
metabCombiner 2.0: Disparate Multi-Dataset Feature Alignment for LC-MS Metabolomics

 , ,
, ,  , ,
, ,  , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
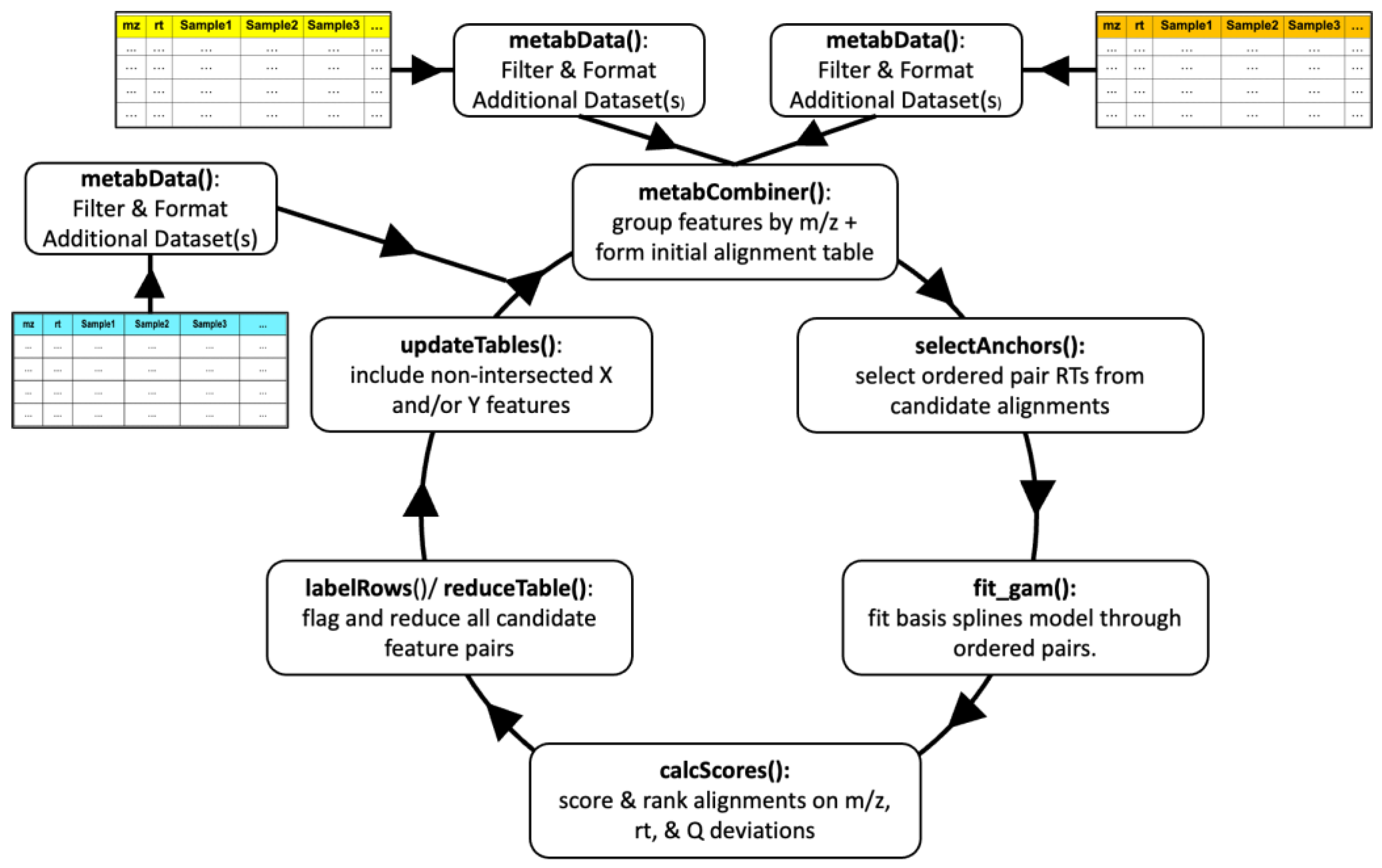
2.1. Workflow Overview
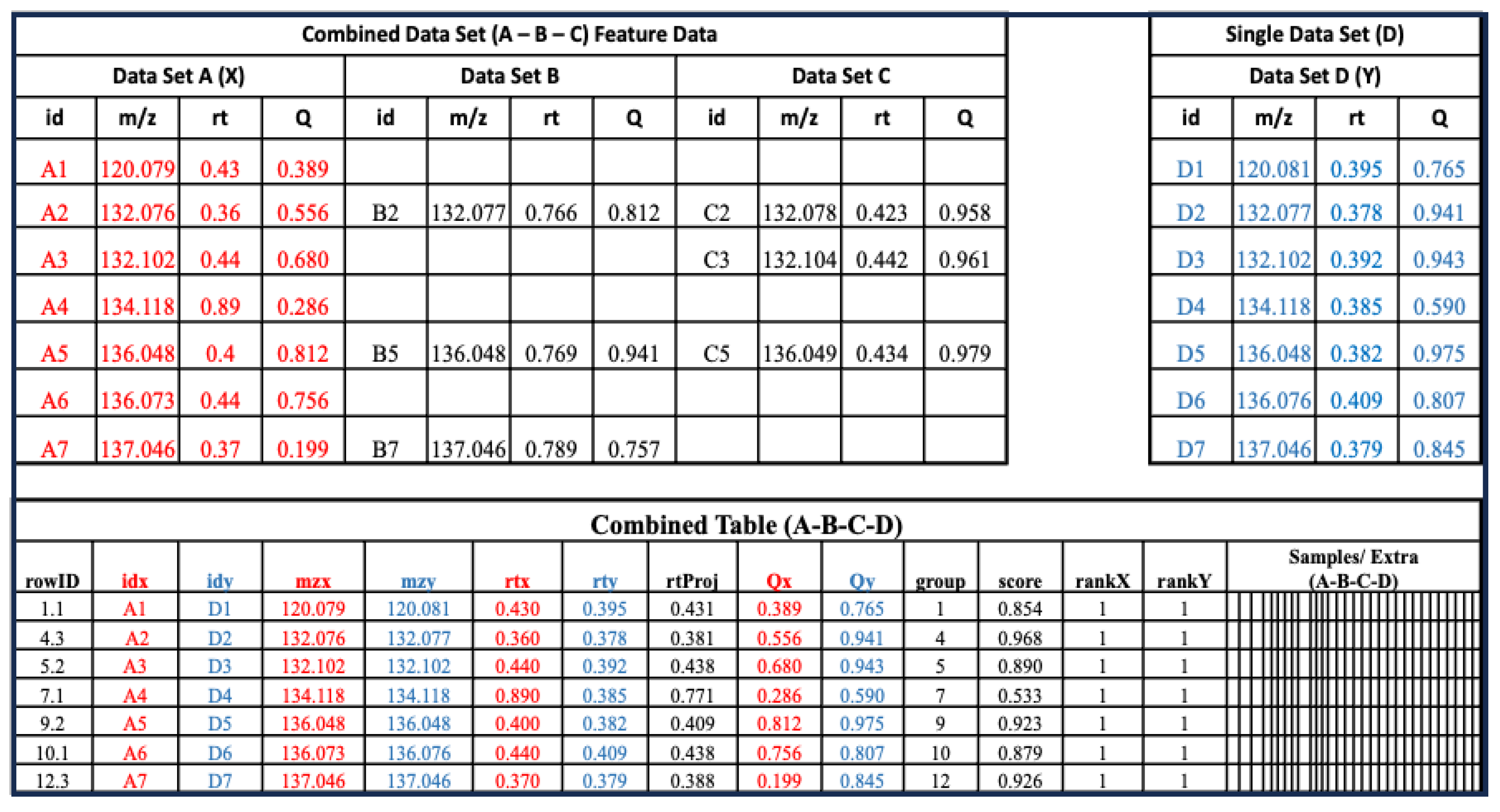
2.2. Terminology
2.3. Initial Data Processing
2.4. Combined Dataset Construction
2.5. RT Mapping and Score Calculation
2.6. Aligned Feature Table Annotation and Reduction
2.7. Assembling Aligned and Non-Aligned Features
2.8. batchCombine: Extension to Multi-Batch LC-MS Alignment Tasks
2.9. metabCombiner Online
2.10. Unknown Lipids Consortium Study
2.10.1. Study Design and Experimental Methods
2.10.2. Data Processing and Metabolite Identification
2.10.3. Stepwise Alignment with metabCombiner
2.11. Early Life Exposures in Mexico to Eevironmental Toxicants (ELEMENT)
2.11.1. Study Design and Experimental Methods
2.11.2. Data Pre-Processing
2.11.3. Alignment with batchCombine
3. Results
3.1. Aligning Multi-Laboratory Untargeted Lipidomics with metabCombiner 2.0
3.1.1. Unknown Lipids Study
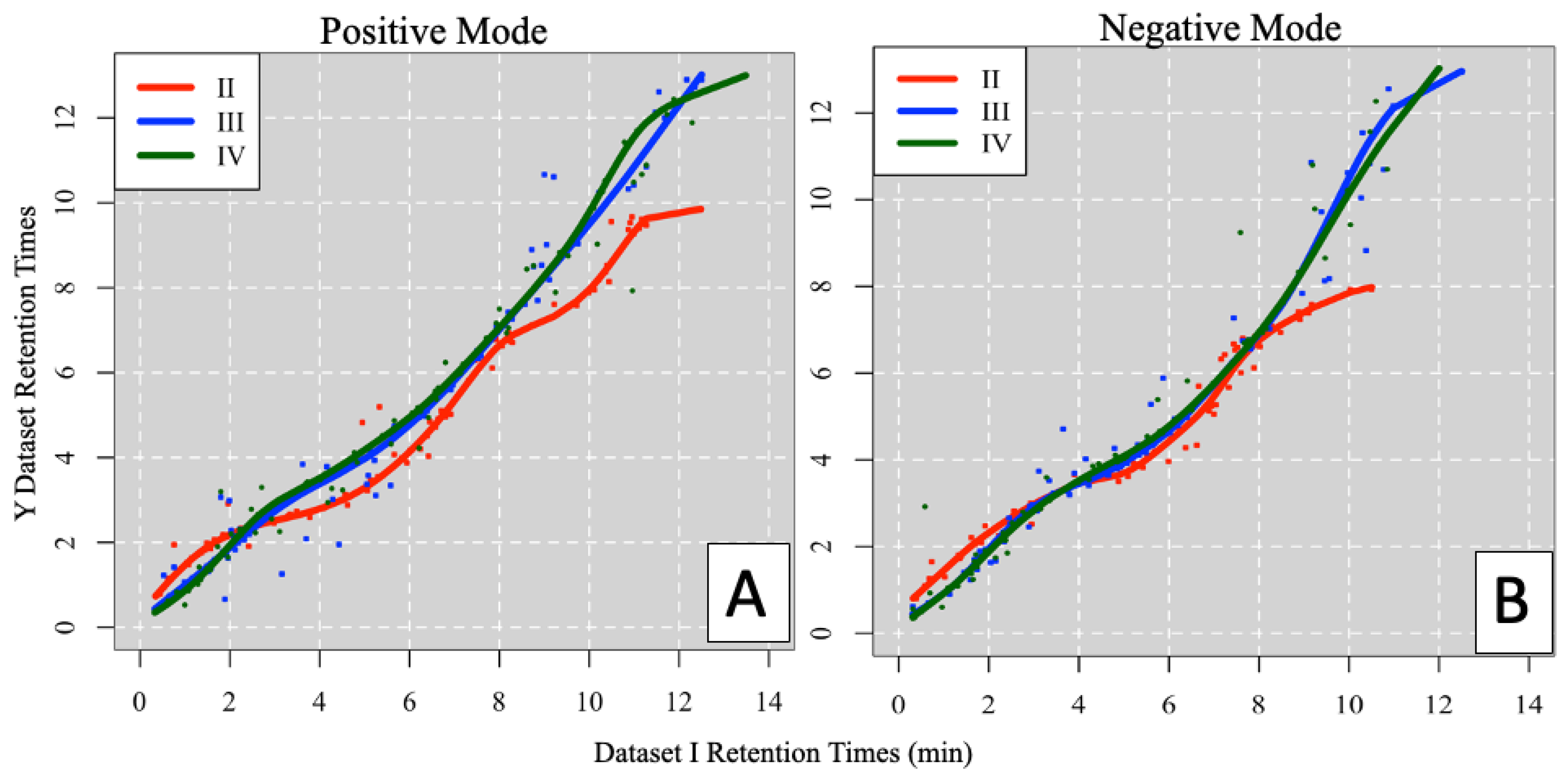
3.1.2. m/z Grouping and RT Mapping Results
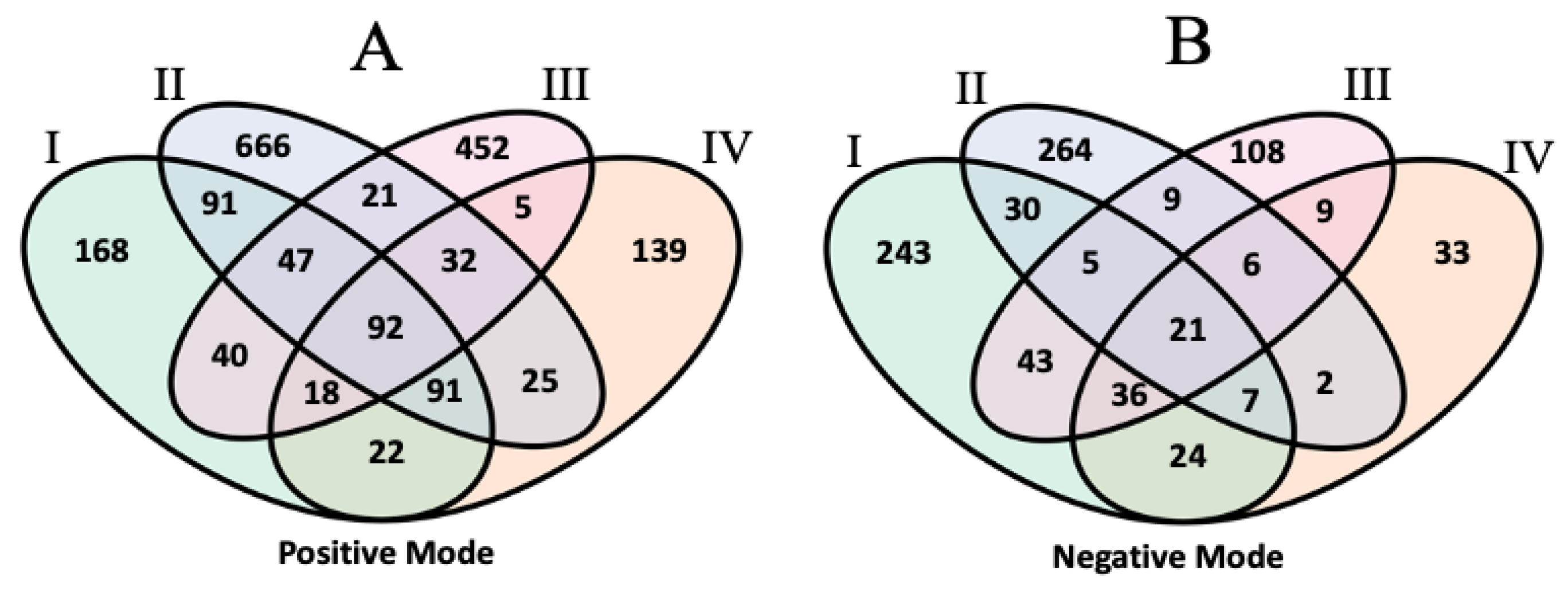
3.1.3. Inter-Laboratory Alignment Results
3.1.4. Comparative Lipid Annotation Analysis
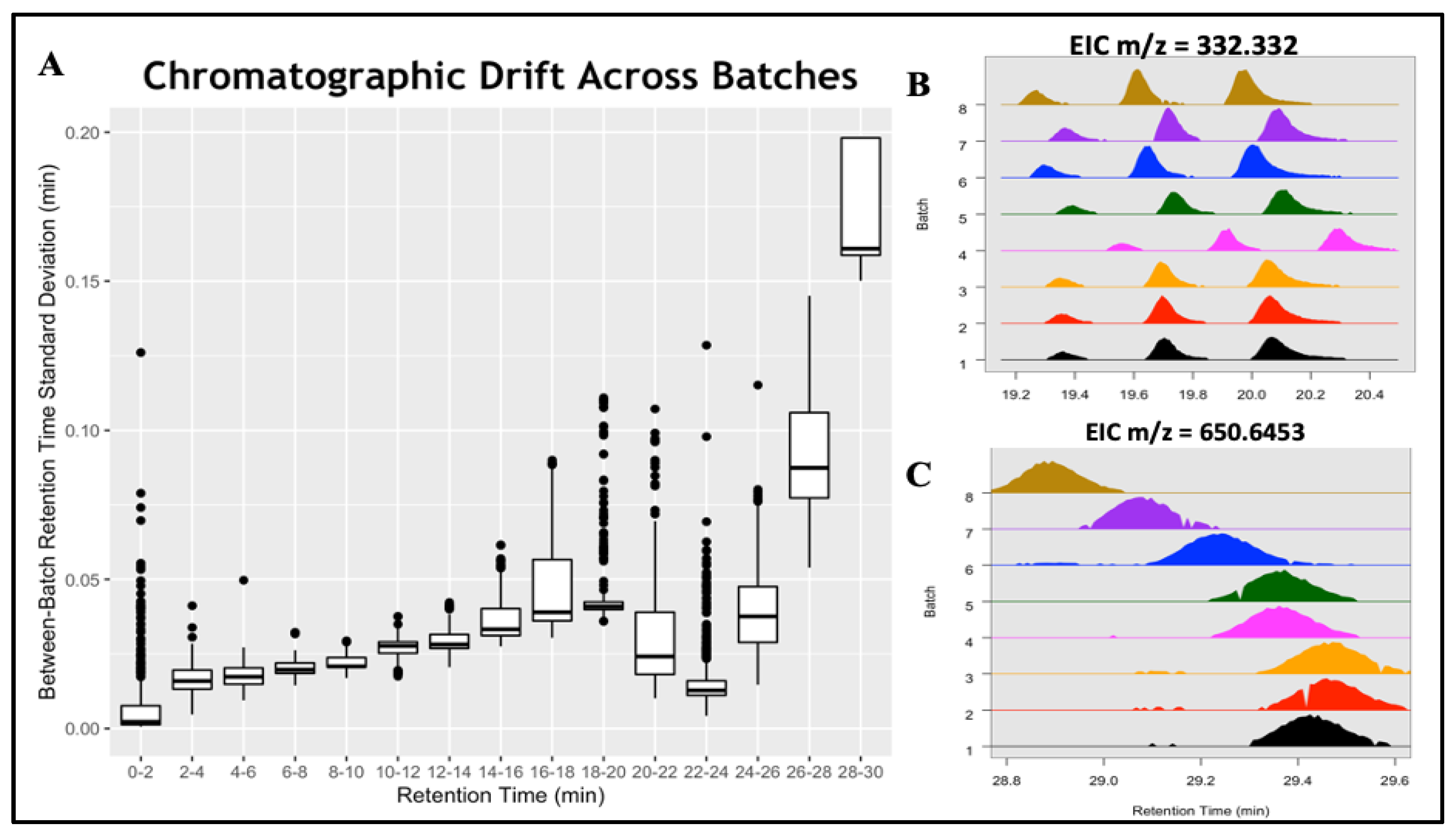
3.2. Alignment of the Multi-Batch ELEMENT Study with batchCombine
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schrimpe-Rutledge, A.C.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A. Untargeted Metabolomics Strategies—Challenges and Emerging Directions. J. Am. Soc. Mass. Spectrom. 2016, 27, 1897–1905. [Google Scholar] [CrossRef]
- Smith, R.; Ventura, D.; Prince, J.T. LC-MS Alignment in Theory and Practice: A Comprehensive Algorithmic Review. Brief. Bioinform. 2015, 16, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Boswell, P.G.; Schellenberg, J.R.; Carr, P.W.; Cohen, J.D.; Hegeman, A.D. A Study on Retention “Projection” as a Supplementary Means for Compound Identification by Liquid Chromatography-Mass Spectrometry Capable of Predicting Retention with Different Gradients, Flow Rates, and Instruments. J. Chromatogr. A 2011, 1218, 6732–6741. [Google Scholar] [CrossRef] [PubMed]
- Christin, C.; Smilde, A.K.; Hoefsloot, H.C.J.; Suits, F.; Bischoff, R.; Horvatovich, P.L. Optimized Time Alignment Algorithm for LC−MS Data: Correlation Optimized Warping Using Component Detection Algorithm-Selected Mass Chromatograms. Anal. Chem. 2008, 80, 7012–7021. [Google Scholar] [CrossRef] [PubMed]
- Abate-Pella, D.; Freund, D.M.; Ma, Y.; Simón-Manso, Y.; Hollender, J.; Broeckling, C.D.; Huhman, D.V.; Krokhin, O.V.; Stoll, D.R.; Hegeman, A.D.; et al. Retention Projection Enables Accurate Calculation of Liquid Chromatographic Retention Times across Labs and Methods. J. Chromatogr. A 2015, 1412, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Brunius, C.; Shi, L.; Landberg, R. Large-Scale Untargeted LC-MS Metabolomics Data Correction Using between-Batch Feature Alignment and Cluster-Based within-Batch Signal Intensity Drift Correction. Metabolomics 2016, 12, 173. [Google Scholar] [CrossRef]
- Liu, Q.; Walker, D.; Uppal, K.; Liu, Z.; Ma, C.; Tran, V.; Li, S.; Jones, D.P.; Yu, T. Addressing the Batch Effect Issue for LC/MS Metabolomics Data in Data Preprocessing. Sci. Rep. 2020, 10, 13856. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-T.; Wang, Y.; Wang, Y.; Ebbels, T.; Karaman, I.; Graça, G.; Pinto, R.; Herrington, D.M.; Wang, Y.; Yu, G. Targeted Realignment of LC-MS Profiles by Neighbor-Wise Compound-Specific Graphical Time Warping with Misalignment Detection. Bioinformatics 2020, 36, 2862–2871. [Google Scholar] [CrossRef]
- Hsu, Y.-H.H.; Churchhouse, C.; Pers, T.H.; Mercader, J.M.; Metspalu, A.; Fischer, K.; Fortney, K.; Morgen, E.K.; Gonzalez, C.; Gonzalez, M.E.; et al. PAIRUP-MS: Pathway Analysis and Imputation to Relate Unknowns in Profiles from Mass Spectrometry-Based Metabolite Data. PLoS Comput. Biol. 2019, 15, e1006734. [Google Scholar] [CrossRef]
- Mak, T.D.; Goudarzi, M.; Laiakis, E.C.; Stein, S.E. Disparate Metabolomics Data Reassembler: A Novel Algorithm for Agglomerating Incongruent LC-MS Metabolomics Datasets. Anal. Chem. 2020, 92, 5231–5239. [Google Scholar] [CrossRef]
- Climaco Pinto, R.; Karaman, I.; Lewis, M.R.; Hällqvist, J.; Kaluarachchi, M.; Graça, G.; Chekmeneva, E.; Durainayagam, B.; Ghanbari, M.; Ikram, M.A.; et al. Finding Correspondence between Metabolomic Features in Untargeted Liquid Chromatography–Mass Spectrometry Metabolomics Datasets. Anal. Chem. 2022, 94, 5493–5503. [Google Scholar] [CrossRef]
- Habra, H.; Kachman, M.; Bullock, K.; Clish, C.; Evans, C.R.; Karnovsky, A. metabCombiner: Paired Untargeted LC-HRMS Metabolomics Feature Matching and Concatenation of Disparately Acquired Data Sets. Anal. Chem. 2021, 93, 5028–5036. [Google Scholar] [CrossRef] [PubMed]
- Perng, W.; Tamayo-Ortiz, M.; Tang, L.; Sánchez, B.N.; Cantoral, A.; Meeker, J.D.; Dolinoy, D.C.; Roberts, E.F.; Martinez-Mier, E.A.; Lamadrid-Figueroa, H.; et al. Early Life Exposure in Mexico to ENvironmental Toxicants (ELEMENT) Project. BMJ Open 2019, 9, e030427. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular Framework for Processing, Visualizing, and Analyzing Mass Spectrometry-Based Molecular Profile Data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-Independent MS/MS Deconvolution for Comprehensive Metabolome Analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Conway, C.; Rempfert, K.R.; Kyle, J.E.; Colby, S.M.; Gaul, D.A.; Habra, H.; Kong, F.; Bloodsworth, K.J.; Allen, D.; et al. The Unknown Lipids Project: Harmonized Methods Improve Compound Identification and Data Reproducibility in an Inter-Laboratory Untargeted Lipidomics Study. biorXiv 2023. [Google Scholar] [CrossRef]
- Myers, O.D.; Sumner, S.J.; Li, S.; Barnes, S.; Du, X. One Step Forward for Reducing False Positive and False Negative Compound Identifications from Mass Spectrometry Metabolomics Data: New Algorithms for Constructing Extracted Ion Chromatograms and Detecting Chromatographic Peaks. Anal. Chem. 2017, 89, 8696–8703. [Google Scholar] [CrossRef]
- Rodríguez-Carmona, Y.; Meijer, J.L.; Zhou, Y.; Jansen, E.C.; Perng, W.; Banker, M.; Song, P.X.K.; Téllez-Rojo, M.M.; Cantoral, A.; Peterson, K.E. Metabolomics Reveals Sex-specific Pathways Associated with Changes in Adiposity and Muscle Mass in a Cohort of Mexican Adolescents. Pediatr. Obes. 2022, 17, e12887. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A Cross-Platform Toolkit for Mass Spectrometry and Proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Böttcher, C.; Neumann, S. Highly Sensitive Feature Detection for High Resolution LC/MS. BMC Bioinform. 2008, 9, 504. [Google Scholar] [CrossRef] [PubMed]
- Hohrenk, L.L.; Itzel, F.; Baetz, N.; Tuerk, J.; Vosough, M.; Schmidt, T.C. Comparison of Software Tools for Liquid Chromatography–High-Resolution Mass Spectrometry Data Processing in Nontarget Screening of Environmental Samples. Anal. Chem. 2020, 92, 1898–1907. [Google Scholar] [CrossRef]
- Clark, T.N.; Houriet, J.; Vidar, W.S.; Kellogg, J.J.; Todd, D.A.; Cech, N.B.; Linington, R.G. Interlaboratory Comparison of Untargeted Mass Spectrometry Data Uncovers Underlying Causes for Variability. J. Nat. Prod. 2021, 84, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Liebisch, G.; Vizcaíno, J.A.; Köfeler, H.; Trötzmüller, M.; Griffiths, W.J.; Schmitz, G.; Spener, F.; Wakelam, M.J.O. Shorthand Notation for Lipid Structures Derived from Mass Spectrometry. J. Lipid Res. 2013, 54, 1523–1530. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
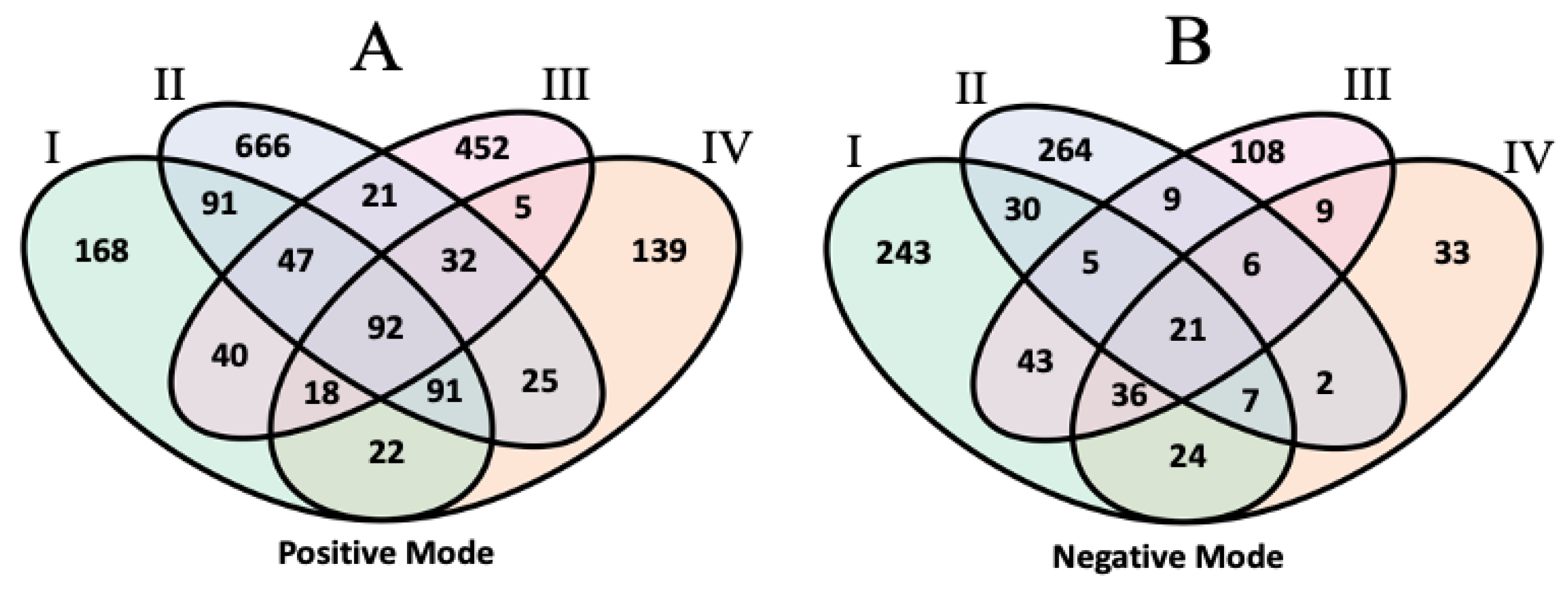
| Positive | Negative | |||||
|---|---|---|---|---|---|---|
| Detected Across Four Datasets | 601 | 215 | ||||
| Detected in I, II, and III | 155 | 30 | ||||
| Detected in I, II, and IV | 280 | 115 | ||||
| Detected in I, III, and IV | 580 | 533 | ||||
| Detected in I and II | 156 | 211 | ||||
| Detected in I and III | 1053 | 398 | ||||
| Detected in I and IV | 743 | 670 | ||||
| Detected in I only | 5202 | 3691 | ||||
| II | III | IV | II | III | IV | |
| Detected Outside I | 2617 | 22,516 | 17,165 | 1676 | 6132 | 7402 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habra, H.; Meijer, J.L.; Shen, T.; Fiehn, O.; Gaul, D.A.; Fernández, F.M.; Rempfert, K.R.; Metz, T.O.; Peterson, K.E.; Evans, C.R.; et al. metabCombiner 2.0: Disparate Multi-Dataset Feature Alignment for LC-MS Metabolomics. Metabolites 2024, 14, 125. https://doi.org/10.3390/metabo14020125
Habra H, Meijer JL, Shen T, Fiehn O, Gaul DA, Fernández FM, Rempfert KR, Metz TO, Peterson KE, Evans CR, et al. metabCombiner 2.0: Disparate Multi-Dataset Feature Alignment for LC-MS Metabolomics. Metabolites. 2024; 14(2):125. https://doi.org/10.3390/metabo14020125
Chicago/Turabian StyleHabra, Hani, Jennifer L. Meijer, Tong Shen, Oliver Fiehn, David A. Gaul, Facundo M. Fernández, Kaitlin R. Rempfert, Thomas O. Metz, Karen E. Peterson, Charles R. Evans, and et al. 2024. "metabCombiner 2.0: Disparate Multi-Dataset Feature Alignment for LC-MS Metabolomics" Metabolites 14, no. 2: 125. https://doi.org/10.3390/metabo14020125
APA StyleHabra, H., Meijer, J. L., Shen, T., Fiehn, O., Gaul, D. A., Fernández, F. M., Rempfert, K. R., Metz, T. O., Peterson, K. E., Evans, C. R., & Karnovsky, A. (2024). metabCombiner 2.0: Disparate Multi-Dataset Feature Alignment for LC-MS Metabolomics. Metabolites, 14(2), 125. https://doi.org/10.3390/metabo14020125