Family A and B DNA Polymerases in Cancer: Opportunities for Therapeutic Interventions


Abstract
1. Introduction
2. Family A DNA Polymerases
2.1. DNA Polymerase θ
2.2. DNA Polymerase ν
3. Family B DNA Polymerases
3.1 DNA Polymerase α
3.2 DNA Polymerase δ and ε
3.3. Polymerase ζ
4. Potential Therapeutic Interventions
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tubbs, A.; Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Wood, R.D. Quality control by DNA repair. Science 1999, 286, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Bessman, M.J.; Kornberg, A.; Lehman, I.R.; Simms, E.S. Enzymic synthesis of deoxyribonucleic acid. Biochim. Biophys. Acta 1956, 21, 197–198. [Google Scholar] [PubMed]
- Kornberg, A. Biologic synthesis of deoxyribonucleic acid. Science 1960, 131, 1503–1508. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, A.; Lieberman, I.; Simms, E.S. Enzymatic synthesis and properties of 5-phosphoribosylpyrophosphate. J. Biol. Chem. 1955, 215, 389–402. [Google Scholar] [PubMed]
- Lehman, I.R. Discovery of DNA polymerase. J. Biol. Chem. 2003, 278, 34733–34738. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C. The eureka enzyme: The discovery of DNA polymerase. Nat. Rev. Mol. Cell Biol. 2006, 7, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Delarue, M.; Poch, O.; Tordo, N.; Moras, D.; Argos, P. An attempt to unify the structure of polymerases. Protein Eng. 1990, 3, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Ishino, Y.; Komori, K.; Cann, I.K.; Koga, Y. A novel DNA polymerase family found in archaea. J. Bacteriol. 1998, 180, 2232–2236. [Google Scholar] [PubMed]
- Braithwaite, D.K.; Ito, J. Compilation, alignment, and phylogenetic relationships of DNA polymerases. Nucleic Acids Res. 1993, 21, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Cann, I.K.; Ishino, Y. Archaeal DNA replication: Identifying the pieces to solve a puzzle. Genetics 1999, 152, 1249–1267. [Google Scholar] [PubMed]
- Ohmori, H.; Friedberg, E.C.; Fuchs, R.P.; Goodman, M.F.; Hanaoka, F.; Hinkle, D.; Kunkel, T.A.; Lawrence, C.W.; Livneh, Z.; Nohmi, T.; et al. The y-family of DNA polymerases. Mol. Cell 2001, 8, 7–8. [Google Scholar] [CrossRef]
- Patel, P.H.; Loeb, L.A. Getting a grip on how DNA polymerases function. Nat. Struct. Biol. 2001, 8, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Sandalli, C.; Singh, K.; Modak, M.J.; Ketkar, A.; Canakci, S.; Demir, I.; Belduz, A.O. A new DNA polymerase i from geobacillus caldoxylosilyticus tk4: Cloning, characterization, and mutational analysis of two aromatic residues. Appl. Microbiol. Biotechnol. 2009, 84, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Koonin, E.V.; Leipe, D.D.; Aravind, L. Origin and evolution of the archaeo-eukaryotic primase superfamily and related palm–domain proteins: Structural insights and new members. Nucleic Acids Res. 2005, 33, 3875–3896. [Google Scholar] [CrossRef] [PubMed]
- Ito, J.; Braithwaite, D.K. Compilation and alignment of DNA polymerase sequences. Nucleic Acids Res. 1991, 19, 4045–4057. [Google Scholar] [CrossRef] [PubMed]
- Baltimore, D. RNA-dependent DNA polymerase in virions of rna tumour viruses. Nature 1970, 226, 1209–1211. [Google Scholar] [CrossRef] [PubMed]
- Temin, H.M.; Mizutani, S. RNA-dependent DNA polymerase in virions of rous sarcoma virus. Nature 1970, 226, 1211–1213. [Google Scholar] [CrossRef] [PubMed]
- Klenow, H.; Henningsen, I. Selective elimination of the exonuclease activity of the deoxyribonucleic acid polymerase from escherichia coli b by limited proteolysis. Proc. Natl. Acad. Sci. USA 1970, 65, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Polesky, A.H.; Dahlberg, M.E.; Benkovic, S.J.; Grindley, N.D.; Joyce, C.M. Side chains involved in catalysis of the polymerase reaction of DNA polymerase I from Escherichia coli. J. Biol. Chem. 1992, 267, 8417–8428. [Google Scholar] [PubMed]
- Polesky, A.H.; Steitz, T.A.; Grindley, N.D.; Joyce, C.M. Identification of residues critical for the polymerase activity of the klenow fragment of DNA polymerase I from Escherichia coli. J. Biol. Chem. 1990, 265, 14579–14591. [Google Scholar] [PubMed]
- Astatke, M.; Ng, K.; Grindley, N.D.; Joyce, C.M. A single side chain prevents escherichia coli DNA polymerase I (klenow fragment) from incorporating ribonucleotides. Proc. Natl. Acad. Sci. USA 1998, 95, 3402–3407. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.M., Jr.; Grindley, N.D.; Joyce, C.M. Interaction of DNA polymerase I (klenow fragment) with the single-stranded template beyond the site of synthesis. Biochemistry 2003, 42, 2373–2385. [Google Scholar] [CrossRef] [PubMed]
- Joyce, C.M.; Steitz, T.A. Function and structure relationships in DNA polymerases. Annu. Rev. Biochem. 1994, 63, 777–822. [Google Scholar] [CrossRef] [PubMed]
- Bermek, O.; Grindley, N.D.; Joyce, C.M. Prechemistry nucleotide selection checkpoints in the reaction pathway of DNA polymerase I and roles of Glu710 and Tyr766. Biochemistry 2013, 52, 6258–6274. [Google Scholar] [CrossRef] [PubMed]
- Lam, W.C.; Thompson, E.H.; Potapova, O.; Sun, X.C.; Joyce, C.M.; Millar, D.P. 3′-5′ exonuclease of klenow fragment: Role of amino acid residues within the single-stranded DNA binding region in exonucleolysis and duplex DNA melting. Biochemistry 2002, 41, 3943–3951. [Google Scholar] [CrossRef] [PubMed]
- Pandey, V.N.; Williams, K.R.; Stone, K.L.; Modak, M.J. Photoaffinity labeling of the thymidine triphosphate binding domain in Escherichia coli DNA polymerase I: Identification of histidine-881 as the site of cross-linking. Biochemistry 1987, 26, 7744–7748. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.D.; Pandey, V.N.; Modak, M.J. Properties of tyrosine 766→serine mutant of Escherichia coli DNA polymerase I: Template-specific effects. Biochemistry 1994, 33, 11868–11874. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Modak, M.J. A unified DNA- and dNTP-binding mode for DNA polymerases. Trends Biochem. Sci. 1998, 23, 277–281. [Google Scholar] [CrossRef]
- Tuske, S.; Singh, K.; Kaushik, N.; Modak, M.J. The J-helix of Escherichia coli DNA polymerase I (Klenow fragment) regulates polymerase and 3′-5′-exonuclease functions. J. Biol. Chem. 2000, 275, 23759–23768. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Modak, M.J. Presence of 18-a long hydrogen bond track in the active site of escherichia coli DNA polymerase i (klenow fragment). Its requirement in the stabilization of enzyme-template-primer complex. J. Biol. Chem. 2003, 278, 11289–11302. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Modak, M.J. Contribution of polar residues of the J-helix in the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I (Klenow fragment): Q677 regulates the removal of terminal mismatch. Biochemistry 2005, 44, 8101–8110. [Google Scholar] [CrossRef] [PubMed]
- Kukreti, P.; Singh, K.; Ketkar, A.; Modak, M.J. Identification of a new motif required for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I (Klenow fragment): The RRRY motif is necessary for the binding of single-stranded DNA substrate and the template strand of the mismatched duplex. J. Biol. Chem. 2008, 283, 17979–17990. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Srivastava, A.; Patel, S.S.; Modak, M.J. Participation of the fingers subdomain of Escherichia coli DNA polymerase I in the strand displacement synthesis of DNA. J. Biol. Chem. 2007, 282, 10594–10604. [Google Scholar] [CrossRef] [PubMed]
- Joyce, C.M.; Grindley, N.D. Construction of a plasmid that overproduces the large proteolytic fragment (Klenow fragment) of DNA polymerase i of Escherichia coli. Proc. Natl. Acad. Sci. USA 1983, 80, 1830–1834. [Google Scholar] [CrossRef] [PubMed]
- Ollis, D.L.; Kline, C.; Steitz, T.A. Domain of E. coli DNA polymerase I showing sequence homology to T7 DNA polymerase. Nature 1985, 313, 818–819. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A. Conformational coupling in DNA polymerase fidelity. Annu. Rev. Biochem. 1993, 62, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Korolev, S.; Waksman, G. Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of thermus aquaticus DNA polymerase I: Structural basis for nucleotide incorporation. EMBO J. 1998, 17, 7514–7525. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chopra, R.; Verdine, G.L.; Harrison, S.C. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: Implications for drug resistance. Science 1998, 282, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Doublie, S.; Tabor, S.; Long, A.M.; Richardson, C.C.; Ellenberger, T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 a resolution. Nature 1998, 391, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Kohlstaedt, L.A.; Wang, J.; Friedman, J.M.; Rice, P.A.; Steitz, T.A. Crystal structure at 3.5 a resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 1992, 256, 1783–1790. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Das, K.; Hsiou, Y.; Sarafianos, S.G.; Clark, A.D., Jr.; Jacobo-Molina, A.; Tantillo, C.; Hughes, S.H.; Arnold, E. Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 a resolution. J. Mol. Biol. 1998, 284, 1095–1111. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Smerdon, S.J.; Jager, J.; Kohlstaedt, L.A.; Rice, P.A.; Friedman, J.M.; Steitz, T.A. Structural basis of asymmetry in the human immunodeficiency virus type 1 reverse transcriptase heterodimer. Proc. Natl. Acad. Sci. USA 1994, 91, 7242–7246. [Google Scholar] [CrossRef] [PubMed]
- Jacobo-Molina, A.; Clark, A.D., Jr.; Williams, R.L.; Nanni, R.G.; Clark, P.; Ferris, A.L.; Hughes, S.H.; Arnold, E. Crystals of a ternary complex of human immunodeficiency virus type 1 reverse transcriptase with a monoclonal antibody Fab fragment and double-stranded DNA diffract x-rays to 3.5-a resolution. Proc. Natl. Acad. Sci. USA 1991, 88, 10895–10899. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, H.; Sawaya, M.R.; Kumar, A.; Wilson, S.H.; Kraut, J. Structures of ternary complexes of rat DNA polymerase β, α DNA template-primer, and ddCTP. Science 1994, 264, 1891–1903. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R.; Pelletier, H.; Kumar, A.; Wilson, S.H.; Kraut, J. Crystal structure of rat DNA polymerase β: Evidence for a common polymerase mechanism. Science 1994, 264, 1930–1935. [Google Scholar] [CrossRef] [PubMed]
- Hubscher, U.; Maga, G.; Spadari, S. Eukaryotic DNA polymerases. Annu. Rev. Biochem. 2002, 71, 133–163. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Washington, M.T. Translesion synthesis: Insights into the selection and switching of DNA polymerases. Genes 2017, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.S.; Takata, K.; Wood, R.D. DNA polymerases and cancer. Nat. Rev. Cancer 2011, 11, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Rudd, S.G.; Bianchi, J.; Doherty, A.J. Primpol—A new polymerase on the block. Mol. Cell. Oncol. 2014, 1, e960754. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C.; Longley, M.J. Mitochondrial genome maintenance in health and disease. DNA Repair 2014, 19, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C. Inherited mitochondrial diseases of DNA replication. Annu. Rev. Med. 2008, 59, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.J.; Naviaux, R.K.; Brunetti-Pierri, N.; Zhang, Q.; Schmitt, E.S.; Truong, C.; Milone, M.; Cohen, B.H.; Wical, B.; Ganesh, J.; et al. Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum. Mutat. 2008, 29, E150–E172. [Google Scholar] [CrossRef] [PubMed]
- Saneto, R.P.; Naviaux, R.K. Polymerase gamma disease through the ages. Dev. Disabil. Res. Rev. 2010, 16, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Genetics: Mitochondrial DNA in evolution and disease. Nature 2016, 535, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Krasich, R.; Copeland, W.C. DNA polymerases in the mitochondria: A critical review of the evidence. Front. Biosci. 2017, 22, 692–709. [Google Scholar]
- Wood, R.D.; Doublie, S. DNA polymerase θ (POLQ), double-strand break repair, and cancer. DNA Repair 2016, 44, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Knobel, P.A.; Marti, T.M. Translesion DNA synthesis in the context of cancer research. Cancer Cell Int. 2011, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Malaby, A.W.; Martin, S.K.; Wood, R.D.; Doublie, S. Expression and structural analyses of human DNA polymerase θ (POLQ). Methods Enzymol. 2017, 592, 103–121. [Google Scholar] [PubMed]
- Black, S.J.; Kashkina, E.; Kent, T.; Pomerantz, R.T. DNA polymerase θ: A unique multifunctional end-joining machine. Genes 2016, 7, 67. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.A.; Cooper, C.D.; Aitkenhead, H.; Gileadi, O. Structure of the helicase domain of DNA polymerase theta reveals a possible role in the microhomology-mediated end-joining pathway. Structure 2015, 23, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Zahn, K.E.; Averill, A.M.; Aller, P.; Wood, R.D.; Doublie, S. Human DNA polymerase θ grasps the primer terminus to mediate DNA repair. Nat. Struct. Mol. Biol. 2015, 22, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Marini, F.; Wood, R.D. POLQ (Pol θ), a DNA polymerase and DNA-dependent ATPase in human cells. Nucleic Acids Res. 2003, 31, 6117–6126. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Longley, M.J.; Sharief, F.S.; Hou, E.W.; Copeland, W.C.; Wilson, S.H. Human DNA polymerase theta possesses 5′-drp lyase activity and functions in single-nucleotide base excision repair in vitro. Nucleic Acids Res. 2009, 37, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Masutani, C.; Yang, L.W.; Schuffert, A.; Iwai, S.; Bahar, I.; Wood, R.D. High-efficiency bypass of DNA damage by human DNA polymerase Q. EMBO J. 2004, 23, 4484–4494. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.E.; Washington, M.T.; Haracska, L.; Prakash, S.; Prakash, L. Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature 2000, 406, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Ukai, A.; Maruyama, T.; Mochizuki, S.; Ouchida, R.; Masuda, K.; Kawamura, K.; Tagawa, M.; Kinoshita, K.; Sakamoto, A.; Tokuhisa, T.; et al. Role of DNA polymerase θ in tolerance of endogenous and exogenous DNA damage in mouse B cells. Genes Cells 2006, 11, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Shima, N.; Hartford, S.A.; Duffy, T.; Wilson, L.A.; Schimenti, K.J.; Schimenti, J.C. Phenotype-based identification of mouse chromosome instability mutants. Genetics 2003, 163, 1031–1040. [Google Scholar] [PubMed]
- Goff, J.P.; Shields, D.S.; Seki, M.; Choi, S.; Epperly, M.W.; Dixon, T.; Wang, H.; Bakkenist, C.J.; Dertinger, S.D.; Torous, D.K.; et al. Lack of DNA polymerase θ (POLQ) radiosensitizes bone marrow stromal cells in vitro and increases reticulocyte micronuclei after total-body irradiation. Radiat. Res. 2009, 172, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.S.; Prevo, R.; Lee, Y.F.; Helleday, T.; Muschel, R.J.; Taylor, S.; Yoshimura, M.; Hickson, I.D.; Bernhard, E.J.; McKenna, W.G. A small interfering RNA screen of genes involved in DNA repair identifies tumor-specific radiosensitization by POLQ knockdown. Cancer Res. 2010, 70, 2984–2993. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Bahar, R.; Seimiya, M.; Chiyo, M.; Wada, A.; Okada, S.; Hatano, M.; Tokuhisa, T.; Kimura, H.; Watanabe, S.; et al. DNA polymerase θ is preferentially expressed in lymphoid tissues and upregulated in human cancers. Int. J. Cancer 2004, 109, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Casademunt, J.; Jimenez-Aquino, J.I.; Sancho, J.M. Decay of unstable states in the presence of colored noise and random initial conditions. I. Theory of nonlinear relaxation times. Phys. Rev. A 1989, 40, 5905–5914. [Google Scholar] [CrossRef]
- Lemee, F.; Bergoglio, V.; Fernandez-Vidal, A.; Machado-Silva, A.; Pillaire, M.J.; Bieth, A.; Gentil, C.; Baker, L.; Martin, A.L.; Leduc, C.; et al. DNA polymerase θ up-regulation is associated with poor survival in breast cancer, perturbs DNA replication, and promotes genetic instability. Proc. Natl. Acad. Sci. USA 2010, 107, 13390–13395. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.S.; Harris, A.L.; Prevo, R.; Helleday, T.; McKenna, W.G.; Buffa, F.M. Overexpression of POLQ confers a poor prognosis in early breast cancer patients. Oncotarget 2010, 1, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Allera-Moreau, C.; Rouquette, I.; Lepage, B.; Oumouhou, N.; Walschaerts, M.; Leconte, E.; Schilling, V.; Gordien, K.; Brouchet, L.; Delisle, M.B.; et al. DNA replication stress response involving PLK1, CDC6, POLQ, RAD51 and CLASPIN upregulation prognoses the outcome of early/mid-stage non-small cell lung cancer patients. Oncogenesis 2012, 1, e30. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.; O′Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Beard, W.A.; Wilson, S.H. Structures of human DNA polymerases ν and θ expose their end game. Nat. Struct. Mol. Biol. 2015, 22, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Takata, K.; Shimizu, T.; Iwai, S.; Wood, R.D. Human DNA polymerase N (POLN) is a low fidelity enzyme capable of error-free bypass of 5s-thymine glycol. J. Biol. Chem. 2006, 281, 23445–23455. [Google Scholar] [CrossRef] [PubMed]
- Shivapurkar, N.; Sood, S.; Wistuba, I.I.; Virmani, A.K.; Maitra, A.; Milchgrub, S.; Minna, J.D.; Gazdar, A.F. Multiple regions of chromosome 4 demonstrating allelic losses in breast carcinomas. Cancer Res. 1999, 59, 3576–3580. [Google Scholar] [PubMed]
- Takata, K.I.; Reh, S.; Yousefzadeh, M.J.; Zelazowski, M.J.; Bhetawal, S.; Trono, D.; Lowery, M.G.; Sandoval, M.; Takata, Y.; Lu, Y.; et al. Analysis of DNA polymerase ν function in meiotic recombination, immunoglobulin class-switching, and DNA damage tolerance. PLoS Genet. 2017, 13, e1006818. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Gao, Y.; Yang, W. How a homolog of high-fidelity replicases conducts mutagenic DNA synthesis. Nat. Struct. Mol. Biol. 2015, 22, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Mitaxov, V.; Waksman, G. Structure-based design of Taq DNA polymerases with improved properties of dideoxynucleotide incorporation. Proc. Natl. Acad. Sci. USA 1999, 96, 9491–9496. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.C.; Sheaff, R.J.; Kuchta, R.D. Interactions of calf thymus DNA polymerase α with primer/templates. Nucleic Acids Res. 1995, 23, 4109–4115. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, Y.I.; Frahm, C.; Nick McElhinny, S.A.; Niimi, A.; Suzuki, M.; Kunkel, T.A. Evidence that errors made by DNA polymerase α are corrected by DNA polymerase δ. Curr. Biol. 2006, 16, 202–207. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, S.D.; Kunkel, T.A. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008, 18, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Perrino, F.W.; Loeb, L.A. Hydrolysis of 3′-terminal mispairs in vitro by the 3′-5′ exonuclease of DNA polymerase δ permits subsequent extension by DNA polymerase α. Biochemistry 1990, 29, 5226–5231. [Google Scholar] [CrossRef] [PubMed]
- Preston, B.D.; Albertson, T.M.; Herr, A.J. DNA replication fidelity and cancer. Semin. Cancer Biol. 2010, 20, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.; Eckert, K.A. Eukaryotic replicative DNA polymerases. Nucleic Acid Polym. 2014, 17–41. [Google Scholar]
- Larrea, A.A.; Lujan, S.A.; Nick McElhinny, S.A.; Mieczkowski, P.A.; Resnick, M.A.; Gordenin, D.A.; Kunkel, T.A. Genome-wide model for the normal eukaryotic DNA replication fork. Proc. Natl. Acad. Sci. USA 2010, 107, 17674–17679. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.E.; Klassen, R.; Prakash, L.; Prakash, S. A major role of DNA polymerase δ in replication of both the leading and lagging DNA strands. Mol. Cell 2015, 59, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Burgers, P.M.J.; Gordenin, D.; Kunkel, T.A. Who is leading the replication fork, Pol ε or Pol δ? Mol. Cell 2016, 61, 492–493. [Google Scholar] [CrossRef] [PubMed]
- Lujan, S.A.; Williams, J.S.; Pursell, Z.F.; Abdulovic-Cui, A.A.; Clark, A.B.; Nick McElhinny, S.A.; Kunkel, T.A. Mismatch repair balances leading and lagging strand DNA replication fidelity. PLoS Genet. 2012, 8, e1003016. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Monnat, R.J., Jr. DNA polymerases and human disease. Nat. Rev. Genet. 2008, 9, 594–604. [Google Scholar] [CrossRef] [PubMed]
- da Costa, L.T.; Liu, B.; el-Deiry, W.; Hamilton, S.R.; Kinzler, K.W.; Vogelstein, B.; Markowitz, S.; Willson, J.K.; de la Chapelle, A.; Downey, K.M.; et al. Polymerase δ variants in RER colorectal tumours. Nat. Genet. 1995, 9, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Flohr, T.; Dai, J.C.; Buttner, J.; Popanda, O.; Hagmuller, E.; Thielmann, H.W. Detection of mutations in the DNA polymerase δ gene of human sporadic colorectal cancers and colon cancer cell lines. Int. J. Cancer 1999, 80, 919–929. [Google Scholar] [CrossRef]
- Popanda, O.; Flohr, T.; Fox, G.; Thielmann, H.W. A mutation detected in DNA polymerase δ cDNA from Novikoff hepatoma cells correlates with abnormal catalytic properties of the enzyme. J. Cancer Res. Clin. Oncol. 1999, 125, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Bernad, A.; Blanco, L.; Lazaro, J.M.; Martin, G.; Salas, M. A conserved 3′→5′ exonuclease active site in prokaryotic and eukaryotic DNA polymerases. Cell 1989, 59, 219–228. [Google Scholar] [CrossRef]
- Derbyshire, V.; Pinsonneault, J.K.; Joyce, C.M. Structure-function analysis of 3′→5′-exonuclease of DNA polymerases. Methods Enzymol. 1995, 262, 363–385. [Google Scholar] [PubMed]
- Shevelev, I.V.; Hubscher, U. The 3′→5′ exonucleases. Nat. Rev. Mol. Cell Biol. 2002, 3, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Fox, G.; Popanda, O.; Edler, L.; Thielmann, H.W. Preferential inhibition of DNA polymerases α, δ, and ε from Novikoff hepatoma cells by inhibitors of cell proliferation. J. Cancer Res. Clin. Oncol. 1996, 122, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Fox, G.; Popanda, O.; Thielmann, H.W. Evidence for reduced copying fidelity of DNA polymerases α, δ, and ε from Novikoff hepatoma cells. J. Cancer Res. Clin. Oncol. 1997, 123, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, F.J.; Degtyareva, N.P.; Kokoska, R.J.; Petes, T.D. Reduced levels of DNA polymerase δ induce chromosome fragile site instability in yeast. Mol. Cell. Biol. 2008, 28, 5359–5368. [Google Scholar] [CrossRef] [PubMed]
- Kokoska, R.J.; Stefanovic, L.; DeMai, J.; Petes, T.D. Increased rates of genomic deletions generated by mutations in the yeast gene encoding DNA polymerase δ or by decreases in the cellular levels of DNA polymerase δ. Mol. Cell. Biol. 2000, 20, 7490–7504. [Google Scholar] [CrossRef] [PubMed]
- Hoang, L.N.; McConechy, M.K.; Kobel, M.; Anglesio, M.; Senz, J.; Maassen, M.; Kommoss, S.; Meng, B.; Postovit, L.; Kelemen, L.E.; et al. Polymerase ε exonuclease domain mutations in ovarian endometrioid carcinoma. Int. J. Gynecol. Cancer 2015, 25, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Tomlinson, I. Replicative DNA polymerase mutations in cancer. Curr. Opin. Genet. Dev. 2014, 24, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Mertz, T.M.; Harcy, V.; Roberts, S.A. Risks at the DNA replication fork: Effects upon carcinogenesis and tumor heterogeneity. Genes 2017, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, R.; Miyashita, K.; Inoue, M.; Shimamoto, A.; Yan, Z.; Egashira, A.; Oki, E.; Kakeji, Y.; Oda, S.; Maehara, Y. Concurrent genetic alterations in DNA polymerase proofreading and mismatch repair in human colorectal cancer. Eur. J. Hum. Genet. 2011, 19, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Shamoo, Y.; Steitz, T.A. Building a replisome from interacting pieces: Sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex. Cell 1999, 99, 155–166. [Google Scholar] [CrossRef]
- Henninger, E.E.; Pursell, Z.F. DNA polymerase ε and its roles in genome stability. IUBMB Life 2014, 66, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Cancer Genome Atlas Research, N.; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Choi, M.; Overton, J.D.; Bellone, S.; Roque, D.M.; Cocco, E.; Guzzo, F.; English, D.P.; Varughese, J.; Gasparrini, S.; et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 2916–2921. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.A.; Wang, L. From human genome to cancer genome: The first decade. Genome Res. 2013, 23, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Church, D.N.; Briggs, S.E.; Palles, C.; Domingo, E.; Kearsey, S.J.; Grimes, J.M.; Gorman, M.; Martin, L.; Howarth, K.M.; Hodgson, S.V.; et al. DNA polymerase ε and δ exonuclease domain mutations in endometrial cancer. Hum. Mol. Genet. 2013, 22, 2820–2828. [Google Scholar] [CrossRef] [PubMed]
- Albertson, T.M.; Ogawa, M.; Bugni, J.M.; Hays, L.E.; Chen, Y.; Wang, Y.; Treuting, P.M.; Heddle, J.A.; Goldsby, R.E.; Preston, B.D. DNA polymerase ε and δ proofreading suppress discrete mutator and cancer phenotypes in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 17101–17104. [Google Scholar] [CrossRef] [PubMed]
- Rayner, E.; van Gool, I.C.; Palles, C.; Kearsey, S.E.; Bosse, T.; Tomlinson, I.; Church, D.N. A panoply of errors: Polymerase proofreading domain mutations in cancer. Nat. Rev. Cancer 2016, 16, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Shlien, A.; Campbell, B.B.; de Borja, R.; Alexandrov, L.B.; Merico, D.; Wedge, D.; Van Loo, P.; Tarpey, P.S.; Coupland, P.; Behjati, S.; et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat. Genet. 2015, 47, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.R.; Lawrence, C.W.; Hinkle, D.C. Thymine-thymine dimer bypass by yeast DNA polymerase ζ. Science 1996, 272, 1646–1649. [Google Scholar] [CrossRef] [PubMed]
- Murakumo, Y.; Roth, T.; Ishii, H.; Rasio, D.; Numata, S.; Croce, C.M.; Fishel, R. A human REV7 homolog that interacts with the polymerase ζ catalytic subunit hREV3 and the spindle assembly checkpoint protein hMAD2. J. Biol. Chem. 2000, 275, 4391–4397. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.; Prakash, L. Translesion DNA synthesis in eukaryotes: A one- or two-polymerase affair. Genes Dev. 2002, 16, 1872–1883. [Google Scholar] [CrossRef] [PubMed]
- Makarova, A.V.; Stodola, J.L.; Burgers, P.M. A four-subunit DNA polymerase ζ complex containing Pol δ accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res. 2012, 40, 11618–11626. [Google Scholar] [CrossRef] [PubMed]
- Baranovskiy, A.G.; Lada, A.G.; Siebler, H.M.; Zhang, Y.; Pavlov, Y.I.; Tahirov, T.H. DNA polymerase δ and ζ switch by sharing accessory subunits of DNA polymerase δ. J. Biol. Chem. 2012, 287, 17281–17287. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.E.; Prakash, L.; Prakash, S. Pol31 and Pol32 subunits of yeast DNA polymerase δ are also essential subunits of DNA polymerase ζ. Proc. Natl. Acad. Sci. USA 2012, 109, 12455–12460. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, E.; Hanafusa, T.; Kamei, K.; Song, I.; Tomida, J.; Hashimoto, H.; Vaziri, C.; Ohmori, H. Identification of a novel REV1-interacting motif necessary for DNA polymerase κ function. Genes Cells 2009, 14, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Prakash, L.; Prakash, S. Error-free replicative bypass of (6-4) photoproducts by DNA polymerase ζ in mouse and human cells. Genes Dev. 2010, 24, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Shachar, S.; Ziv, O.; Avkin, S.; Adar, S.; Wittschieben, J.; Reissner, T.; Chaney, S.; Friedberg, E.C.; Wang, Z.; Carell, T.; et al. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009, 28, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Szuts, D.; Marcus, A.P.; Himoto, M.; Iwai, S.; Sale, J.E. Rev1 restrains DNA polymerase ζ to ensure frame fidelity during translesion synthesis of UV photoproducts in vivo. Nucleic Acids Res. 2008, 36, 6767–6780. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.G.; Tsaalbi-Shtylik, A.; Hendriks, G.; Verspuy, J.; Gali, H.; Haracska, L.; de Wind, N. Mammalian polymerase ζ is essential for post-replication repair of UV-induced DNA lesions. DNA Repair 2009, 8, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Bhatia, G.; Prakash, S.; Prakash, L. Error-free replicative bypass of thymine glycol by the combined action of DNA polymerases κ and ζ in human cells. Proc. Natl. Acad. Sci. USA 2010, 107, 14116–14121. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Shah, N.A.; Joiner, A.M.; Roberts, K.H.; Canman, C.E. DNA polymerase ζ is a major determinant of resistance to platinum-based chemotherapeutic agents. Mol. Pharmacol. 2012, 81, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Doles, J.; Hemann, M.T.; Walker, G.C. Error-prone translesion synthesis mediates acquired chemoresistance. Proc. Natl. Acad. Sci. USA 2010, 107, 20792–20797. [Google Scholar] [CrossRef] [PubMed]
- Wittschieben, J.P.; Reshmi, S.C.; Gollin, S.M.; Wood, R.D. Loss of DNA polymerase ζ causes chromosomal instability in mammalian cells. Cancer Res. 2006, 66, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Wittschieben, J.P.; Patil, V.; Glushets, V.; Robinson, L.J.; Kusewitt, D.F.; Wood, R.D. Loss of DNA polymerase ζ enhances spontaneous tumorigenesis. Cancer Res. 2010, 70, 2770–2778. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Li, X.; Owens, K.M.; Vanniarajan, A.; Liang, P.; Singh, K.K. Human REV3 DNA polymerase ζ localizes to mitochondria and protects the mitochondrial genome. PLoS ONE 2015, 10, e0140409. [Google Scholar] [CrossRef] [PubMed]
- Varadi, V.; Bevier, M.; Grzybowska, E.; Johansson, R.; Enquist, K.; Henriksson, R.; Butkiewicz, D.; Pamula-Pilat, J.; Tecza, K.; Hemminki, K.; et al. Genetic variation in genes encoding for polymerase ζ subunits associates with breast cancer risk, tumour characteristics and survival. Breast Cancer Res. Treat. 2011, 129, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Nishikawa, H. Roles of regulatory T cells in cancer immunity. Int. Immunol. 2016, 28, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O'Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crino, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Sloan, E.A.; Ring, K.L.; Willis, B.C.; Modesitt, S.C.; Mills, A.M. PD-L1 expression in mismatch repair-deficient endometrial carcinomas, including lynch syndrome-associated and MLH1 promoter hypermethylated tumors. Am. J. Surg. Pathol. 2017, 41, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- van Gool, I.C.; Eggink, F.A.; Freeman-Mills, L.; Stelloo, E.; Marchi, E.; de Bruyn, M.; Palles, C.; Nout, R.A.; de Kroon, C.D.; Osse, E.M.; et al. POLE proofreading mutations elicit an antitumor immune response in endometrial cancer. Clin. Cancer Res. 2015, 21, 3347–3355. [Google Scholar] [CrossRef] [PubMed]
- Howitt, B.E.; Shukla, S.A.; Sholl, L.M.; Ritterhouse, L.L.; Watkins, J.C.; Rodig, S.; Stover, E.; Strickland, K.C.; D'Andrea, A.D.; Wu, C.J.; et al. Association of polymerase e-mutated and microsatellite-instable endometrial cancers with neoantigen load, number of tumor-infiltrating lymphocytes, and expression of PD-1 and PD-l1. JAMA Oncol. 2015, 1, 1319–1323. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wei, Q.; Wang, J.; Huang, X.; Li, C.; Zheng, Q.; Cao, J.; Jia, Z. DNA polymerases as targets for gene therapy of hepatocellular carcinoma. BMC Cancer 2015, 15, 325. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.H.; Clark, A.B.; Slebos, R.J.; Al-Refai, H.; Taylor, J.A.; Kunkel, T.A.; Resnick, M.A.; Gordenin, D.A. Cadmium is a mutagen that acts by inhibiting mismatch repair. Nat. Genet. 2003, 34, 326–329. [Google Scholar] [CrossRef] [PubMed]
- Mertz, T.M.; Sharma, S.; Chabes, A.; Shcherbakova, P.V. Colon cancer-associated mutator DNA polymerase δ variant causes expansion of dNTP pools increasing its own infidelity. Proc. Natl. Acad. Sci. USA 2015, 112, E2467–2476. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.N.; Marjavaara, L.; Knowels, G.M.; Schultz, E.M.; Fox, E.J.; Chabes, A.; Herr, A.J. dNTP pool levels modulate mutator phenotypes of error-prone DNA polymerase ε variants. Proc. Natl. Acad. Sci. USA 2015, 112, E2457–2466. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]


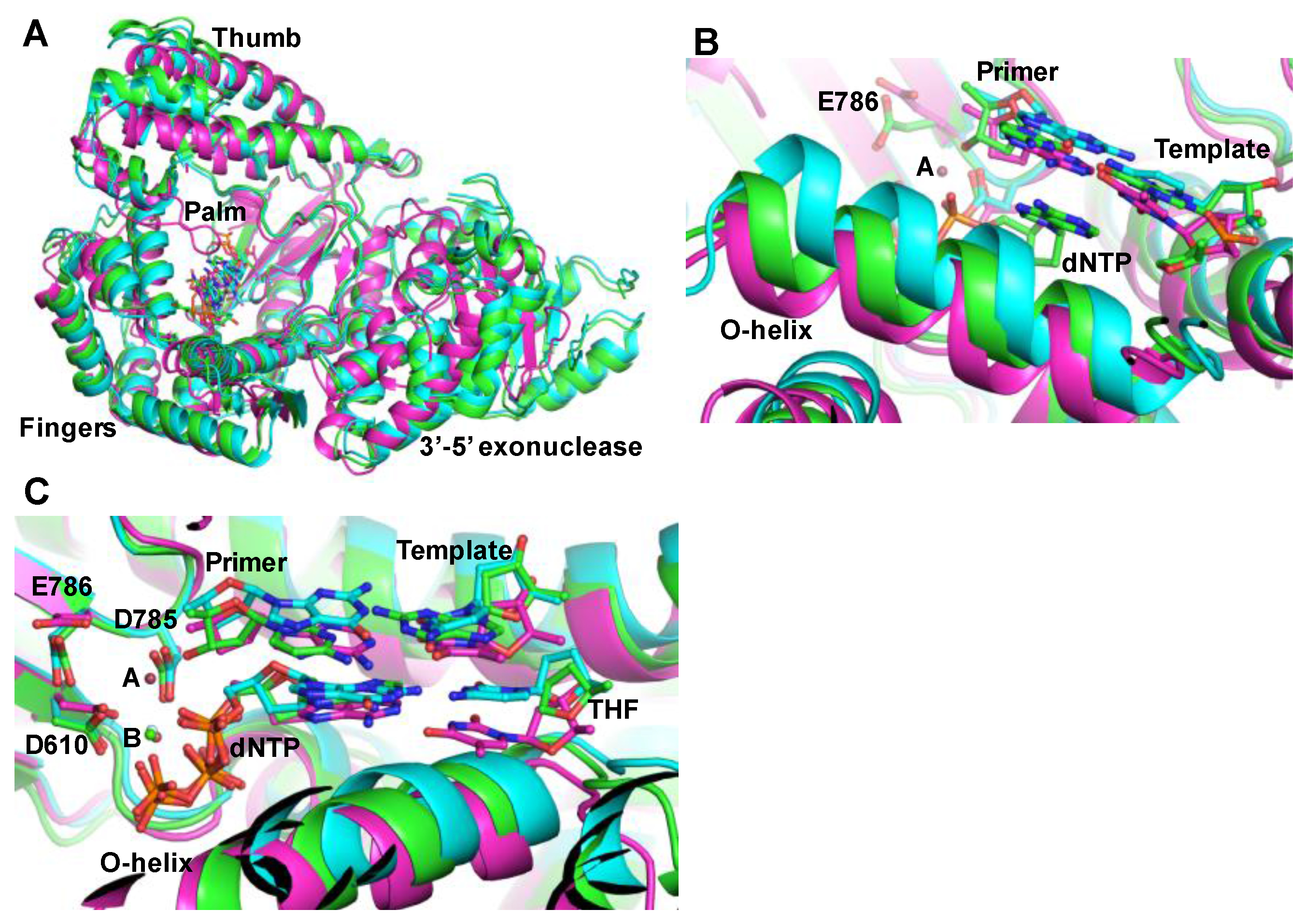{kind=link}
| Family | Prokaryotic a | Eukaryotic | Archaea | Virus |
|---|---|---|---|---|
| A | Pol I | Pol γ, θ, ν | T3, T5, T7 pol | |
| B | Pol II | Pol α, δ, ε, ζ | Pol BI, BII | RB69, T4 pol |
| C | Pol III | |||
| D | Pol D | |||
| X | Pol β, λ, μ | |||
| Y | Pol IV, V | Pol η, ι, κ | ||
| RT | hTERT | Telomerase | Reverse Transcriptase | |
| AEP | Prim-pol | poxviruses, asfarviruses, iridoviruses, phycodnaviruses mimivirus |
| Polymerase δ | Predisposition to the Cancer Type | Polymerase ε | Predisposition to the Cancer Type |
|---|---|---|---|
| C319Y | Multiple myeloma and Glioblastoma | D275V | Endometrial |
| D316G | Colorectal, endometrial and breast | E277 | Endometrial |
| D316H | Colorectal, breast, and mesothelioma | P286R/H/S | Colorectal |
| L474P | Colorectal and endometrial | S297F | Ovarian |
| R409W | Colorectal | F367S in | Colorectal |
| S478N | Colorectal and endometrial | V411L | Colorectal |
| P327L | Multiple adenomas | L424V | Colorectal |
| P436R/S | Colorectal | ||
| M444K | Colorectal | ||
| A456P | Colorectal | ||
| S459F | Colorectal |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shanbhag, V.; Sachdev, S.; Flores, J.A.; Modak, M.J.; Singh, K. Family A and B DNA Polymerases in Cancer: Opportunities for Therapeutic Interventions. Biology 2018, 7, 5. https://doi.org/10.3390/biology7010005
Shanbhag V, Sachdev S, Flores JA, Modak MJ, Singh K. Family A and B DNA Polymerases in Cancer: Opportunities for Therapeutic Interventions. Biology. 2018; 7(1):5. https://doi.org/10.3390/biology7010005
Chicago/Turabian StyleShanbhag, Vinit, Shrikesh Sachdev, Jacqueline A. Flores, Mukund J. Modak, and Kamalendra Singh. 2018. "Family A and B DNA Polymerases in Cancer: Opportunities for Therapeutic Interventions" Biology 7, no. 1: 5. https://doi.org/10.3390/biology7010005
APA StyleShanbhag, V., Sachdev, S., Flores, J. A., Modak, M. J., & Singh, K. (2018). Family A and B DNA Polymerases in Cancer: Opportunities for Therapeutic Interventions. Biology, 7(1), 5. https://doi.org/10.3390/biology7010005