Ligand-Based and Structured-Based In Silico Repurposing Approaches to Predict Inhibitors of SARS-CoV-2 Mpro Protein

 ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Section
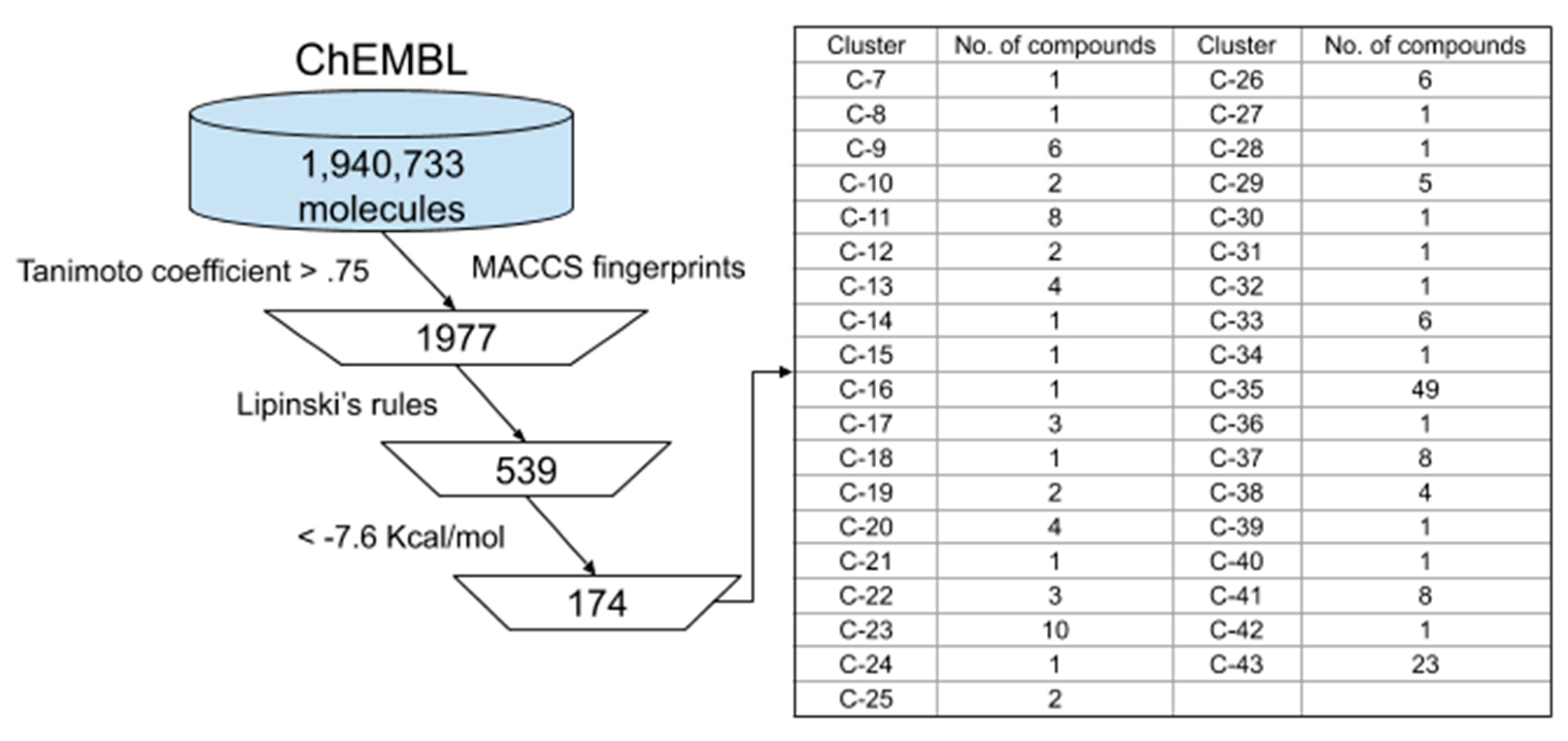
2.1. Data Preparation
2.2. Molecular Docking
2.3. Interaction Pattern Calculation
3. Results and Discussion
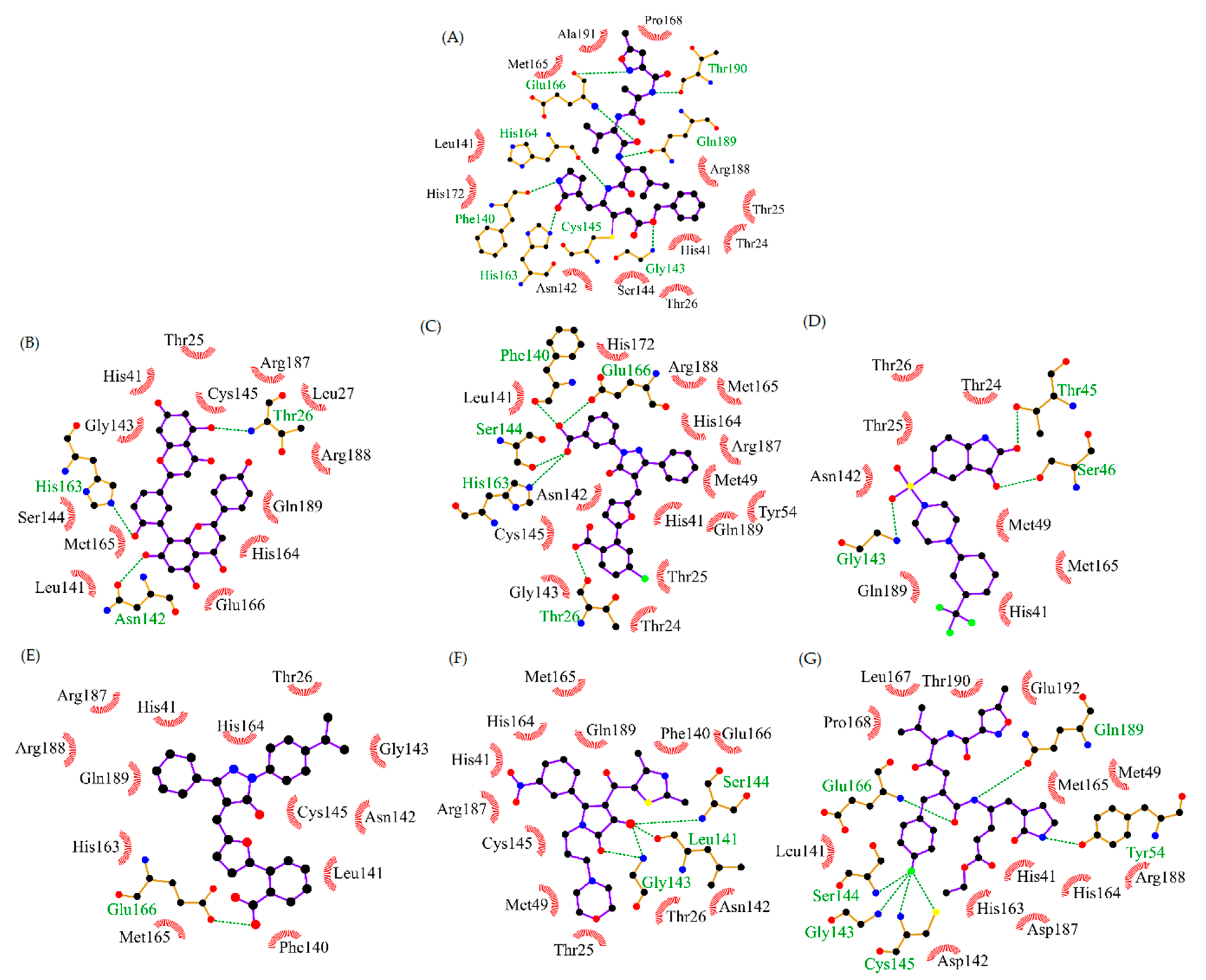
3.1. Screening of Known CoV Inhibitors
3.2. Ligand-Based ChEMBL Database Screening
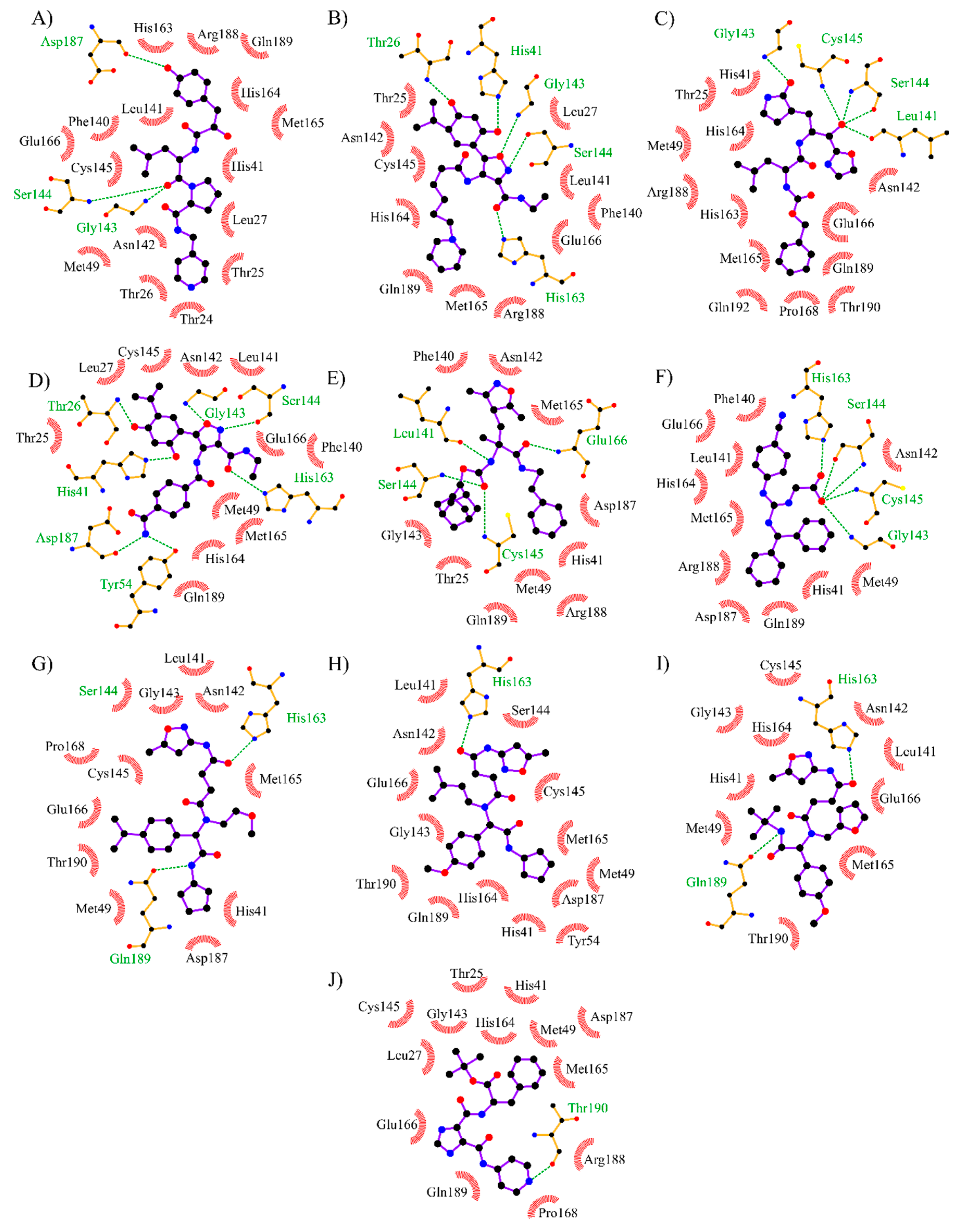
3.3. Virtual Screening of Approved/Experimental Drugs
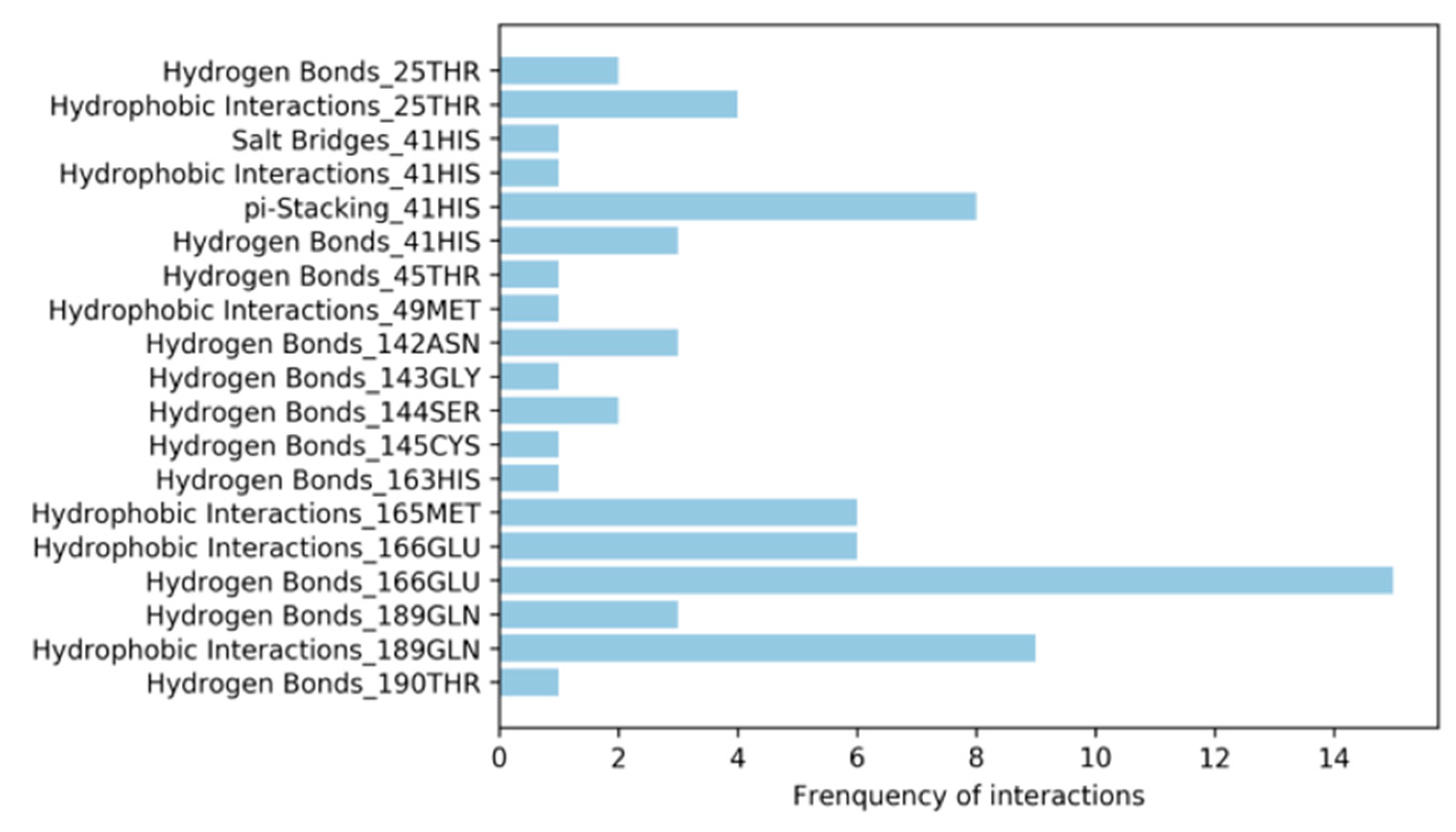
3.4. Analysis of Protein-Ligand Interactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cascella, M.; Rajnik, M.; Cuomo, A.; Dulebohn, S.C.; Di Napoli., R. Features, Evaluation and Treatment Coronavirus (COVID-19); StatPearls, 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK554776/ (accessed on 15 October 2020).
- Chen, L.R.; Wang, Y.C.; Yi, W.L.; Chou, S.Y.; Chen, S.F.; Lee, T.L.; Wu, Y.T.; Kuo, C.J.; Chen, T.S.S.; Juang, S.H. Synthesis and evaluation of isatin derivatives as effective SARS coronavirus 3CL protease inhibitors. Bioorganic Med. Chem. Lett. 2005, 15, 3058–3062. [Google Scholar] [CrossRef]
- Mousavizadeh, L.; Ghasemi, S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J. Microbiol. Immunol. Infect. 2020, 10. [Google Scholar] [CrossRef]
- Shyr, Z.A.; Gorshkov, K.; Chen, C.Z.; Zheng, W. Drug Discovery Strategies for SARS-CoV-2. J. Pharmacol. Exp. Ther. 2020, 375, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Kumar, Y.; Singh, H.; Patel, C.N. In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J. Infect. Public Health 2020, 13, 1210–1223. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhang, L.; Liu, X.; Li, F.; Ma, R.; Zhu, Z.; Zhang, J.; Wu, J.; Shi, Y.; Pan, Y.; et al. Repurposing low-molecular-weight drugs against the main protease of severe acute respiratory syndrome coronavirus 2. J. Phys. Chem. Lett. 2020, 11, 7267–7272. [Google Scholar] [CrossRef]
- Meyer-Almes, F.J. Repurposing approved drugs as potential inhibitors of 3CL-protease of SARS-CoV-2: Virtual screening and structure based drug design. Comput. Biol. Chem. 2020, 88, 107351. [Google Scholar] [CrossRef]
- Touret, F.; Gilles, M.; Barral, K.; Nougairède, A.; van Helden, J.; Decroly, E.; de Lamballerie, X.; Coutard, B. In vitro screening of a FDA approved chemical library reveals potential inhibitors of SARS-CoV-2 replication. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Sharma, S.; Deep, S. In-Silico Drug Repurposing for Targeting SARS-CoV-2 Mpro. 2020. Available online: https://chemrxiv.org/ndownloader/files/22457816 (accessed on 15 August 2020).
- Smith, M.; Smith, J.C. Repurposing Therapeutics for COVID-19: Supercomputer-Based Docking to the SARS-CoV-2 Viral Spike Protein and Viral Spike Protein-Human ACE2 Interface. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Jin, Y.; Yang, H.; Ji, W.; Wu, W.; Chen, S.; Zhang, W.; Duan, G. Virology, epidemiology, pathogenesis, and control of covid-19. Viruses 2020, 12, 372. [Google Scholar] [CrossRef]
- Kumar, V.; Tan, K.; Wang, Y.; Lin, S.; Liang, P. Identification, synthesis and evaluation of SARS-CoV and MERS-CoV 3C-like protease inhibitors. Bioorganic Med. Chem. 2016, 24, 3035–3042. [Google Scholar] [CrossRef]
- Cavasotto, C.N.; Di Filippo, J.I. In silico Drug Repurposing for COVID-19: Targeting SARS-CoV-2 Proteins through Docking and Consensus Ranking. Mol. Inform. 2020, 2000115, 1–9. [Google Scholar] [CrossRef]
- Ferraz, W.R.; Gomes, R.A.S.; Novaes, A.L.; Goulart Trossini, G.H. Ligand and structure-based virtual screening applied to the SARS-CoV-2 main protease: An in silico repurposing study. Future Med. Chem. 2020. [Google Scholar] [CrossRef]
- Wang, J. Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) through Computational Drug Repurposing Study. J. Chem. Inf. Model. 2020, 60, 3277–3286. [Google Scholar] [CrossRef] [PubMed]
- Battisti, V.; Wieder, O.; Garon, A.; Seidel, T.; Urban, E.; Langer, T. A Computational Approach to Identify Potential Novel Inhibitors against the Coronavirus SARS-CoV-2. Mol. Inform. 2020, 39, 1–8. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. Dogsitescorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2010, 30, 2785–2791. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Autodock vina: Improving the speed and accuracy of docking. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar]
- Culleta, G.; Gulotta, M.R.; Perricone, U.; Zappalà, M.; Almerico, A.M.; Tutone, M. Exploring the SARS-CoV-2 Proteome in the Search of Potential Inhibitors via Structure-Based Pharmacophore Modeling/Docking Approach. Computation 2020, 8, 77. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Khan, M.F.; Alam, M.M.; Verma, G.; Akhtar, W.; Akhter, M.; Shaquiquzzaman, M. The therapeutic voyage of pyrazole and its analogs: A review. Eur. J. Med. Chem. 2016, 120, 170–201. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.H. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: Peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef]
- Singh, P.; Sharma, A.; Nandi, S.P. Identification of Potent Inhibitors of COVID-19 Main Protease Enzyme by Molecular Docking Study. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Liu, W.; Zhu, H.M.; Niu, G.J.; Shi, E.Z.; Chen, J.; Sun, B.; Chen, W.Q.; Zhou, H.G.; Yang, C. Synthesis, modification and docking studies of 5-sulfonyl isatin derivatives as SARS-CoV 3C-like protease inhibitors. Bioorganic Med. Chem. 2014, 22, 292–302. [Google Scholar] [CrossRef]
- Kumari, A.; Singh, R.K. Morpholine as ubiquitous pharmacophore in medicinal chemistry: Deep insight into the structure-activity relationship (SAR). Bioorg. Chem. 2020, 96, 103578. [Google Scholar] [CrossRef]
- Shie, J.J.; Fang, J.M.; Kuo, T.H.; Kuo, C.J.; Liang, P.H.; Huang, H.J.; Wu, Y.T.; Jan, J.T.; Cheng, Y.S.E.; Wong, C.H. Inhibition of the severe acute respiratory syndrome 3CL protease by peptidomimetic α,β-unsaturated esters. Bioorganic Med. Chem. 2005, 13, 5240–5252. [Google Scholar] [CrossRef]
- Sandgren, V.; Agback, T.; Johansson, P.O.; Lindberg, J.; Kvarnström, I.; Samuelsson, B.; Belda, O.; Dahlgren, A. Highly potent macrocyclic BACE-1 inhibitors incorporating a hydroxyethylamine core: Design, synthesis and X-ray crystal structures of enzyme inhibitor complexes. Bioorganic Med. Chem. 2012, 20, 4377–4389. [Google Scholar] [CrossRef][Green Version]
- Terracciano, S.; Bruno, I.; D’Amico, E.; Bifulco, G.; Zampella, A.; Sepe, V.; Smith, C.D.; Riccio, R. Synthetic and pharmacological studies on new simplified analogues of the potent actin-targeting Jaspamide. Bioorganic Med. Chem. 2008, 16, 6580–6588. [Google Scholar] [CrossRef]
- Wu, C.Y.; Jan, J.T.; Ma, S.H.; Kuo, C.J.; Juan, H.F.; Cheng, Y.S.E.; Hsu, H.H.; Huang, H.C.; Wu, D.; Brik, A.; et al. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 10012–10017. [Google Scholar] [CrossRef] [PubMed]
- Gassel, M.; Breitenlechner, C.B.; König, N.; Huber, R.; Engh, R.A.; Bossemeyer, D. The protein kinase C inhibitor bisindolyl maleimide 2 binds with reversed orientations to different conformations of protein kinase A. J. Biol. Chem. 2004, 279, 23679–23690. [Google Scholar] [CrossRef] [PubMed]
- Witters, H.; Freyberger, A.; Smits, K.; Vangenechten, C.; Lofink, W.; Weimer, M.; Bremer, S.; Ahr, P.H.J.; Berckmans, P. The assessment of estrogenic or anti-estrogenic activity of chemicals by the human stably transfected estrogen sensitive MELN cell line: Results of test performance and transferability. Reprod. Toxicol. 2010, 30, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 16, 1–18. [Google Scholar]
- Hall, D.C.J.; Ji, H.F. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Med. Infect. Dis. 2020, 35, 101646. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juárez-Saldívar, A.; Lara-Ramírez, E.E.; Reyes-Espinosa, F.; Paz-González, A.D.; Villalobos-Rocha, J.C.; Rivera, G. Ligand-Based and Structured-Based In Silico Repurposing Approaches to Predict Inhibitors of SARS-CoV-2 Mpro Protein. Sci. Pharm. 2020, 88, 54. https://doi.org/10.3390/scipharm88040054
Juárez-Saldívar A, Lara-Ramírez EE, Reyes-Espinosa F, Paz-González AD, Villalobos-Rocha JC, Rivera G. Ligand-Based and Structured-Based In Silico Repurposing Approaches to Predict Inhibitors of SARS-CoV-2 Mpro Protein. Scientia Pharmaceutica. 2020; 88(4):54. https://doi.org/10.3390/scipharm88040054
Chicago/Turabian StyleJuárez-Saldívar, Alfredo, Edgar E. Lara-Ramírez, Francisco Reyes-Espinosa, Alma D. Paz-González, Juan Carlos Villalobos-Rocha, and Gildardo Rivera. 2020. "Ligand-Based and Structured-Based In Silico Repurposing Approaches to Predict Inhibitors of SARS-CoV-2 Mpro Protein" Scientia Pharmaceutica 88, no. 4: 54. https://doi.org/10.3390/scipharm88040054
APA StyleJuárez-Saldívar, A., Lara-Ramírez, E. E., Reyes-Espinosa, F., Paz-González, A. D., Villalobos-Rocha, J. C., & Rivera, G. (2020). Ligand-Based and Structured-Based In Silico Repurposing Approaches to Predict Inhibitors of SARS-CoV-2 Mpro Protein. Scientia Pharmaceutica, 88(4), 54. https://doi.org/10.3390/scipharm88040054