
Inferring Gene Regulatory Networks from RNA-seq Data Using Kernel Classification

Abstract
Simple Summary
Abstract
1. Introduction
2. Methods
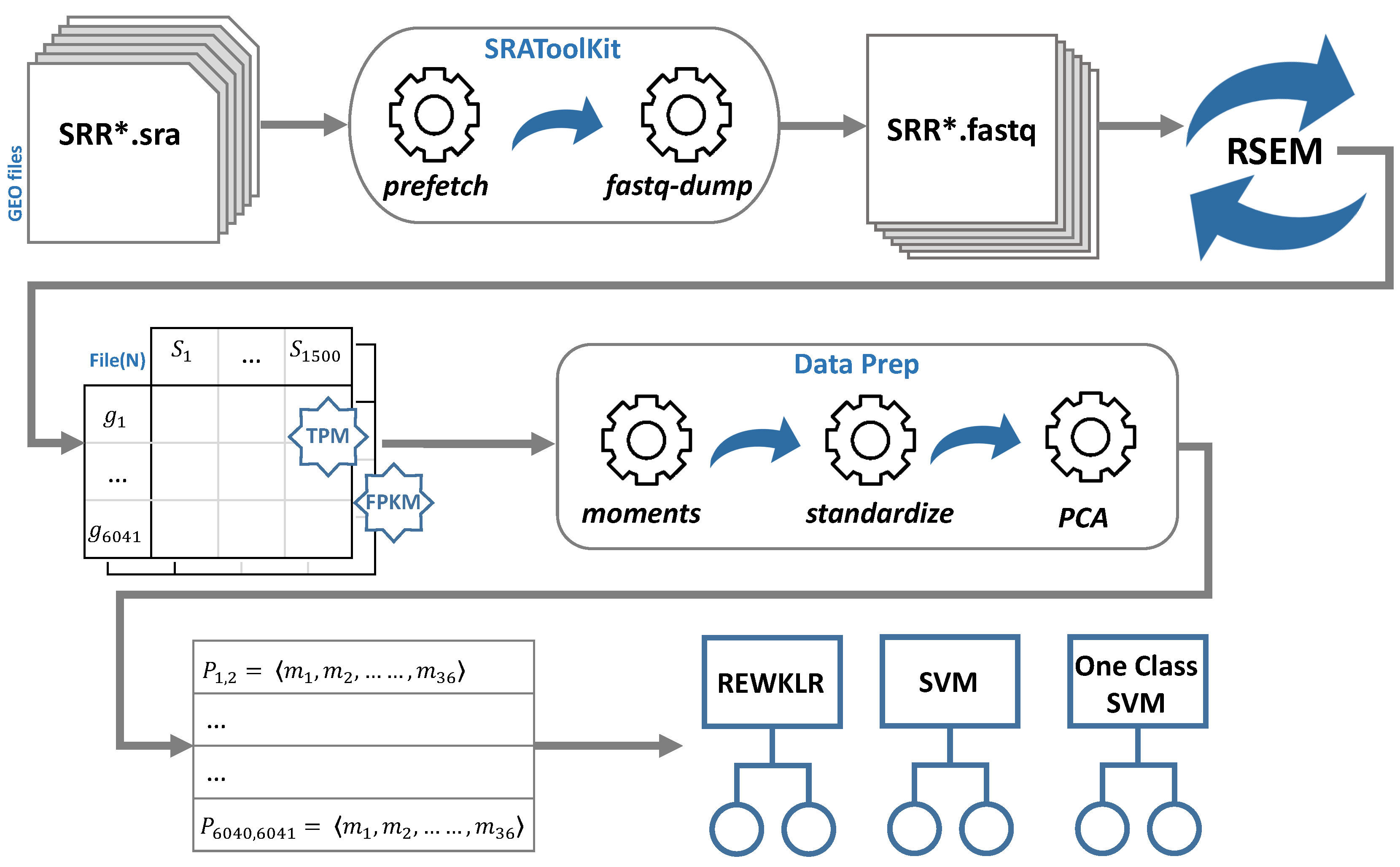
2.1. RNA-seq Data Analysis
- GEO database: The Gene Expression Omnibus (GEO) was used to obtain the S. cerevisiae RNA-seq expression data [12]. We used three main GEO platforms to download RNA-seq records (GPL19756, GPL17342, and GPL13821). Each of these platform files contains different experimentation conditions (series) and various samples for each series. To obtain these samples and specifically those representing RNA-seq data delivered by using Illumina sequencing technology, we used the BioProject ID that links the GEO sample to the correspondent Sequence Read Archive (SRA) accession IDs [13]. We randomly selected and downloaded around 1500 SRA accession IDs.
- SRA-Toolkit: This is another well-designed service by NCBI. SRA-Toolkit is a collection of tools to utilize SRA data [14]. We mainly used two operations; namely prefetch and fastq-dump. Prefetch was used to download sequence files in the compressed SRA format using the SRA accession IDs obtained from the previous step. These sequence files were then used by the fastq-dump sub-tool to retrieve the SRA fastQ files. This process required a significant amount of time and disk space. At this point, all the files were prepared for mapping and transcript quantification to obtain the reads for each gene.
- RSEM tool: Finally, the fastQ files were fed to the RNA-seq by Expectation Maximization (RSEM) package tool [15]. RSEM is an open-source software tool for gene quantification using single-end or paired-end RNA-seq data. It also utilizes Bowtie; a powerful and efficient alignment software [16]. We used the main functions showcased in the typical RSEM workflow: rsem-prepare-reference and rsem-calculate-expression.The reference was prepared by obtaining the yeast FASTA-formatted file from Ensembl genome browser release 82 [17]. Then, RSEM takes as input the fastQ files downloaded previously using SRA-Toolkit. The tool maps each fastQ file and outputs the calculated expression as gene read counts. The read counts are normalized by default with Transcripts Per Kilobase Million (TPM) and Fragments Per Kilobase Million (FPKM) normalization metrics. In Section 3, we showcase the effect of the normalization method on the prediction accuracy for the gene–gene association. For each normalization metric, we merged all the read counts according to their gene correspondence to create a matrix table of genes (rows) and sample counts (columns). The output files of this step are referred to as File(N), where N denotes the normalization method (TPM or FPKM).
2.2. Data Preparation for Classification
2.2.1. Microarray
2.2.2. RNA-seq
2.3. Training Data and Models
2.4. Kernel Classification Methods
- REWKLR is identified as a rare-event classifier where the unrelated pairs outnumber the related ones. This captures the natural dynamics of genetic data and considers the sparsity of the data. In particular, the data in this article exhibit the genetic data nature where the percentages of zeros (unrelated pairs) exceed that of ones (related pairs). REWKLR embeds the kernel in a typical Logistic Regression model to reflect the nonlinearity of data separation. The performance of REWKLR was shown to compete with other classifiers such as SVM [25,26,27]. The parameters optimized for REWKLR are the regularization variable () and the dual variable ().
- SVM is one of the widely used supervised learning models. It efficiently uses the kernel trick to separate two or more classes with nonlinear classification. It is also one of the highly effective models to handle a sparse number of features in a high-dimensional representation. However, it might not always be the optimal solution for capturing rare events [28]. Other methods such as REWKLR [26] and hexagonal cellular networks [29] consider the rarity of events in classification. We used the Gaussian radial basis function (RBF), a commonly used kernel with SVM, to train and predict our data. The hyperparameters optimized for training are the penalty parameter of the error term (C), and the Gamma parameter ().
- One-class SVM is shown to be the version of SVM that detects rare events more sufficiently [30,31]. To validate that, we tested our data with this model since the negative class might not be deterministic as stated previously in Section 2.3. Hence, we trained the positive class data features. We used the RBF kernel in one-class SVM as well. The parameters optimized in this model, however, are the Gamma parameter () and the Nu parameter () which acts both as a lower bound and an upper bound for the number of features.
3. Results and Discussion
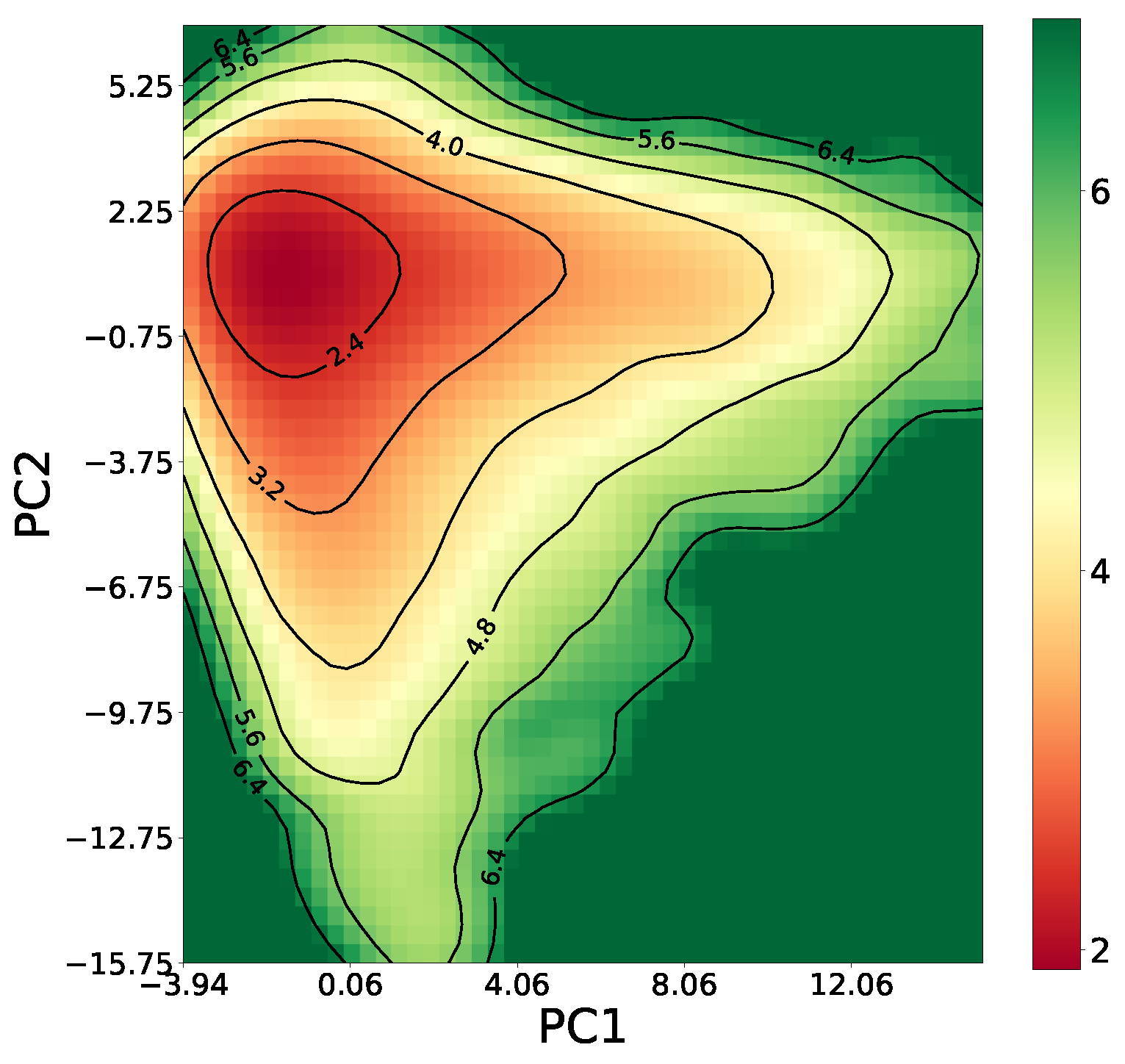
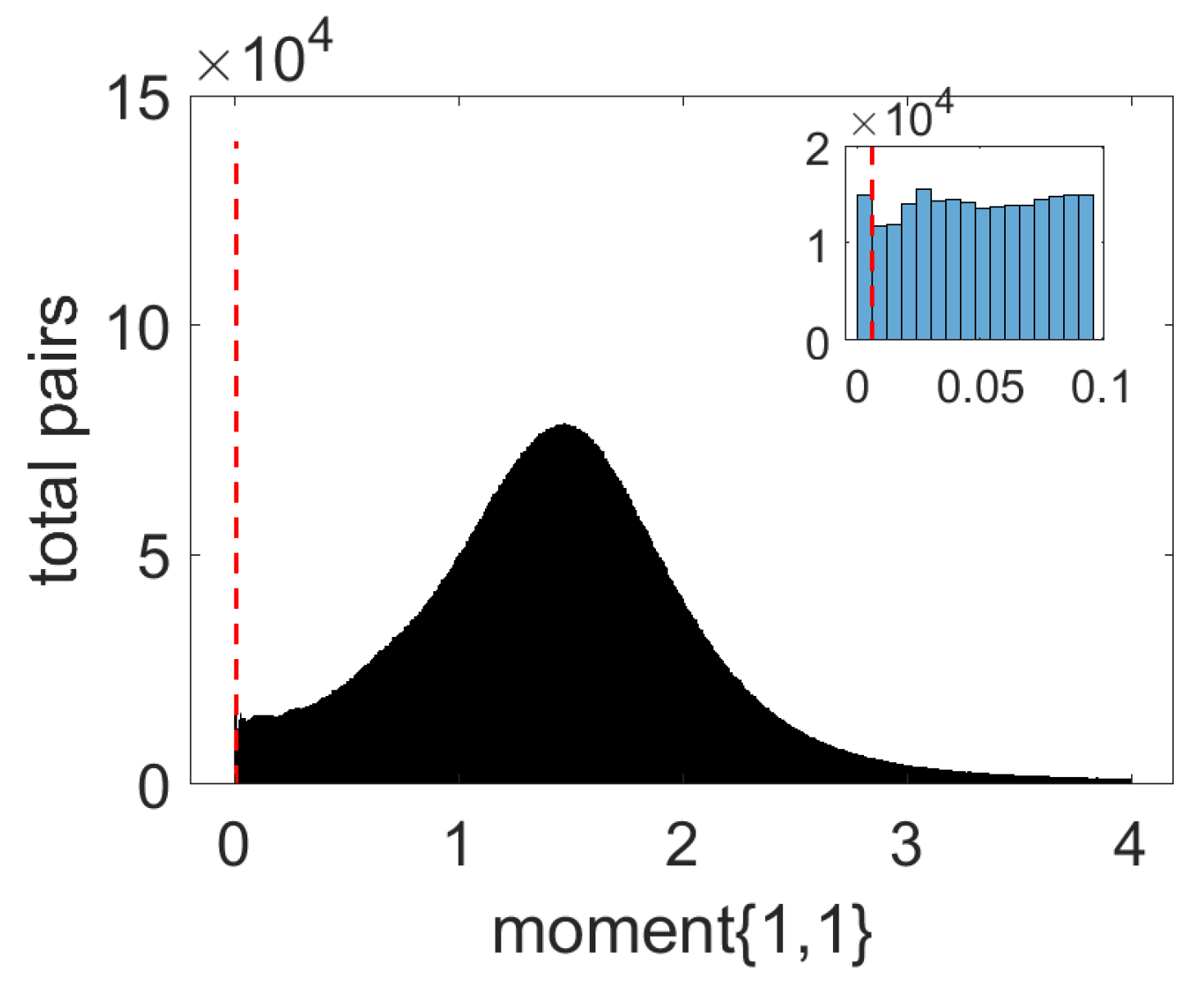
3.1. Data Preparation and System Specification
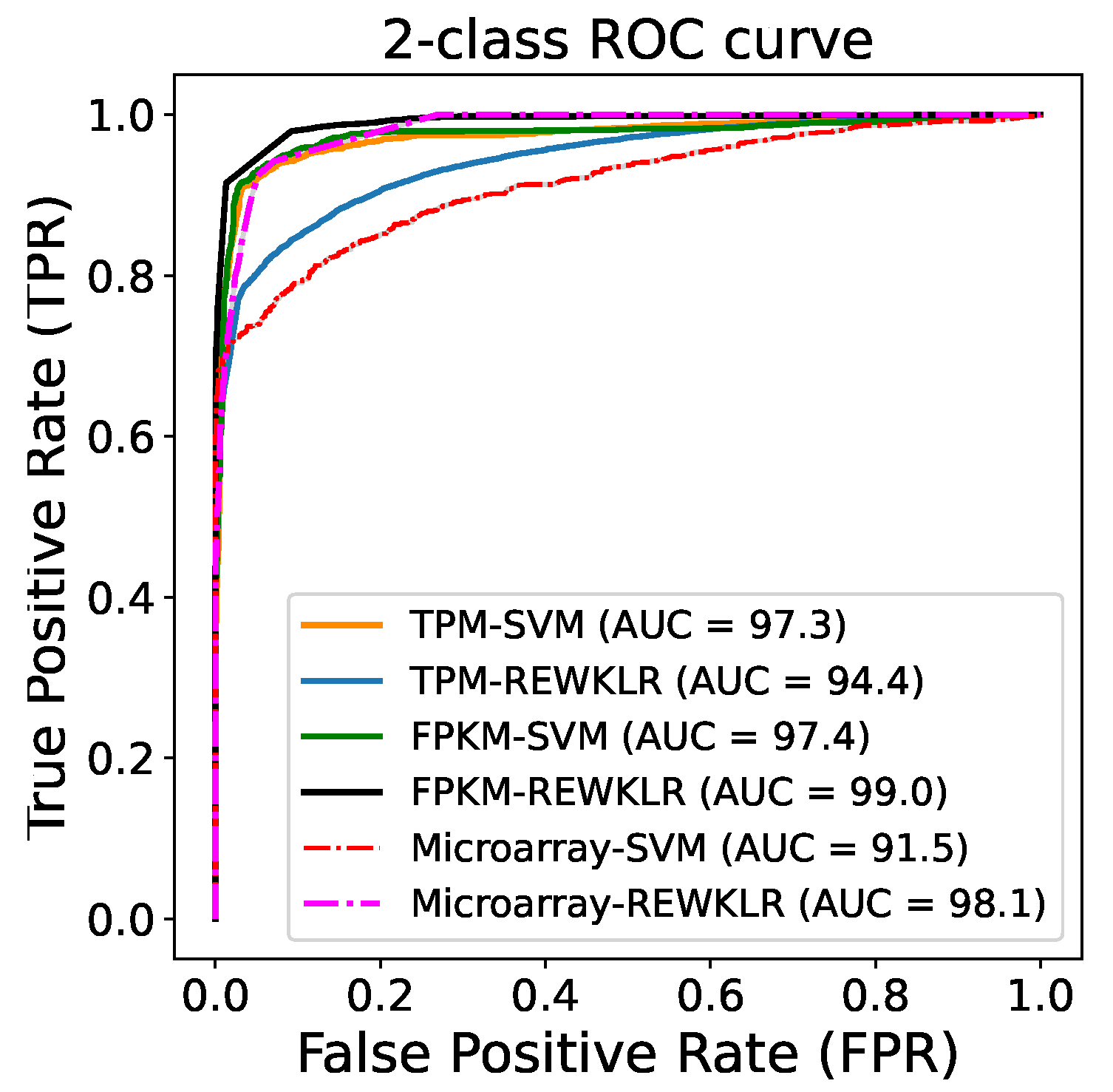
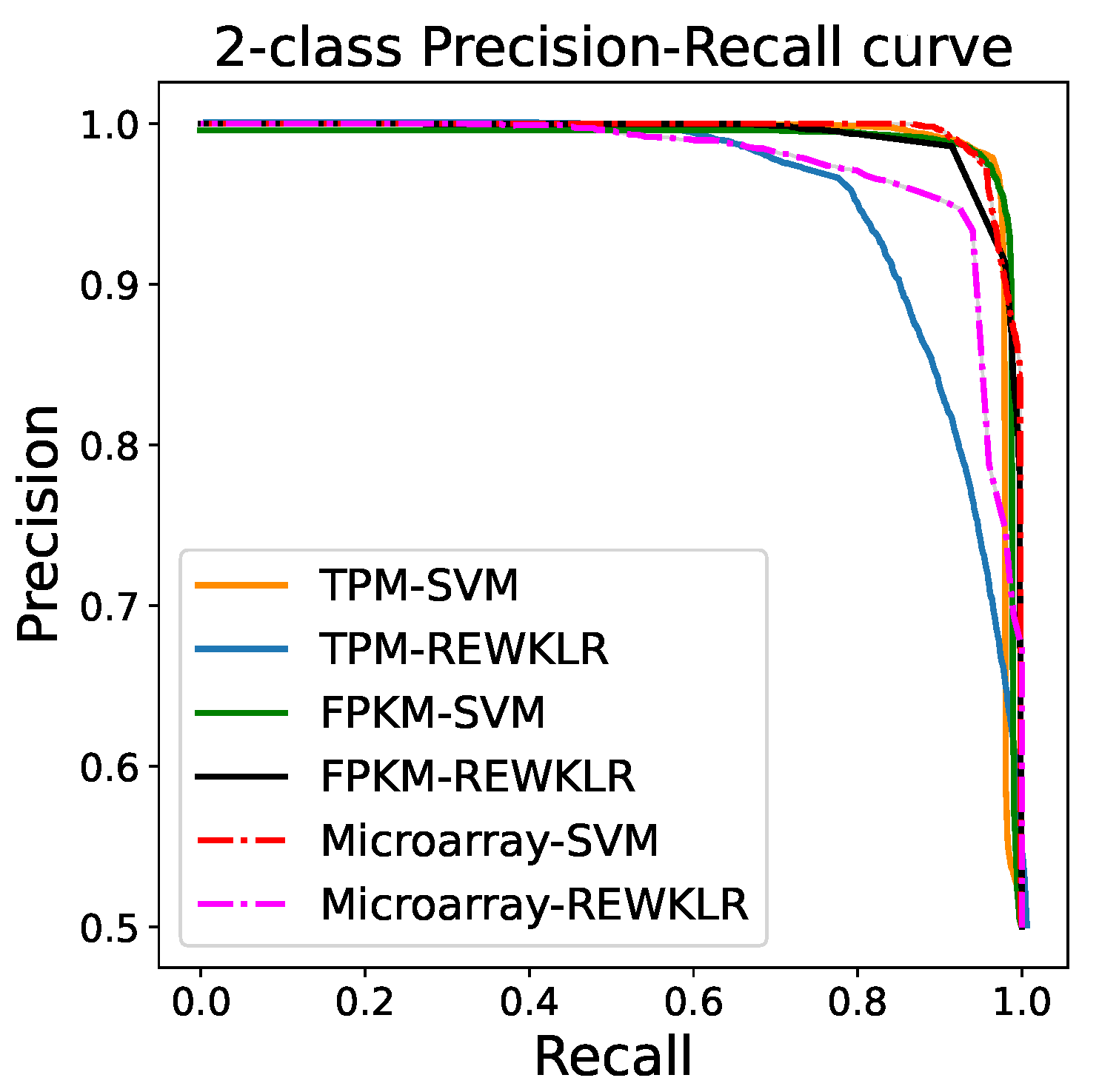
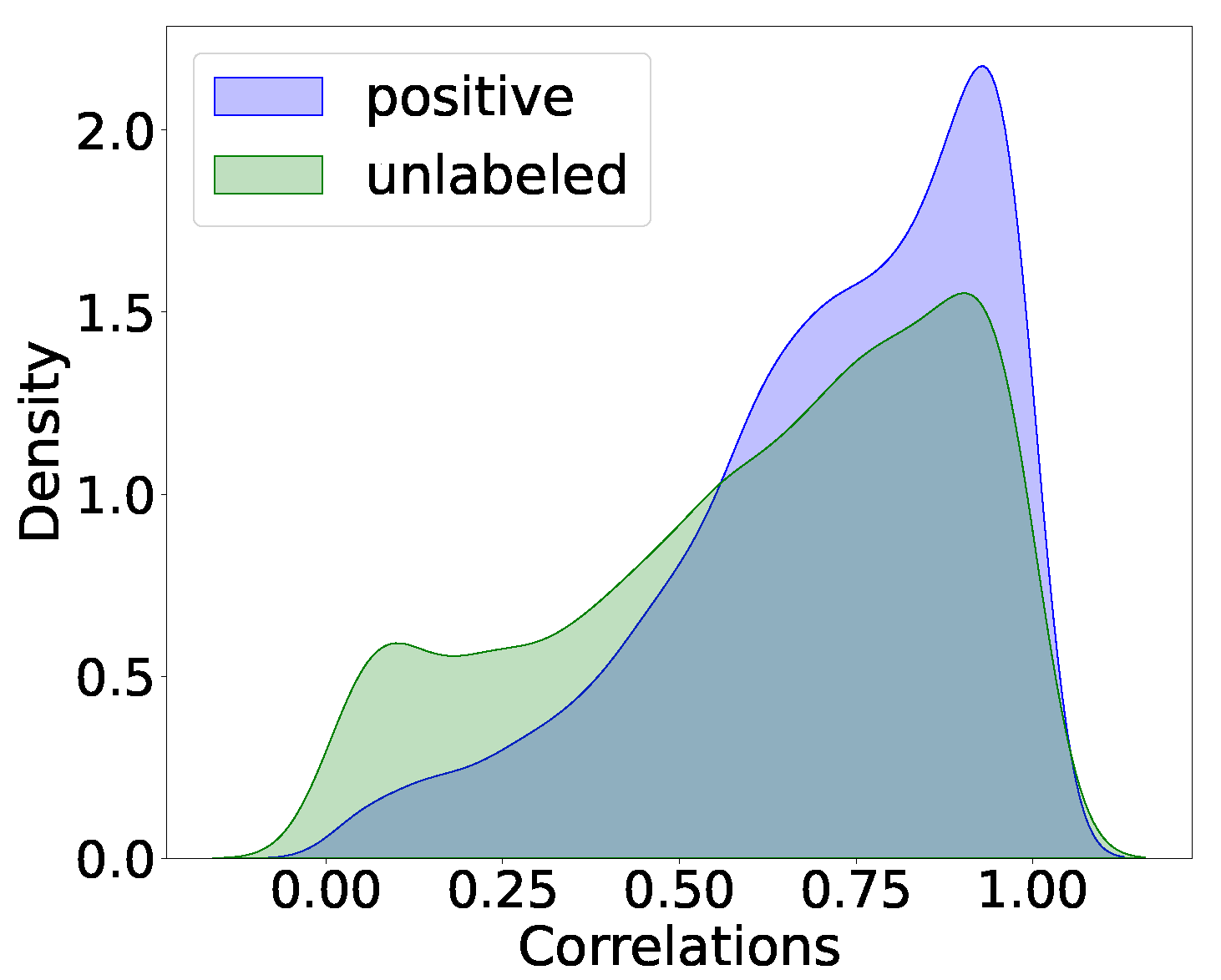
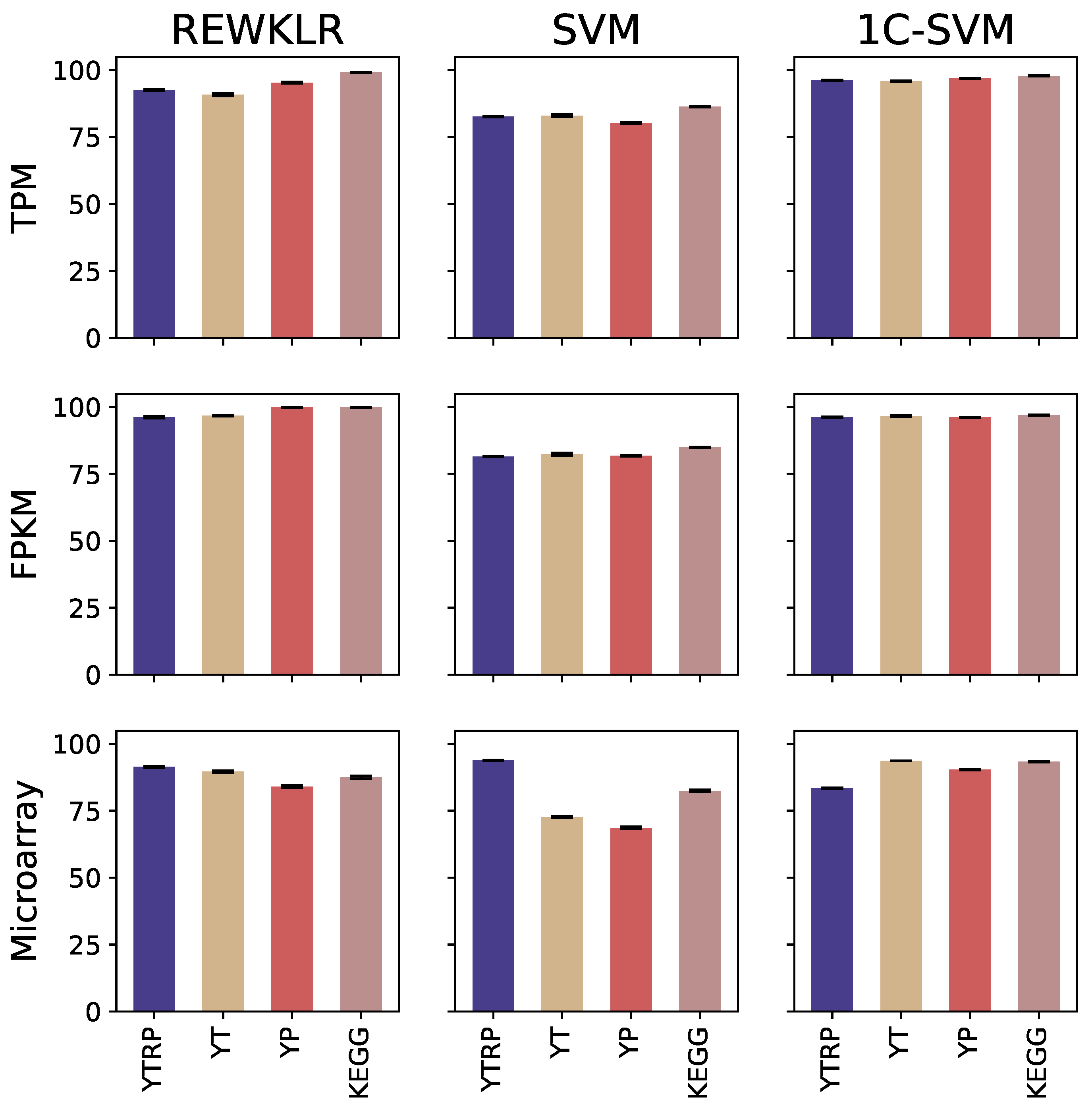
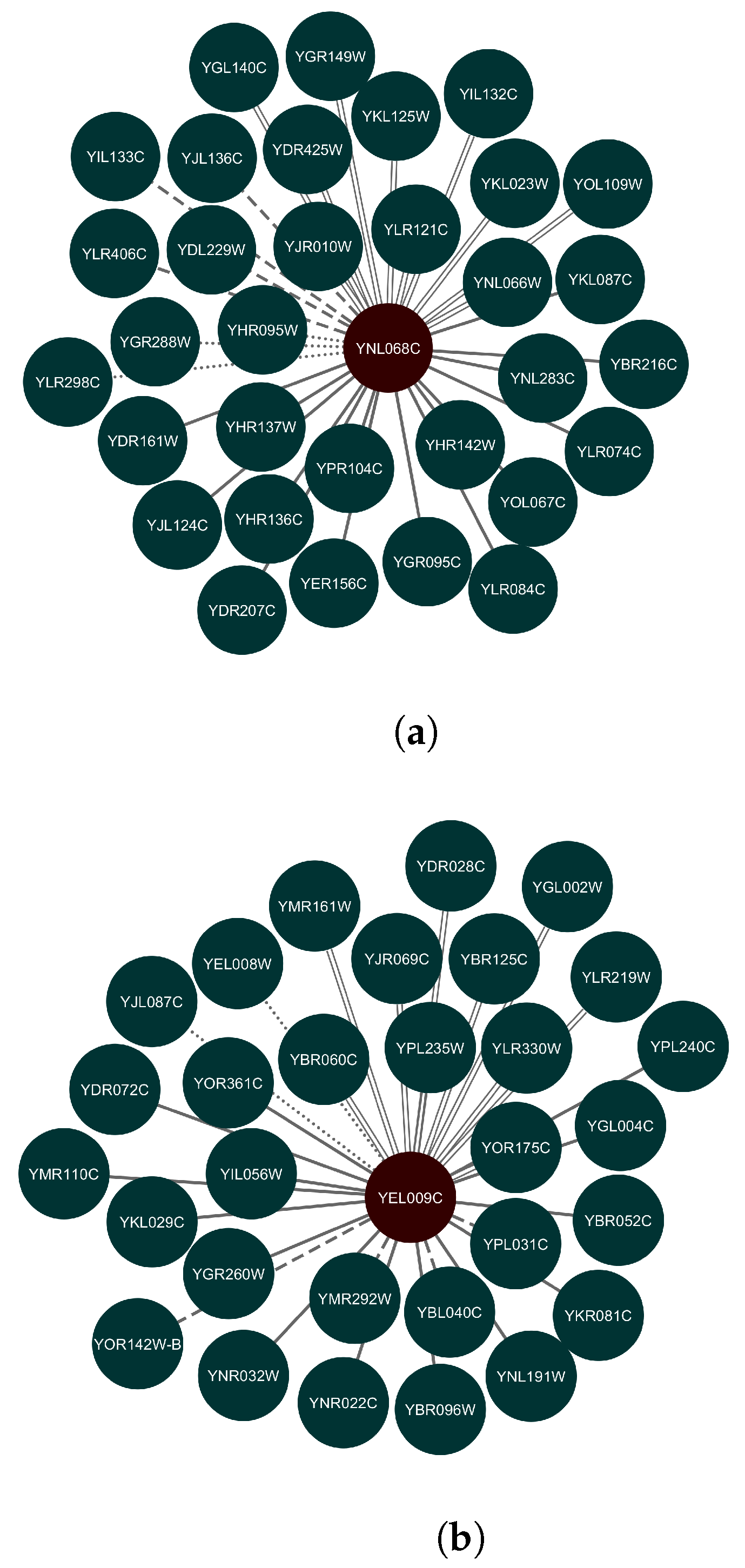
3.2. Prediction Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| YEASTRACT | Yeast Search for Transcriptional Regulators and Consensus Tracking |
| YTRP | Yeast Transcriptional Regulatory Pathway |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
References
- Rai, M.; Tycksen, E.; Sandell, L.; Brophy, R. Advantages of RNA-seq compared to RNA microarrays for transcriptome profiling of anterior cruciate ligament tears. J. Orthop. Res. 2018, 36, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.; Zegar, C.; Giordano, A. Advantages and limitations of microarray technology in human cancer. Oncogene 2003, 22, 6497–6507. [Google Scholar] [CrossRef] [PubMed]
- Koltai, H.; Weingarten-Baror, C. Specificity of DNA microarray hybridization: Characterization, effectors and approaches for data correction. Nucleic Acids Res. 2008, 36, 2395–2405. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Ballouz, S.; Verleyen, W.; Gillis, J. Guidance for RNA-seq co-expression network construction and analysis: Safety in numbers. Bioinformatics 2015, 31, 2123–2130. [Google Scholar] [CrossRef]
- Johnson, K.; Krishnan, A. Robust normalization and transformation techniques for constructing gene coexpression networks from RNA-seq data. Genome Biol. 2022, 23, 1–26. [Google Scholar] [CrossRef]
- Shahjaman, M.; Mollah, M.; Rahman, M.; Islam, S.; Mollah, M. Robust identification of differentially expressed genes from RNA-seq data. Genomics 2020, 112, 2000–2010. [Google Scholar] [CrossRef]
- Zhang, W.; Yu, Y.; Hertwig, F.; Thierry-Mieg, J.; Zhang, W.; Thierry-Mieg, D.; Wang, J.; Furlanello, C.; Devanarayan, V.; Cheng, J. Others Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol. 2015, 16, 1–12. [Google Scholar] [CrossRef]
- Giorgi, F.; Del Fabbro, C.; Licausi, F. Comparative study of RNA-seq-and microarray-derived coexpression networks in Arabidopsis thaliana. Bioinformatics 2013, 29, 717–724. [Google Scholar] [CrossRef]
- Su, Z.; Fang, H.; Hong, H.; Shi, L.; Zhang, W.; Zhang, W.; Zhang, Y.; Dong, Z.; Lancashire, L.; Bessarabova, M.; et al. An investigation of biomarkers derived from legacy microarray data for their utility in the RNA-seq era. Genome Biol. 2014, 15, 1–25. [Google Scholar] [CrossRef]
- Al-Aamri, A.; Taha, K.; Maalouf, M.; Kudlicki, A.; Homouz, D. Inferring Causation in Yeast gene association Networks with Kernel Logistic Regression. Evol. Bioinform. 2020, 16, 1–6. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Shumway, M.; Cochrane, G.; Sugawara, H. Archiving next generation sequencing data. Nucleic Acids Res. 2010, 38, D870–D871. [Google Scholar] [CrossRef]
- SRA Toolkit Development Team Sequence Read Archive Toolkit. Available online: https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software (accessed on 10 August 2022).
- Li, B.; Dewey, C. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.; Armean, I.; Austine-Orimoloye, O.; Azov, A.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- Consortium, U. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J. A User’s Guide to Principal Components; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar]
- Teixeira, M.; Monteiro, P.; Jain, P.; Tenreiro, S.; Fernandes, A.; Mira, N.; Alenquer, M.; Freitas, A.; Oliveira, A.; Sá-Correia, I. The YEASTRACT database: A tool for the analysis of transcription regulatory associations in Saccharomyces cerevisiae. Nucleic Acids Res. 2006, 34, D446–D451. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Wang, C.; Wang, Y.; Wu, W. YTRP: A repository for yeast transcriptional regulatory pathways. Database 2014, 2014, bau014. [Google Scholar] [CrossRef]
- Harbison, C.; Gordon, D.; Lee, T.; Rinaldi, N.; Macisaac, K.; Danford, T.; Hannett, N.; Tagne, J.; Reynolds, D.; Yoo, J.; et al. Transcriptional regulatory code of a eukaryotic genome. Nature 2004, 431, 99–104. [Google Scholar] [CrossRef]
- Cherry, J.; Hong, E.; Amundsen, C.; Balakrishnan, R.; Binkley, G.; Chan, E.; Christie, K.; Costanzo, M.; Dwight, S.; Engel, S.; et al. Saccharomyces Genome Database: The genomics resource of budding yeast. Nucleic Acids Res. 2012, 40, D700–D705. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2022, 51, D587–D592. [Google Scholar] [CrossRef]
- Maalouf, M.; Humouz, D.; Kudlicki, A. Robust weighted kernel logistic regression to predict gene-gene regulatory association. IIE Annu. Conf. Proc. 2014, 2014, 1356–1360. [Google Scholar]
- Maalouf, M.; Trafalis, T. Robust weighted kernel logistic regression in imbalanced and rare events data. Comput. Stat. Data Anal. 2011, 55, 168–183. [Google Scholar] [CrossRef]
- Maalouf, M.; Homouz, D. Kernel ridge regression using truncated newton method. Knowl.-Based Syst. 2014, 71, 339–344. [Google Scholar] [CrossRef]
- Köknar-Tezel, S.; Latecki, L. Improving SVM classification on imbalanced data sets in distance spaces. IEEE Int. Conf. Data Min. 2009, 2009, 259–267. [Google Scholar]
- Azeem, M.; Jamil, M.; Shang, Y. Notes on the localization of generalized hexagonal cellular networks. Mathematics 2023, 11, 844. [Google Scholar] [CrossRef]
- Schölkopf, B.; Williamson, R.; Smola, A.; Shawe-Taylor, J.; Platt, J. Support vector method for novelty detection. Adv. Neural Inf. Process. Syst. 1999, 12, 1–7. [Google Scholar]
- Guerbai, Y.; Chibani, Y.; Hadjadji, B. The effective use of the One-Class SVM classifier for reduced training samples and its application to handwritten signature verification. Int. Conf. Multimed. Comput. Syst. 2014, 2014, 362–366. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Razaghi-Moghadam, Z.; Nikoloski, Z. Supervised learning of gene-regulatory networks based on graph distance profiles of transcriptomics data. NPJ Syst. Biol. Appl. 2020, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Kc, K.; Li, R.; Cui, F.; Yu, Q.; Haake, A. GNE: A deep learning framework for gene network inference by aggregating biological information. BMC Syst. Biol. 2019, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Marbach, D.; Costello, J.C.; Küffner, R.; Vega, N.M.; Prill, R.J.; Camacho, D.M.; Allison, K.R.; Kellis, M.; Collins, J.J.; Stolovitzky, G. Wisdom of crowds for robust gene network inference. Nat. Methods 2012, 9, 796–804. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| REWKLR | tpm | fpkm | micro |
| Class Accuracy 0 | 94 | 94 | 97 |
| Class Accuracy 1 | 83 | 95 | 75 |
| 2.4 | 0.2 | 0.01 | |
| 0.2 | 0.1 | 1.5 | |
| SVM | tpm | fpkm | micro |
| Class Accuracy 0 | 99 | 99 | 93.5 |
| Class Accuracy 1 | 86 | 85 | 99 |
| 4 | 3.8 | 3.1 | |
| C | 18 | 20 | 3.0 |
| One-class SVM | tpm | fpkm | micro |
| Class Accuracy 1 | 96.6 | 97.5 | 94.8 |
| 1.8 | 1.5 | 0.1 | |
| 0.03 | 0.03 | 0.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Aamri, A.; Kudlicki, A.S.; Maalouf, M.; Taha, K.; Homouz, D. Inferring Gene Regulatory Networks from RNA-seq Data Using Kernel Classification. Biology 2023, 12, 518. https://doi.org/10.3390/biology12040518
Al-Aamri A, Kudlicki AS, Maalouf M, Taha K, Homouz D. Inferring Gene Regulatory Networks from RNA-seq Data Using Kernel Classification. Biology. 2023; 12(4):518. https://doi.org/10.3390/biology12040518
Chicago/Turabian StyleAl-Aamri, Amira, Andrzej S. Kudlicki, Maher Maalouf, Kamal Taha, and Dirar Homouz. 2023. "Inferring Gene Regulatory Networks from RNA-seq Data Using Kernel Classification" Biology 12, no. 4: 518. https://doi.org/10.3390/biology12040518
APA StyleAl-Aamri, A., Kudlicki, A. S., Maalouf, M., Taha, K., & Homouz, D. (2023). Inferring Gene Regulatory Networks from RNA-seq Data Using Kernel Classification. Biology, 12(4), 518. https://doi.org/10.3390/biology12040518