
Antiphospholipid Syndrome—Diagnostic and Methodologic Approach



Abstract
1. Introduction
2. Antiphospholipid Antibodies
3. Pathophysiology of Antiphospholipid Syndrome
4. Varieties of the Antiphospholipid Syndrome
5. Classification of Antiphospholipid Syndrome
6. Catastrophic Antiphospholipid Syndrome
7. Laboratory Diagnostics of Antiphospholipid Syndrome
7.1. Standard Laboratory Criteria
7.2. Lupus Anticoagulant Testing
7.3. ACL and aβ2-GPI Detection
7.4. Risk Stratification and Interpretation
7.5. Pre-Analytical Considerations
8. Treatment of Antiphospholipid Syndrome
8.1. General Principles of Treatment
8.2. Primary Prevention
8.3. Secondary Prevention
8.4. Obstetric and Catastrophic APS
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| aβ2-GPI | Anti-β2-glycoprotein I antibody |
| aCL | Anticardiolipin antibody |
| ACR | American College of Rheumatology |
| aPL | Antiphospholipid antibody |
| APS | Antiphospholipid syndrome |
| APTT | Activated partial thromboplastin time |
| ASA | Acetylsalicylic acid |
| ASTRO-APS | Apixaban for Secondary Prevention of Thromboembolism Among Patients with Antiphospholipid Syndrome |
| BAL | Bronchoalveolar lavage |
| β2GPI | β2-glycoprotein I |
| DOAC | Direct oral coagulation inhibitor |
| dRVVT | Dilute Russell viper venom test |
| ELISA | Enzyme-linked immunosorbent assay |
| eNOS | Endothelial nitric oxide synthase |
| EULAR | European Alliance of Associations for Rheumatology |
| HELLP | Hemolysis, elevated liver enzymes, and low platelet count syndrome |
| HLA | Human leukocyte antigen |
| ICAM-1 | Intercellular adhesion molecule 1 |
| IgA | Immunoglobulin A |
| IgG | Immunoglobulin G |
| IgM | Immunoglobulin M |
| IL-8 | Interleukin 8 |
| INR | International normalized ratio |
| ISTH | International Society on Thrombosis and Hemostasis |
| LA | Lupus anticoagulant |
| LMWH | Low-molecular-weight heparin |
| MAPK | Mitogen-activated protein kinase |
| NET | Neutrophil extracellular trap |
| NF-κB | nuclear factor kappa B |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PAPS | Primary antiphospholipid syndrome |
| SAPS | Secondary antiphospholipid syndrome |
| SCT | Sillica Clotting Time |
| SIRS | Systemic inflammatory response |
| SLE | Systemic lupus erythematosus |
| SN-APS | Seronegative antiphospholipid syndrome |
| TF | Tissue factor |
| TFPI | Tissue factor pathway inhibitor |
| TNF-α | Tumor necrosis factor alpha |
| VCAM-1 | Vascular cell adhesion molecule |
| VKA | Vitamin K antagonist |
| VTE | Venous thromboembolism |
| vWF | von Willebrand factor |
References
- Ambati, A.; Knight, J.S.; Zuo, Y. Antiphospholipid syndrome (APS) management: A 2023 update and practical algorithm-based approach. Curr. Opin. Rheumatol. 2023, 35, 149–160. [Google Scholar] [CrossRef]
- Chaturvedi, S.; McCrae, K. The antiphospholipid syndrome: Still an enigma. Hematol. Am. Soc. Hematol. Educ. Program. 2015, 2015, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Khogeer, H.; Altahan, S.; Alrehaily, A.; Sheikh, A.; Awartani, K.; Al-Kaff, M.; Saleh, S.; Alzahrani, H.; Alfattani, A.; Owaidah, T. The Diagnostic Value of New Additional Antiphospholipid Antibodies in Antiphospholipid Syndrome. Ann. Clin. Lab. Sci. 2021, 51, 552–556. [Google Scholar]
- Knight, J.S.; Kanthi, Y. Mechanisms of immunothrombosis and vasculopathy in antiphospholipid syndrome. Semin. Immunopathol. 2022, 44, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.; Derksen, R.H.; De Groot, P.G.; Koike, T.; Meroni, P.L.; et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306. [Google Scholar] [CrossRef]
- Yun, Z.; Duan, L.; Liu, X.; Cai, Q.; Li, C. An update on the biologics for the treatment of antiphospholipid syndrome. Front. Immunol. 2023, 14, 1145145. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Pozzi, N. Understanding the structure of β2-glycoprotein I: New insights and future paths for antiphospholipid syndrome. Blood Vessel. Thromb. Hemost. 2025, 2, 100041. [Google Scholar] [CrossRef]
- Pérez, D.; Stojanovich, L.; Naranjo, L.; Stanisavljevic, N.; Bogdanovic, G.; Serrao, M.; Serrano, A. Presence of Immune Complexes of IgG/IgM Bound to B2-glycoprotein I Is Associated With Non-criteria Clinical Manifestations in Patients With Antiphospholipid Syndrome. Front. Immunol. 2018, 9, 2644. [Google Scholar] [CrossRef]
- Cochery-Nouvellon, É.; Mercier, É.; Bouvier, S.; Balducchi, J.-P.; Quéré, I.; Perez-Martin, A.; Mousty, E.; Letouzey, V.; Gris, J.-C. Obstetric antiphospholipid syndrome: Early variations of angiogenic factors are associated with adverse outcomes. Haematologica 2017, 102, 835–842. [Google Scholar] [CrossRef]
- Dabit, J.Y.; Valenzuela-Almada, M.O.; Vallejo-Ramos, S.; Duarte-García, A. Epidemiology of Antiphospholipid Syndrome in the General Population. Curr. Rheumatol. Rep. 2022, 23, 85. [Google Scholar] [CrossRef]
- Long, Y.; Li, W.; Feng, J.; Ma, Y.; Sun, Y.; Xu, L.; Song, Y.; Liu, C. Follicular helper and follicular regulatory T cell subset imbalance is associated with higher activated B cells and abnormal autoantibody production in primary anti-phospholipid syndrome patients. Clin. Exp. Immunol. 2021, 206, 141–152. [Google Scholar] [CrossRef]
- Cohen, H.; Cuadrado, M.J.; Erkan, D.; Duarte-Garcia, A.; Isenberg, D.A.; Knight, J.S.; Thomas, L.; Ortel, T.L.; Rahman, A.; Salmon, J.E.; et al. 16th International Congress on Antiphospholipid Antibodies Task Force Report on Antiphospholipid Syndrome Treatment Trends. Lupus 2020, 29, 1571–1593. [Google Scholar] [CrossRef]
- Tektonidou, M.G. Antiphospholipid Syndrome Nephropathy: From Pathogenesis to Treatment. Front. Immunol. 2018, 9, 1181. [Google Scholar] [CrossRef] [PubMed]
- Ho, R.C.; Thiaghu, C.; Ong, H.; Lu, Y.; Ho, C.S.; Tam, W.W.; Zhang, M.W. A meta-analysis of serum and cerebrospinal fluid autoantibodies in neuropsychiatric systemic lupus erythematosus. Autoimmun. Rev. 2016, 15, 124–138. [Google Scholar] [CrossRef]
- Gulli, F.; Napodano, C.; Marino, M.; Ciasca, G.; Pocino, K.; Basile, V.; Visentini, M.; Stefanile, A.; Todi, L.; De Spirito, M.; et al. Serum immunoglobulin free light chain levels in systemic autoimmune rheumatic diseases. Clin. Exp. Immunol. 2020, 199, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Sciascia, S.; Khamashta, M.A.; Bertolaccini, M.L. New tests to detect antiphospholipid antibodies: Antiprothrombin (aPT) and anti-phosphatidylserine/prothrombin (aPS/PT) antibodies. Curr. Rheumatol. Rep. 2014, 16, 415. [Google Scholar] [CrossRef]
- Hoxha, A.; Ruffatti, A.; Tonello, M.; Bontadi, A.; Salvan, E.; Banzato, A.; Pengo, V.; Punzi, L. Antiphosphatidylserine/prothrombin antibodies in primary antiphospholipid syndrome. Lupus 2012, 21, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Naranjo, L.; Stojanovich, L.; Djokovic, A.; Andreoli, L.; Tincani, A.; Maślińska, M.; Sciascia, S.; Infantino, M.; Garcinuño, S.; Kostyra-Grabczak, K.; et al. Circulating immune-complexes of IgG/IgM bound to B2-glycoprotein-I associated with complement consumption and thrombocytopenia in antiphospholipid syndrome. Front. Immunol. 2022, 13, 957201. [Google Scholar] [CrossRef]
- Buchholz, I.; McDonnell, T.; Nestler, P.; Tharad, S.; Kulke, M.; Radziszewska, A.; Ripoll, V.M.; Schmidt, F.; Hammer, E.; Toca-Herrera, J.L.; et al. Specific domain V reduction of beta-2-glycoprotein I induces protein flexibility and alters pathogenic antibody binding. Sci. Rep. 2021, 11, 4542. [Google Scholar] [CrossRef]
- Lopez, L.R.; Santos, M.E.; Espinoza, L.R.; La Rosa, F.G. Clinical significance of immunoglobulin A versus immunoglobulins G and M anti-cardiolipin antibodies in patients with systemic lupus erythematosus. Correlation with thrombosis, thrombocytopenia, and recurrent abortion. Am. J. Clin. Pathol. 1992, 98, 449–454. [Google Scholar] [CrossRef]
- Lóczi, L.; Kappelmayer, J.; Tarr, T.; Bagoly, Z. Antiphospholipid syndrome and the risk of myocardial infarction: Current evidence and uncertaines. Kardiol. Pol. 2020, 78, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Chayoua, W.; Kelchtermans, H.; Moore, G.W.; Musiał, J.; Wahl, D.; de Laat, B.; Devreese, K.M. Identification of high thrombotic risk triple-positive antiphospholipid syndrome patients is dependent on anti-cardiolipin and anti-beta2glycoprotein I antibody detection assays. J. Thromb. Haemost. 2018, 16, 2016–2023. [Google Scholar] [CrossRef]
- Devreese, K.M. Noncriteria antiphospholipid antibodies in antiphospholipid syndrome. Int. J. Lab. Hematol. 2024, 46 (Suppl. 1), 34–42. [Google Scholar] [CrossRef]
- Pelkmans, L.; de Laat, B. Antibodies against domain I of β2-glycoprotein I: The one and only? Lupus 2012, 21, 769–772. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, T.; Wincup, C.; Buchholz, I.; Pericleous, C.; Giles, I.; Ripoll, V.; Cohen, H.; Delcea, M.; Rahman, A. The role of beta-2-glycoprotein I in health and disease associating structure with function: More than just APS. Blood Rev. 2020, 39, 100610. [Google Scholar] [CrossRef]
- Agar, C.; van Os, G.M.; Morgelin, M.; Sprenger, R.R.; Marquart, J.A.; Urbanus, R.T. Beta2-glycoprotein I can exist in 2 conformations: Implications for our understanding of the antiphospholipid syndrome. Blood 2010, 116, 1336–1343. [Google Scholar] [CrossRef]
- Rauch, J.; Salem, D.; Subang, R.; Kuwana, M.; Levine, J. β2-Glycoprotein I-Reactive T Cells in Autoimmune Disease. Front. Immunol. 2018, 9, 2836. [Google Scholar] [CrossRef]
- Cheng, S.; Wang, H.; Zhou, H. The Role of TLR4 on B Cell Activation and Anti- β2GPI Antibody Production in the Antiphospholipid Syndrome. J. Immunol. Res. 2016, 2016, 1719720. [Google Scholar] [CrossRef]
- Sadeghi, M.; Dehnavi, S.; Jamialahmadi, T.; Johnston, T.P.; Sahebkar, A. Neutrophil extracellular trap: A key player in the pathogenesis of autoimmune diseases. Int. Immunopharmacol. 2023, 116, 109843. [Google Scholar] [CrossRef] [PubMed]
- Heikal, N.M.; Jaskowski, T.D.; Malmberg, E.; Lakos, G.; Branch, D.W.; Tebo, A.E. Laboratory evaluation of anti-phospholipid syndrome: A preliminary prospective study of phosphatidylserine/prothrombin antibodies in an at-risk patient cohort. Clin. Exp. Immunol. 2014, 180, 218–2216. [Google Scholar] [CrossRef]
- Dobrowolski, C.; Erkan, D. Treatment of antiphospholipid syndrome beyond anticoagulation. Clin Immunol. 2019, 206, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Laird, S.; Li, T.; Ledger, W. Preliminary prospective study of the endocrinology of conception cycles and early pregnancy in women with antiphospholipid syndrome treated with low molecular weight heparin. Fertil. Steril. 2006, 85, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Okuma, H.; Kitagawa, Y.; Yasuda, T.; Tokuoka, K.; Takagi, S. Comparison between single antiplatelet therapy and combination of antiplatelet and anticoagulation therapy for secondary prevention in ischemic stroke patients with antiphospholipid syndrome. Int. J. Med. Sci. 2010, 7, 15–18. [Google Scholar] [CrossRef]
- Marinho, A.; Delgado Alves, J.; Fortuna, J.; Faria, R.; Almeida, I.; Alves, G.; Correia, J.A.; Campar, A.; Brandão, M.; Crespo, J.; et al. Biological therapy in systemic lupus erythematosus, antiphospholipid syndrome, and Sjögren’s syndrome: Evidence and practice-based guidance. Front. Immunol. 2023, 14, 1117699. [Google Scholar] [CrossRef]
- de Groot, P.G.; de Laat, B. Mechanisms of thrombosis in systemic lupus erythematosus and antiphospholipid syndrome. Best. Pract. Res. Clin. Rheumatol. 2017, 31, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pintó, I.; Espinosa, G.; Cervera, R. Catastrophic antiphospholipid syndrome: The current management approach. Best. Pract. Res. Clin. Rheumatol. 2016, 30, 239–249. [Google Scholar] [CrossRef]
- Cervera, R. Update on the Diagnosis, Treatment, and Prognosis of the Catastrophic Antiphospholipid Syndrome. Curr Rheumatol Rep. 2010, 12, 70–76. [Google Scholar] [CrossRef]
- Kapitany, A.; Tarr, T.; Gyetvai, A.; Szodoray, P.; Tumpek, J.; Poor, G.; Szegedi, G.; Sipka, S.; Kiss, E. Human leukocyte antigen-DRB1 and -DQB1 genotyping in lupus patients with and without antiphospholipid syndrome. Ann. N. Y. Acad. Sci. 2009, 1173, 545–551. [Google Scholar] [CrossRef]
- Andreoli, L.; Bertsias, G.K.; Agmon-Levin, N.; Brown, S.; Cervera, R.; Costedoat-Chalumeau, N.; Doria, A.; Fischer-Betz, R.; Forger, F.; Moraes-Fontes, M.F.; et al. EULAR recommendations for women’s health and the management of family planning, assisted reproduction, pregnancy and menopause in patients with systemic lupus erythematosus and/or antiphospholipid syndrome. Ann. Rheum. Dis. 2017, 76, 476–485. [Google Scholar] [CrossRef]
- Tektonidou, M.; Andreoli, L.; Limper, M.; Amoura, Z.; Cervera, R.; Costedoat-Chalumeau, N.; Cuadrado, M.J.; Dörner, T.; Ferrer-Oliveras, R.; Hambly, K.; et al. EULAR recommendations for the management of antiphospholipid syndrome in adults. Ann. Rheum. Dis. 2019, 78, 1296–1304. [Google Scholar] [CrossRef]
- Usta, A.; Yayla, M.E.; Uslu, E.; Sezer, S.; Us, E.; Ateş, A.; Turgay, M. The Performance of 2023 American College of Rheumatology (ACR)/European Alliance of Associations for Rheumatology (EULAR) Antiphospholipid Syndrome Classification Criteria in a Real-World Rheumatology Department. Mediterr. J. Hematol. Infect. Dis. 2024, 16, e2024074. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Cohen, H.; Devreese, K.M. Lupus anticoagulant detection in anticoagulated patients. Guidance from the Scientific and Standardization Committee for lupus anticoagulant/antiphospholipid antibodies of the International Society on Thrombosis and Haemostasis. J. Thromb. Haemosis. 2020, 18, 1569–1575. [Google Scholar] [CrossRef]
- Vanhove, B.; Van Hoovels, L.; Broeders, S.; Coucke, W.; Schreurs, M.W.; Bonroy, C.; Schouwers, S.; Vanstokstraeten, R.; Bailleul, E.; Devreese, K.M. Lupus anticoagulant testing in Belgian laboratories: A comparison with the 2020 International Society on Thrombosis and Haemostasis Scientific and Standardization Committee (ISTH-SSC) guidelines. Acta Clin. Belg. 2025, 80, 62–70. [Google Scholar] [CrossRef]
- Chojnowski, K.; Podolak-Dawidziak, M.; Windyga, J. Diagnosis of the prolonged activated partial thromboplastin time (aPTT). Hematologia 2010, 1, 81–86. [Google Scholar]
- Galli, M.; Borrelli, G.; Jacobsen, E.M.; Marfisi, R.M.; Finazzi, G.; Marchioli, R.; Wisloff, F.; Marziali, S.; Morboeuf, O.; Barbui, T. Clinical significance of different antiphospholipid antibodies in the WAPS (warfarin in the antiphospholipid syndrome) study. Blood 2007, 110, 1178–1183. [Google Scholar] [CrossRef]
- Tonello, M.; Bison, E.; Cattini, M.G.; Pontara, E.; Iaccarino, L.; Denas, G.; Cheng, C.; Pengo, V. Anti-phosphatidyl-serine/prothrombin antibodies (aPS/PT) in isolated lupus anticoagulant (LA): Is their presence linked to dual test positivity? Clin. Chem. Lab. Med. 2021, 59, 1950–1953. [Google Scholar] [CrossRef]
- Barbhaiya, M.; Zuily, S.; Ahmadzadeh, Y.; Amigo, C.; Avcin, T.; Bertolaccini, L.; Branch, W.; de Jesus, G.; Devreese, K.; Frances, C.; et al. Development of a New International Antiphospholipid Syndrome Classification Criteria Phase I/II Report: Generation and Reduction of Candidate Criteria. Arthritis Care Res. 2021, 73, 1490–1501. [Google Scholar] [CrossRef]
- Musiał, J. New classification criteria for antiphospholipid syndrome—2023. J. Trans. Med. 2023, 16, 103–109. [Google Scholar] [CrossRef]
- Sciascia, S.; Cuadrado, M.J.; Sanna, G.; Murru, V.; Roccatello, D.; Khamashta, M.A.; Bertolaccini, M.L. Thrombotic risk assessment in systemic lupus erythematosus: Validation of the global antiphospholipid syndrome score in a prospective cohort. Arthritis Care Res. 2014, 66, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Žigon, P.; Boštic, N.; Ambrožič, A.; Rotar, Ž.; Blokar, E.; Ogrič, M.; Čučnik, S. Establishment of ELISA-comparable moderate and high thresholds for anticardiolipin and anti-β2 glycoprotein I chemiluminescent immunoassays according to the 2023 ACR/EULAR APS classification criteria and evaluation of their diagnostic performance. Clin. Chem. Lab. Med. 2024, 63, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.; Hunt, B.J.; Efthymiou, M.; Arachchillage, D.R.; Mackie, I.J.; Clawson, S.; Sylvestre, Y.; Machin, S.J.; Bertolaccini, M.L.; Ruiz-Castellano, M.; et al. Rivaroxaban versus warfarin to treat patients with thrombotic antiphospholipid syndrome, with or without systemic lupus erythematosus (RAPS): A randomised, controlled, open-label, phase 2/3, non-inferiority trial. Lancet Haematol. 2016, 3, e426–e436. [Google Scholar] [CrossRef] [PubMed]
- Bala, M.M.; Celinska-Lowenhoff, M.; Szot, W.; Padjas, A.; Kaczmarczyk, M.; Swierz, M.J.; Undas, A. Antiplatelet and anticoagulant agents for secondary prevention of stroke and other thromboembolic events in people with antiphospholipid syndrome. Cochrane Database Syst. Rev. 2017, 10, CD012169. [Google Scholar] [PubMed]
- Giarretta, I.; Ageno, W.; Dentali, F. Lack of efficacy of direct oral anticoagulants compared to warfarin in antiphospholipid antibody syndrome. Haematologica 2022, 107, 2737–2741. [Google Scholar] [CrossRef] [PubMed]
- Pengo, V.; Hoxha, A.; Andreoli, L.; Tincani, A.; Silvestri, E.; Prisco, D.; Fierro, T.; Gresele, P.; Cafolla, A.; De Micheli, V.; et al. Trial of Rivaroxaban in AntiPhospholipid Syndrome (TRAPS): Two-year outcomes after the study closure. J. Thromb. Haemost. 2021, 19, 531–535. [Google Scholar] [CrossRef]
- Woller, S.; Stevens, S.; Kaplan, D.; Wang, T.-F.; Branch, W.; Groat, D.; Wilson, E.; Armbruster, B.; Aston, V.; Lloyd, J.; et al. Apixaban compared with warfarin to prevent thrombosis in thrombotic antiphospholipid syndrome: A randomized trial. Blood Adv. 2022, 6, 1661–1670. [Google Scholar] [CrossRef]
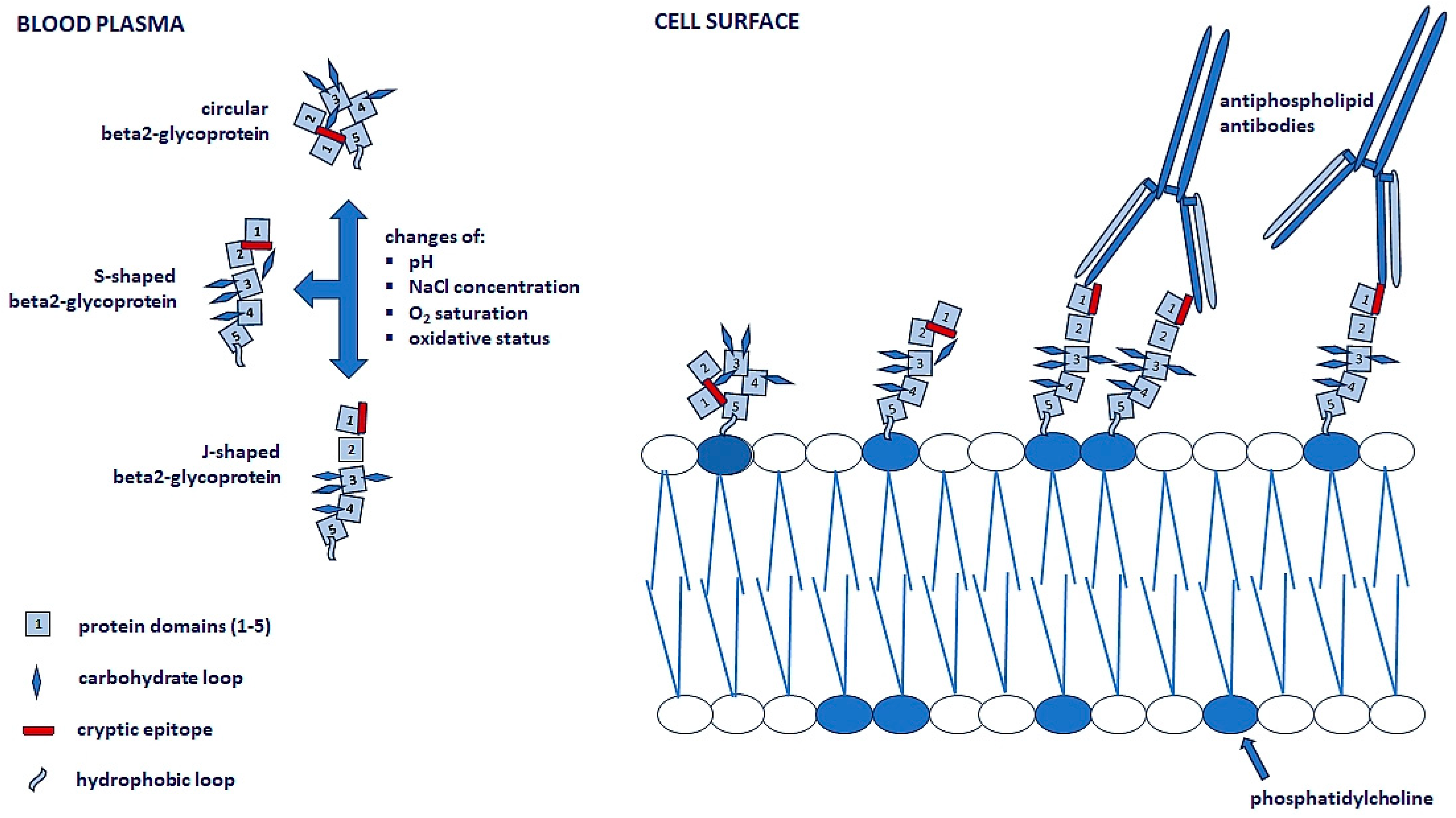
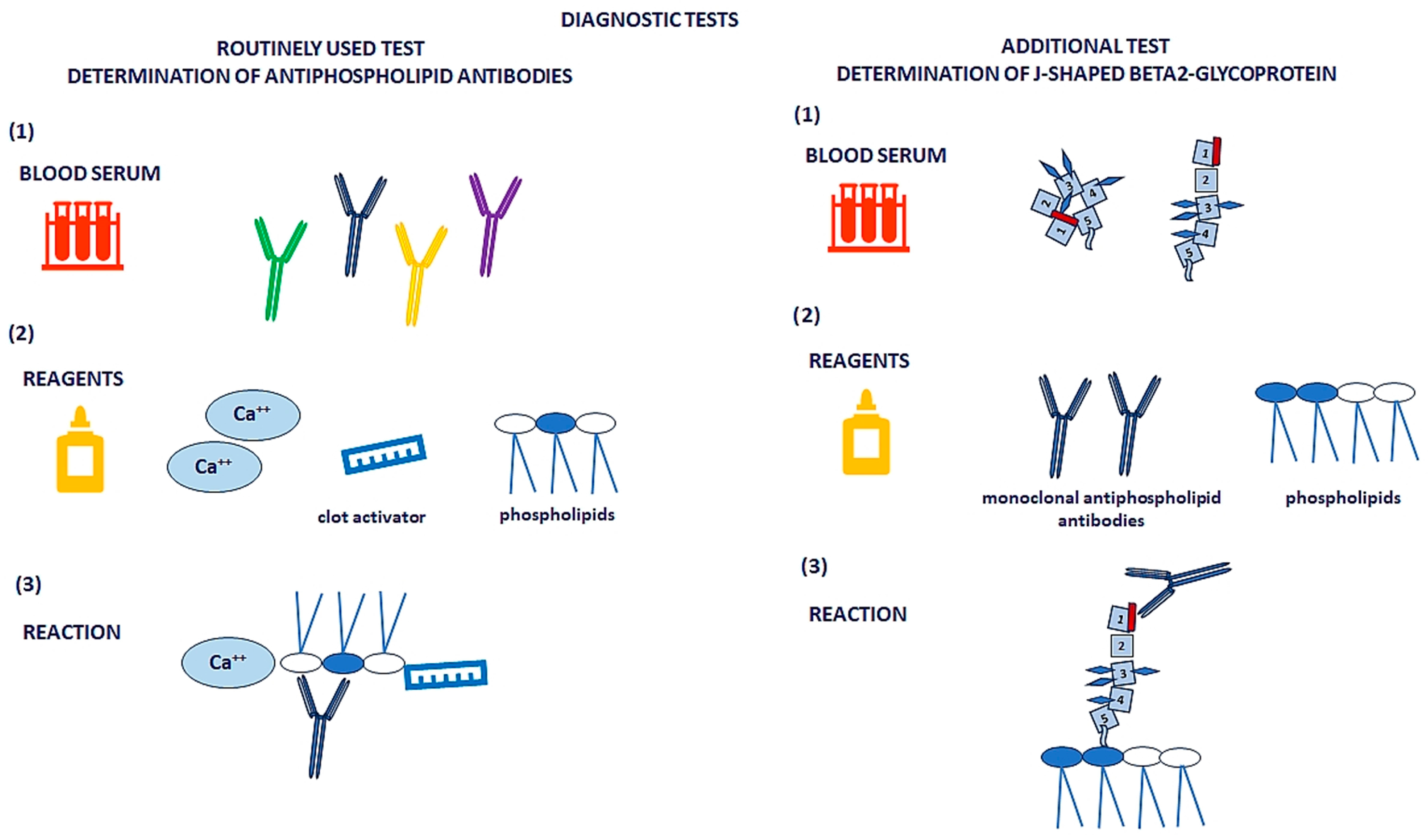

{kind=link}
{kind=link}
| Endothelial cell dysfunction | Antiphospholipid antibody-dependent eNOS inhibition |
| Increased expression of adhesion molecules: ICAM-1, VCAM-1, E-selectin, and others | |
| Increased adhesion of leukocytes to the endothelium | |
| Platelet activation | Increased thromboxane A2 production |
| Increased platelet activation leading to increased glycoprotein IIb/IIIa expression | |
| Disturbance of vWF-dependent platelet adhesion | |
| Increased platelet-derived microparticle formation | |
| Activation of the complement system | Activation of C3, C5 components, and deposition of complement components |
| Cellular Inflammatory processes | Increased expression of TF on monocytes and monocyte-derived microparticles |
| Increased release of IL-8 | |
| Release of NETs | |
| Impairment of anticoagulant mechanisms | Antiphospholipid antibody-dependent anticoagulant properties of annexin V |
| Inhibition of the protein C system | |
| Disturbances of antithrombin function | |
| TFPI inhibition | |
| Inhibition of fibrinolysis/ Abnormal clot structure | Inhibition of plasminogen binding, activation, and activity of plasmin |
| Increased PAI-1 levels | |
| Prothrombotic clot phenotype: denser fibrin networks, low permeability, reduced susceptibility to lysis |
| Autoimmunological disorders | Systemic lupus erythematosus Rheumatoid arthritis Systemic sclerosis S-syndrome Takayasu’s disease Dermatomyositis Autoimmune thyroiditis |
| Virological disorders | Human Immunodeficiency Virus Epstein–Barr Virus Hepatitis C Virus Hepatitis B Virus Rubella virus Mumps virus Parvovirus B19 |
| Bacteriological disorders | Tuberculosis Syphilis Leprosy Rheumatic fever Bacterial endocarditis Klebsiella |
| Parasitic disorders | Malaria Toxoplasmosis |
| Cancers | Lungs Colonial Prostate Cervix Liver Kidneys Esophagus Breast |
| Hematological malignancies | Myeloproliferative: myeloid leukemias, polycythemia vera, myelofibrosis Lymphoproliferative: B-cell leukemia/lymphoma, non-Hodgkin’s and Hodgkin’s lymphomas Paraproteinemias: multiple myeloma, Waldenström’s macroglobulinemia, monoclonal gammopathies |
| Hematological disorders | Sickle cell disease Pernicious anemia Autoimmune thrombocytopenic purpura |
| Drug-induced | Hydralazine Procainamide Quinine Phenothiazine Chlorpromazine |
| Others | Diabetes Inflammatory bowel disease |
| ENTERING CRITERIA At least one documented clinical criterion, included in domains D1–D6, and a positive antiphospholipid antibody (aPL) test result: lupus anticoagulant, moderate or high titer of anticardiolipin or anti-β2-glycoprotein I antibodies, IgG or IgM class, obtained within 3 years of documented clinical criteria. | |
| ↓ | |
| If absent, do not classify as APS, but apply additional criteria. | |
| ↓ | |
| ADDITIONAL CLINICAL AND LABORATORY CRITERIA Do not count a clinical criterion if there is a more probable cause than APS. Only the criterion with the highest weighting (score) should be counted within each domain | |
| Clinical criteria and domain | Weight |
| D1. Large-vessel VTE | |
| - in a patient with a high-risk profile | 1 |
| - in a patient without a high-risk profile | 3 |
| D2. Large-vessel arterial thrombosis | |
| - in a patient with a high cardiovascular risk profile | 2 |
| - in a patient without a high-risk profile | 4 |
| D3. Microvessels | |
| Suspected one of the following: | 5 |
| - livedo racemosa in physical examination | |
| - livedoid vasculopathy in physical examination | |
| - acute/chronic aPL-related nephropathy in physical or laboratory examination | |
| - intravesicular hemorrhage in physical or imaging examination | |
| Established diagnosis of one of the following: | 5 |
| - livedoid vasculopathy in physical examination or histopathology | |
| - acute/chronic aPL-related nephropathy | |
| - intravesicular hemorrhage in BAL or histopathology | |
| - myocardial disease imaging or histopathology | |
| - adrenal hemorrhage imaging or histopathology | |
| D4. Obstetric complications | |
| - consecutive lost ≥ 3 pregnancies at the (pre)embryonic stage (<10 weeks) or | 1 |
| fetal death (10 weeks 0 days—15 weeks 6 days) | |
| - fetal death (16 weeks 0 days—33 weeks 6 days) without the presence of severe preeclampsia or severe placental insufficiency | 1 |
| - severe preeclampsia (<34 weeks 0 days) or severe placental insufficiency | 3 |
| (<34 weeks 0 days) with or without fetal death | |
| - severe preeclampsia (<34 weeks 0 days) and severe placental insufficiency | 4 |
| (<34 weeks 0 days) with or without fetal death | |
| D5. Heart valves | |
| - thickening | 2 |
| - vegetations | 4 |
| D6. Hematology | |
| - thrombocytopenia (20–130 × 109/L) | 2 |
| Laboratory criteria and domain | Weight |
| D7. aPL testing by functional coagulation test (LA test) | |
| - positive LA once occasionally | 1 |
| - positive LA permanently | 5 |
| D8. aPL testing by solid-phase ELISA (aCL or a-β2GPI) is permanently present | |
| - moderately (40–79U)/highly (≥80U) positive aCL and/or a-β2GPI M class | 1 |
| - moderately positive aCL and/or aβ2-GPI IgG class | 4 |
| - highly positive aCL or aβ2-GPI IgG class | 5 |
| - highly positive aCL and aβ2-GPI IgG class | 7 |
| ↓ | |
| SCORE For research purposes, classify as APS if at least 3 points from clinical domains and 3 points from laboratory domains are collected | |
| Criteria 1. Evidence of involvement of ≥3 organs, systems, or tissues. Clinical evidence of vascular occlusion is usually required, confirmed by imaging studies if possible. Renal involvement is defined as a 50% increase in creatinine, severe systemic hypertension (>180/100 mmHg), or proteinuria (>500 mg/24 h). 2. Development of symptoms concurrently or in less than a week. 3. Histopathological confirmation of small-vessel occlusion in at least one organ or tissue. Histopathological confirmation must include significant evidence of thrombosis, although vasculitis may occasionally be present. 4. Laboratory confirmation of aPL (lupus anticoagulant or anticardiolipin antibodies). If the patient has not previously been diagnosed with APS, laboratory confirmation requires at least two detections of aPL within 6 weeks (not necessarily at the time of a relapse), according to the proposed initial criteria for classification of definite APS. |
| A definite diagnosis of CAPS All 4 criteria. |
Probable CAPS
|
| Thrombotic Risk Level | Type of aPL | Presence of Antibodies | Titer of Antibodies |
|---|---|---|---|
| High | Tripositivity or two types of antibodies: anticardiolipin (aCL) IgG or IgM, anti-β2-glycoprotein (aβ2-GPI) IgG or IgM, antiphosphatydylserine/prothrombin complex (aPS/Pt) IgG or IgM, including lupus antibodies (LA) | Long-term | High |
| Moderate | Two types of antibodies: aCL IgG or IgM, anti-β2-glycoprotein IgG or IgM, ACL, and aβ2-GPI aPS/Pt IgG or IgM but without LA | Long-term | High |
| Low | One type of antibody | Transitional | Low |
| Patients <50 years of age |
|
| Venous thromboembolism in an unusual location |
|
| Thrombosis of small vessels (microthrombosis) | |
| Recurrent venous thromboembolism |
|
| Obstetrical failure |
|
| Systemic lupus erythematosus | |
| Immune thrombocytopenia |
|
| Livedo reticularis |
|
| Unexplained, coincidentally detected prolongation of activated partial thromboplastin time (APTT) | |
| High-Risk Profile of Antiphospholipid Antibodies | Systemic Lupus Erythematosus Patients Without Clinical Symptoms | Non-Pregnant Women with a History of Obstetric Antiphospholipid Syndrome | |
|---|---|---|---|
| High-Risk Profile | Low-Risk Profile | Moderate or High-Risk Profile | |
| Low doses of acetylsalicylic acid (75–100 mg/day) | Low doses of acetylsalicylic acid (75–100 mg/day) or hydroxychloroquine | Low doses of acetylsalicylic acid (75–100 mg/day) should be considered | Low doses of acetylsalicylic acid (75–100 mg/day) should be considered after risk assessment |
| Patients with confirmed APS and a first episode of venous thromboembolism |
|
| Patients with confirmed APS and recurrence of venous thromboembolism despite VKA treatment (INR 2.0–3.0) |
|
| Patients with APS and a first episode of arterial thrombosis |
|
| Catastrophic APS |
|
| Obstetric APS |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stańczewska, A.; Szewczyk-Golec, K.; Hołyńska-Iwan, I. Antiphospholipid Syndrome—Diagnostic and Methodologic Approach. Metabolites 2025, 15, 500. https://doi.org/10.3390/metabo15080500
Stańczewska A, Szewczyk-Golec K, Hołyńska-Iwan I. Antiphospholipid Syndrome—Diagnostic and Methodologic Approach. Metabolites. 2025; 15(8):500. https://doi.org/10.3390/metabo15080500
Chicago/Turabian StyleStańczewska, Agata, Karolina Szewczyk-Golec, and Iga Hołyńska-Iwan. 2025. "Antiphospholipid Syndrome—Diagnostic and Methodologic Approach" Metabolites 15, no. 8: 500. https://doi.org/10.3390/metabo15080500
APA StyleStańczewska, A., Szewczyk-Golec, K., & Hołyńska-Iwan, I. (2025). Antiphospholipid Syndrome—Diagnostic and Methodologic Approach. Metabolites, 15(8), 500. https://doi.org/10.3390/metabo15080500