-X | 0.11 nm Range CN B-X and A-X |
|---|---|---|
| PGOPHER | 7115 | 14,349 |
| ExoMol | 10,499 | 78,079 |
| CNv-lsf | 3313 | 3313 |
| Database | 0.11 nm Range CN B-X | 0.11 nm Range CN B-X and A-X |
|---|---|---|
| PGOPHER | 6187 | 9958 |
| ExoMol | 8259 | 28,274 |
| CNv-lsf | 3313 | 3313 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parigger, C.G. Cyanide Molecular Laser-Induced Breakdown Spectroscopy with Current Databases. Atoms 2023, 11, 62. https://doi.org/10.3390/atoms11040062
Parigger CG. Cyanide Molecular Laser-Induced Breakdown Spectroscopy with Current Databases. Atoms. 2023; 11(4):62. https://doi.org/10.3390/atoms11040062
Chicago/Turabian StyleParigger, Christian G. 2023. "Cyanide Molecular Laser-Induced Breakdown Spectroscopy with Current Databases" Atoms 11, no. 4: 62. https://doi.org/10.3390/atoms11040062
APA StyleParigger, C. G. (2023). Cyanide Molecular Laser-Induced Breakdown Spectroscopy with Current Databases. Atoms, 11(4), 62. https://doi.org/10.3390/atoms11040062