
Revealing the Microbiome of Four Different Thermal Springs in Turkey with Environmental DNA Metabarcoding


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Site Description and Sample Collection
2.2. Physicochemical Analysis of Water Samples
2.3. DNA Extraction and PCR Amplifications
2.4. Library Preparations and NGS
2.5. Bioinformatic Analysis
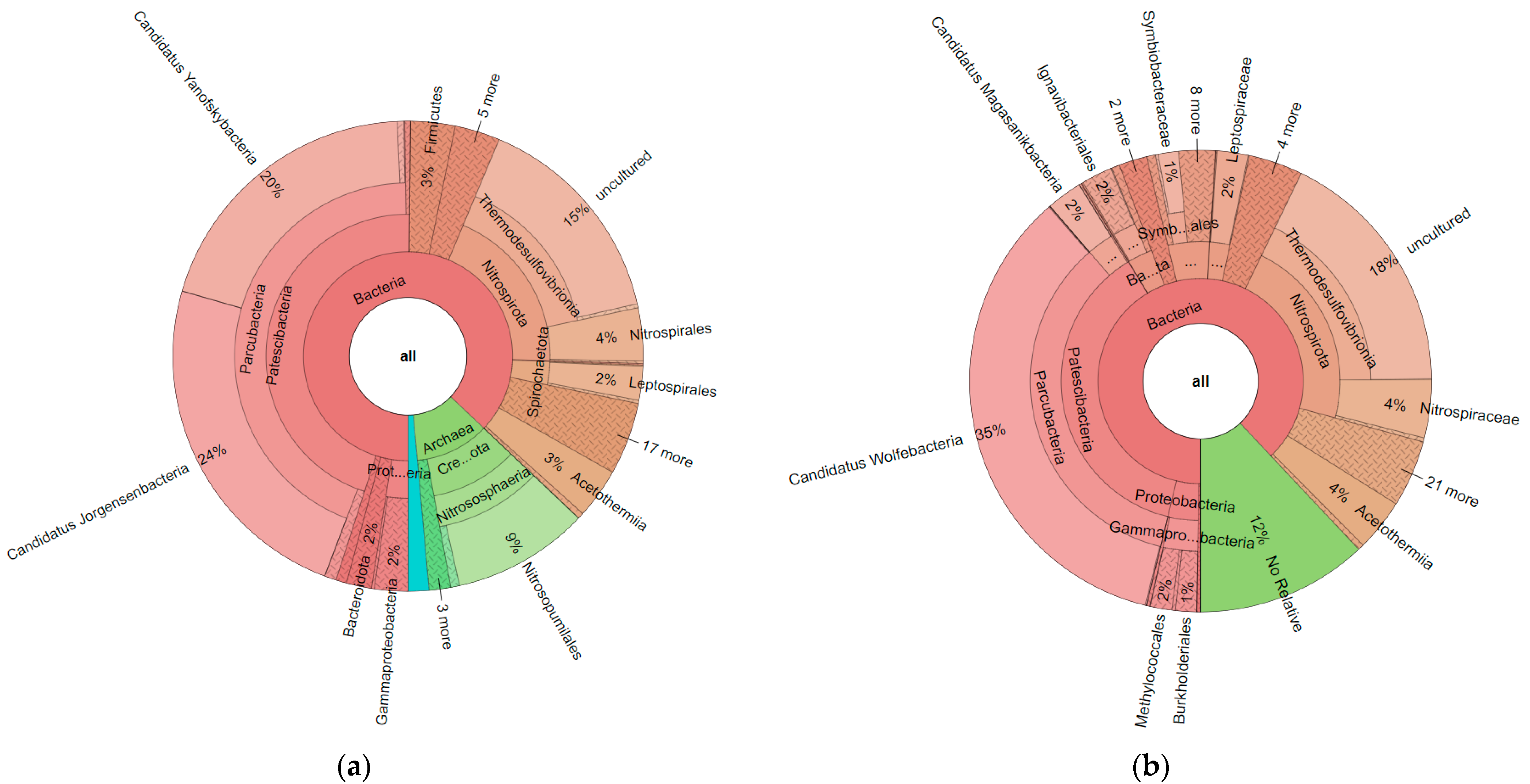
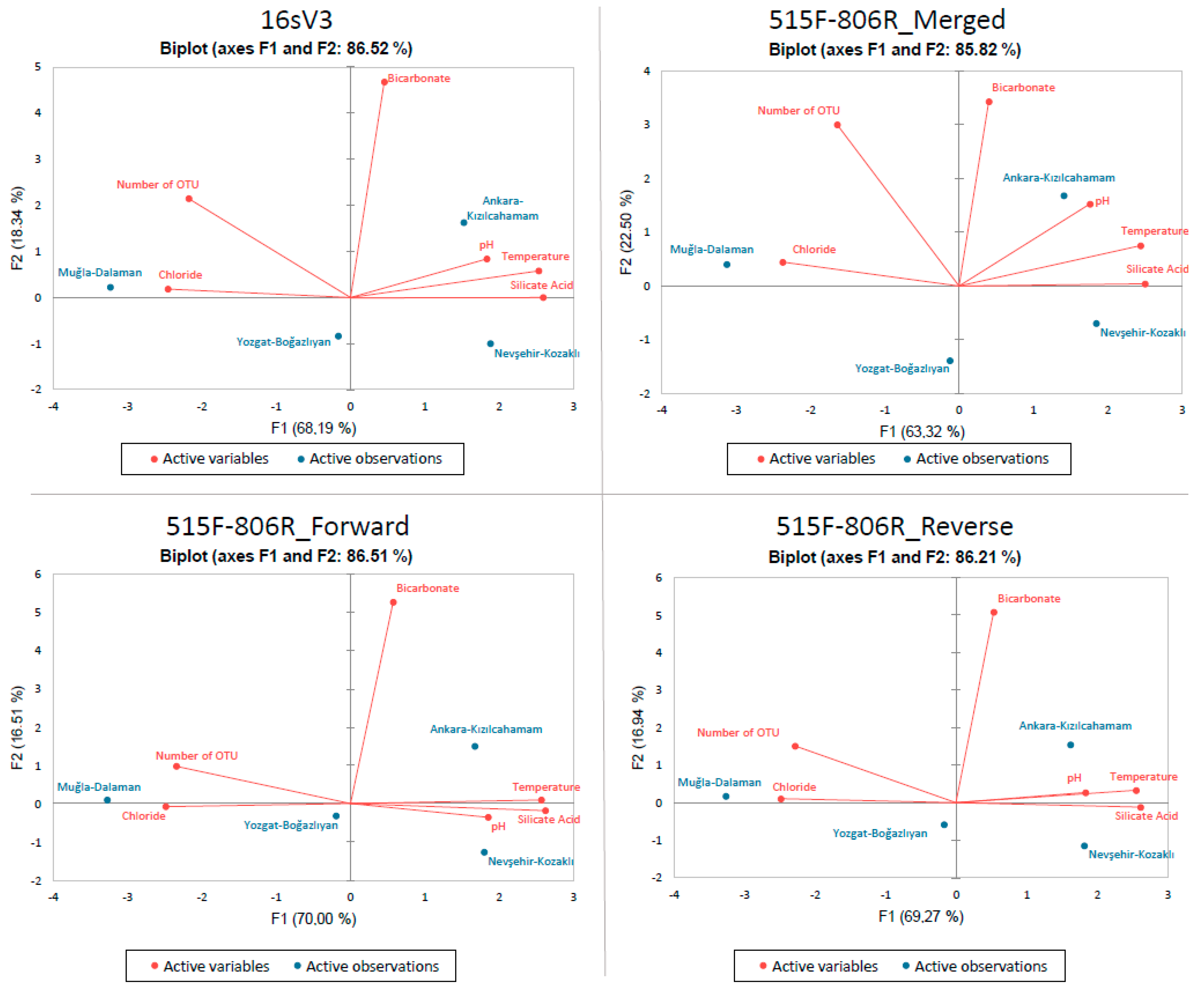
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marchesi, J.R.; Ravel, J. The vocabulary of microbiome research: A proposal. Microbiome 2015, 3, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewin, A.; Wentzel, A.; Valla, S. Metagenomics of microbial life in extreme temperature environments. Curr. Opin. Biotechnol. 2013, 24, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Danilenko, V.; Devyatkin, A.; Marsova, M.; Shibilova, M.; Ilyasov, R.; Shmyrev, V. Common Inflammatory Mechanisms in COVID-19 and Parkinson’s Diseases: The Role of Microbiome, Pharmabiotics and Postbiotics in Their Prevention. J. Inflamm. Res. 2021, 14, 6349–6381. [Google Scholar] [CrossRef]
- Mohajeri, M.H.; Brummer, R.J.M.; Rastall, R.A.; Weersma, R.K.; Harmsen, H.J.M.; Faas, M.; Eggersdorfer, M. The role of the microbiome for human health: From basic science to clinical applications. Eur. J. Nutr. 2018, 57, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foo, J.L.; Ling, H.; Lee, Y.S.; Chang, M.W. Microbiome engineering: Current applications and its future. Biotechnol. J. 2017, 12, 1600099. [Google Scholar] [CrossRef] [Green Version]
- Tighe, S.; Afshinnekoo, E.; Rock, T.M.; McGrath, K.; Alexander, N.; McIntyre, A.; Ahsanuddin, S.; Bezdan, D.; Green, S.J.; Joye, S.; et al. Genomic Methods and Microbiological Technologies for Profiling Novel and Extreme Environments for the Extreme Microbiome Project (XMP). J. Biomol. Tech. 2017, 28, 31–39. [Google Scholar] [CrossRef]
- Bodor, A.; Bounedjoum, N.; Vincze, G.E.; Erdeiné Kis, Á.; Laczi, K.; Bende, G.; Szilágyi, Á.; Kovács, T.; Perei, K.; Rákhely, G. Challenges of unculturable bacteria: Environmental perspectives. Rev. Environ. Sci. Bio/Technol. 2020, 19, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Lopez, O.; Cerdan, M.E.; Gonzalez-Siso, M.I. Hot spring metagenomics. Life 2013, 3, 308–320. [Google Scholar] [CrossRef] [Green Version]
- Rawat, N.; Joshi, G.K. Bacterial community structure analysis of a hot spring soil by next generation sequencing of ribosomal RNA. Genomics 2019, 111, 1053–1058. [Google Scholar] [CrossRef]
- Bukin, Y.S.; Galachyants, Y.P.; Morozov, I.V.; Bukin, S.V.; Zakharenko, A.S.; Zemskaya, T.I. The effect of 16S rRNA region choice on bacterial community metabarcoding results. Sci. Data 2019, 6, 190007. [Google Scholar] [CrossRef] [Green Version]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; Gonzalez, A.; Kosciolek, T.; McCall, L.I.; McDonald, D.; et al. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 2018, 16, 410–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygaard, A.B.; Tunsjo, H.S.; Meisal, R.; Charnock, C. A preliminary study on the potential of Nanopore MinION and Illumina MiSeq 16S rRNA gene sequencing to characterize building-dust microbiomes. Sci. Rep. 2020, 10, 3209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheka, D.; Alabi, N.; Gordon, P.M.K. Oxford nanopore sequencing in clinical microbiology and infection diagnostics. Brief. Bioinform. 2021, 22, bbaa403. [Google Scholar] [CrossRef]
- Thomsen, P.F.; Willerslev, E. Environmental DNA—An emerging tool in conservation for monitoring past and present biodiversity. Biol. Conserv. 2015, 183, 4–18. [Google Scholar] [CrossRef]
- Taberlet, P.; Coissac, E.; Hajibabaei, M.; Rieseberg, L.H. Environmental DNA. Mol. Ecol. 2012, 21, 1789–1793. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, S.; Iguchi, Y.; Shibata, N.; Teramura, I.; Kitagawa, T.; Yamanaka, H. Real-time multiplex PCR for simultaneous detection of multiple species from environmental DNA: An application on two Japanese medaka species. Sci. Rep. 2018, 8, 9138. [Google Scholar] [CrossRef] [Green Version]
- Beng, K.C.; Corlett, R.T. Applications of environmental DNA (eDNA) in ecology and conservation: Opportunities, challenges and prospects. Biodivers. Conserv. 2020, 29, 2089–2121. [Google Scholar] [CrossRef]
- Kress, W.J.; Erickson, D.L. DNA barcodes: Genes, genomics, and bioinformatics. Proc. Natl. Acad. Sci. USA 2008, 105, 2761–2762. [Google Scholar] [CrossRef] [Green Version]
- Chakravorty, S.; Helb, D.; Burday, M.; Connell, N.; Alland, D. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 2007, 69, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Perez, V.; Cortes, J.; Marchant, F.; Dorador, C.; Molina, V.; Cornejo-D’Ottone, M.; Hernandez, K.; Jeffrey, W.; Barahona, S.; Hengst, M.B. Aquatic Thermal Reservoirs of Microbial Life in a Remote and Extreme High Andean Hydrothermal System. Microorganisms 2020, 8, 208. [Google Scholar] [CrossRef] [Green Version]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Lazarevic, V.; Gaïa, N.; Girard, M.; Schrenzel, J. Decontamination of 16S rRNA gene amplicon sequence datasets based on bacterial load assessment by qPCR. BMC Microbiol. 2016, 16, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyer, K.M.; Altrichter, A.E.; Van Horn, D.J.; Takacs-Vesbach, C.D.; Gooseff, M.N.; Barrett, J.E. Environmental controls over bacterial communities in polar desert soils. Ecosphere 2013, 4, art127. [Google Scholar] [CrossRef] [Green Version]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Santos, H.F.; Carmo, F.L.; Duarte, G.; Dini-Andreote, F.; Castro, C.B.; Rosado, A.S.; van Elsas, J.D.; Peixoto, R.S. Climate change affects key nitrogen-fixing bacterial populations on coral reefs. ISME J. 2014, 8, 2272–2279. [Google Scholar] [CrossRef] [Green Version]
- Mußmann, M.; Pjevac, P.; Krüger, K.; Dyksma, S. Genomic repertoire of the Woeseiaceae/JTB255, cosmopolitan and abundant core members of microbial communities in marine sediments. ISME J. 2017, 11, 1276–1281. [Google Scholar] [CrossRef]
- Stressmann, F.A.; Bernal-Bayard, J.; Perez-Pascual, D.; Audrain, B.; Rendueles, O.; Briolat, V.; Bruchmann, S.; Volant, S.; Ghozlane, A.; Häussler, S.; et al. Mining zebrafish microbiota reveals key community-level resistance against fish pathogen infection. ISME J. 2021, 15, 702–719. [Google Scholar] [CrossRef]
- Reitmeier, S.; Hitch, T.C.A.; Treichel, N. Handling of spurious sequences affects the outcome of high-throughput 16S rRNA gene amplicon profiling. ISME Commun. 2021, 1, 31. [Google Scholar] [CrossRef]
- Jimenez, D.J.; Andreote, F.D.; Chaves, D.; Montana, J.S.; Osorio-Forero, C.; Junca, H.; Zambrano, M.M.; Baena, S. Structural and functional insights from the metagenome of an acidic hot spring microbial planktonic community in the Colombian Andes. PLoS ONE 2012, 7, e52069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yvon-Durocher, G.; Allen, A.P.; Cellamare, M.; Dossena, M.; Gaston, K.J.; Leitao, M.; Montoya, J.M.; Reuman, D.C.; Woodward, G.; Trimmer, M. Five Years of Experimental Warming Increases the Biodiversity and Productivity of Phytoplankton. PLoS Biol. 2015, 13, e1002324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panigrahi, S.; Nydahl, A.; Anton, P.; Wikner, J. Strong seasonal effect of moderate experimental warming on plankton respiration in a temperate estuarine plankton community. Estuar. Coast. Shelf Sci. 2013, 135, 269–279. [Google Scholar] [CrossRef]
- Geraldes, P.; Pascoal, C.; Cássio, F. Effects of increased temperature and aquatic fungal diversity on litter decomposition. Fungal Ecol. 2012, 5, 734–740. [Google Scholar] [CrossRef] [Green Version]
- Wohlers, J.; Engel, A.; Zollner, E.; Breithaupt, P.; Jurgens, K.; Hoppe, H.G.; Sommer, U.; Riebesell, U. Changes in biogenic carbon flow in response to sea surface warming. Proc. Natl. Acad. Sci. USA 2009, 106, 7067–7072. [Google Scholar] [CrossRef] [Green Version]
- Graspeuntner, S.; Loeper, N.; Kunzel, S.; Baines, J.F.; Rupp, J. Selection of validated hypervariable regions is crucial in 16S-based microbiota studies of the female genital tract. Sci. Rep. 2018, 8, 9678. [Google Scholar] [CrossRef]
- Youssef, N.; Sheik, C.S.; Krumholz, L.R.; Najar, F.Z.; Roe, B.A.; Elshahed, M.S. Comparison of Species Richness Estimates Obtained Using Nearly Complete Fragments and Simulated Pyrosequencing-Generated Fragments in 16S rRNA Gene-Based Environmental Surveys. Appl. Environ. Microbiol. 2009, 75, 5227–5236. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Qian, P.Y. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS ONE 2009, 4, e7401. [Google Scholar] [CrossRef] [Green Version]
- Zaremba-Niedzwiedzka, K.; Caceres, E.F.; Saw, J.H.; Backstrom, D.; Juzokaite, L.; Vancaester, E.; Seitz, K.W.; Anantharaman, K.; Starnawski, P.; Kjeldsen, K.U.; et al. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 2017, 541, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Carrier, V.; Svenning, M.M.; Gründger, F.; Niemann, H.; Dessandier, P.-A.; Panieri, G.; Kalenitchenko, D. The Impact of Methane on Microbial Communities at Marine Arctic Gas Hydrate Bearing Sediment. Front. Microbiol. 2020, 11, 1932. [Google Scholar] [CrossRef]
- Teske, A.; Wegener, G.; Chanton, J.P.; White, D.; MacGregor, B.; Hoer, D.; de Beer, D.; Zhuang, G.; Saxton, M.A.; Joye, S.B.; et al. Microbial Communities Under Distinct Thermal and Geochemical Regimes in Axial and Off-Axis Sediments of Guaymas Basin. Front. Microbiol. 2021, 12, 633649. [Google Scholar] [CrossRef] [PubMed]
- Clark, A. The thermal limits to life. Int. J. Astrobiol. 2014, 13, 141–154. [Google Scholar] [CrossRef] [Green Version]
- Oschmann, W.; Grasshof, M.; Gudo, M. The early evolution of the planet earth and the origin of life. Senckenbergiana Lethaea 2002, 82, 284–294. [Google Scholar] [CrossRef]
- Cristescu, M.E. From barcoding single individuals to metabarcoding biological communities: Towards an integrative approach to the study of global biodiversity. Trends Ecol. Evol. 2014, 29, 566–571. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Çelik, I.; Keskin, E. Revealing the Microbiome of Four Different Thermal Springs in Turkey with Environmental DNA Metabarcoding. Biology 2022, 11, 998. https://doi.org/10.3390/biology11070998
Çelik I, Keskin E. Revealing the Microbiome of Four Different Thermal Springs in Turkey with Environmental DNA Metabarcoding. Biology. 2022; 11(7):998. https://doi.org/10.3390/biology11070998
Chicago/Turabian StyleÇelik, Işılay, and Emre Keskin. 2022. "Revealing the Microbiome of Four Different Thermal Springs in Turkey with Environmental DNA Metabarcoding" Biology 11, no. 7: 998. https://doi.org/10.3390/biology11070998
APA StyleÇelik, I., & Keskin, E. (2022). Revealing the Microbiome of Four Different Thermal Springs in Turkey with Environmental DNA Metabarcoding. Biology, 11(7), 998. https://doi.org/10.3390/biology11070998