
Potential Methods of Targeting Cellular Aging Hallmarks to Reverse Osteoarthritic Phenotype of Chondrocytes

 , , and
, , and 
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Characterization of OA Chondrocytes
3. Association between OA and Aging
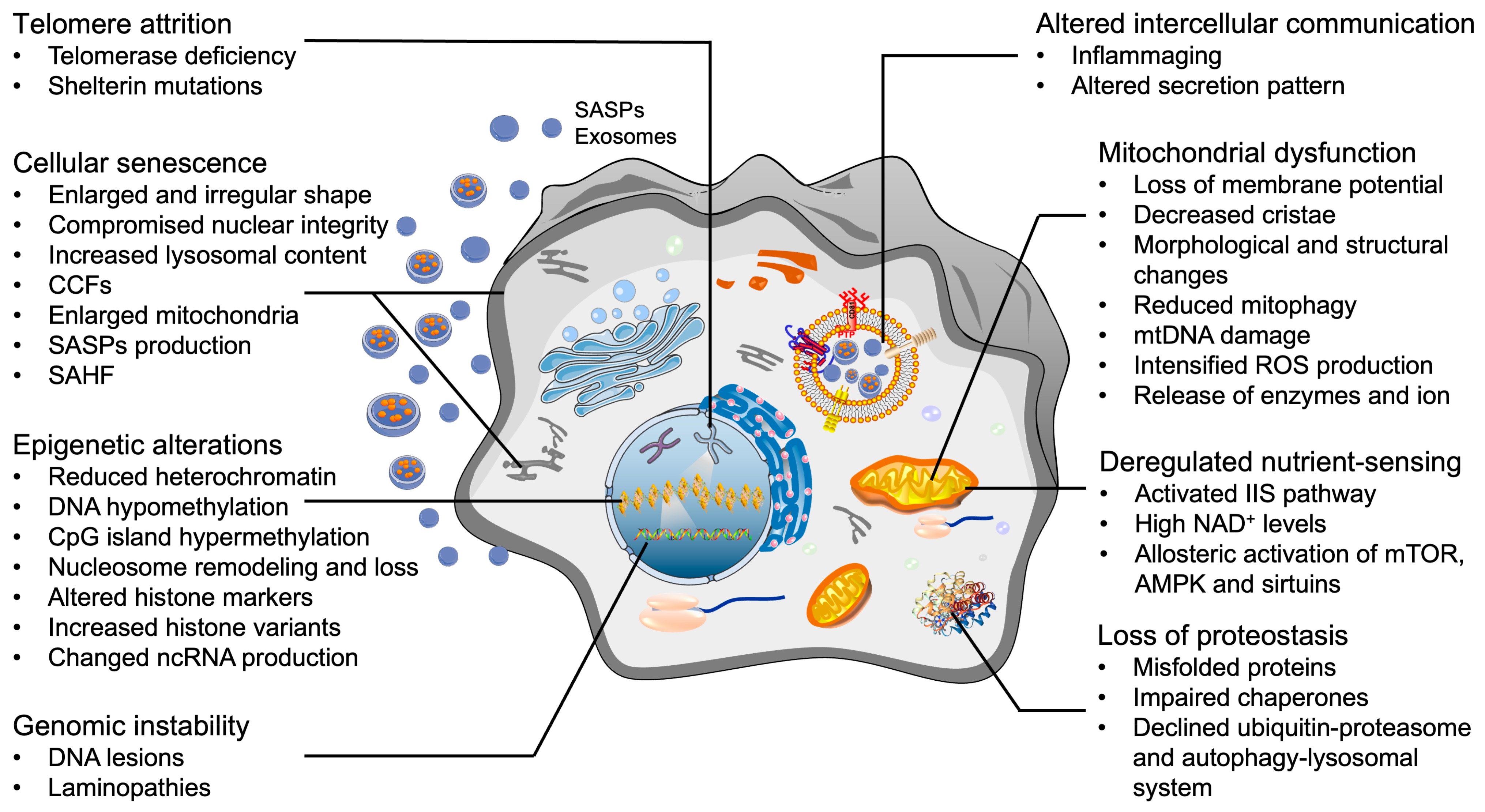
4. Cellular Hallmarks of Aging in OA Chondrocytes
4.1. Telomere Attrition
4.2. Epigenetic Alterations
4.3. Mitochondrial Dysfunction
4.4. Loss of Proteostasis
4.5. Deregulated Nutrient-Sensing
4.6. Genomic Instability
4.7. Cellular Senescence
4.8. Altered Intercellular Communication
5. Potential of Targeting Aging Hallmarks to Reverse OA Chondrocytes
5.1. Increase of Genomic Stability
5.2. Elongation of Telomeres
5.3. Epigenetic Modifications
5.4. Restoration of Mitochondrial Function
5.5. Improvement of Proteostasis
5.6. Metabolism Interventions
5.7. Mitigation of Cellular Senescence
5.8. Reduction of Detrimental Intercellular Communication
6. Healthy Aging of Chondrocytes
7. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| OA | Osteoarthritis |
| AGEs | Advanced glycation end products |
| AMPK | AMP-activated protein kinase |
| ATP | Adenosine-triphosphate |
| ATR | Ataxia-telangiectasia mutated and rad3-related |
| ATRIP | ATR-interacting protein |
| CDK | Cyclin-dependent kinase |
| CR | Caloric restriction |
| DAMPs | Damage-associated molecular patterns |
| DMOADs | Disease-modifying OA drugs |
| ECM | Extracellular matrix |
| eIF | Eukaryotic initiation factor |
| ER | Endoplasmic reticulum |
| ERK | Extracellular regulated protein kinases |
| FOXO | Forkhead box O |
| GAG | Glycosaminoglycan |
| HSF | Heat shock factor |
| IGF | Insulin-like growth factor |
| IIS | Insulin/IGF-1 signaling |
| IL | Interleukin |
| LINE-1 | Long interspersed nuclear element 1 |
| MAPK | Mitogen-activated protein kinase |
| MMPs | Matrix metalloproteinases |
| MSC | Mesenchymal stem cell |
| mtDNA | Mitochondrial DNA |
| NAD | Nicotinamide adenine dinucleotide |
| ncRNAs | Non-coding RNAs |
| Nrf2 | Nuclear factor-erythroid 2-related factor |
| PRR | Pathogen-recognition receptors |
| ROS | Reactive oxygen species |
| RS | Replicative senescence |
| RUNX2 | Runt-related transcription factor 2 |
| SASPs | Senescence-associated secretory phenotypes |
| SA-β-gal | Senescence-associated-β-galactosidase |
| SIPS | Stress-induced premature senescence |
| SIRT | Sirtuin |
| SOD | Superoxide dismutase |
| SPD | Spermidine |
| SQST | Sequestosome |
| TGF-β | Transforming growth factor-β |
| TLR | Toll-like receptors |
| TOR | Target of rapamycin |
References
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Abhishek, A.; Doherty, M. Diagnosis and clinical presentation of osteoarthritis. Rheum Dis Clin. N. Am. 2013, 39, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Safiri, S.; Kolahi, A.A.; Smith, E.; Hill, C.; Bettampadi, D.; Mansournia, M.A.; Hoy, D.; Ashrafi-Asgarabad, A.; Sepidarkish, M.; Almasi-Hashiani, A.; et al. Global, regional and national burden of osteoarthritis 1990-2017: A systematic analysis of the Global Burden of Disease Study 2017. Ann. Rheum. Dis. 2020, 79, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, S.H.; Lim, S.M.; Baek, S.H.; Ha, I.H. Mental health and quality of life of patients with osteoarthritis pain: The sixth Korea National Health and Nutrition Examination Survey (2013–2015). PLoS ONE 2020, 15, e0242077. [Google Scholar] [CrossRef]
- Glyn-Jones, S.; Palmer, A.J.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H.; Carr, A.J. Osteoarthritis. Lancet 2015, 386, 376–387. [Google Scholar] [CrossRef]
- Carr, A.J.; Robertsson, O.; Graves, S.; Price, A.J.; Arden, N.K.; Judge, A.; Beard, D.J. Knee replacement. Lancet 2012, 379, 1331–1340. [Google Scholar] [CrossRef]
- Palazzo, C.; Nguyen, C.; Lefevre-Colau, M.M.; Rannou, F.; Poiraudeau, S. Risk factors and burden of osteoarthritis. Ann. Phys. Rehabil Med. 2016, 59, 134–138. [Google Scholar] [CrossRef]
- Carballo, C.B.; Nakagawa, Y.; Sekiya, I.; Rodeo, S.A. Basic science of articular cartilage. Clin. Sports Med. 2017, 36, 413–425. [Google Scholar] [CrossRef]
- Charlier, E.; Deroyer, C.; Ciregia, F.; Malaise, O.; Neuville, S.; Plener, Z.; Malaise, M.; de Seny, D. Chondrocyte dedifferentiation and osteoarthritis (OA). Biochem. Pharmacol. 2019, 165, 49–65. [Google Scholar] [CrossRef]
- Varela-Eirin, M.; Loureiro, J.; Fonseca, E.; Corrochano, S.; Caeiro, J.R.; Collado, M.; Mayan, M.D. Cartilage regeneration and ageing: Targeting cellular plasticity in osteoarthritis. Ageing Res. Rev. 2018, 42, 56–71. [Google Scholar] [CrossRef]
- Singh, P.; Marcu, K.B.; Goldring, M.B.; Otero, M. Phenotypic instability of chondrocytes in osteoarthritis: On a path to hypertrophy. Ann. N. Y. Acad. Sci. 2019, 1442, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Gratal, P.; Mediero, A.; Sánchez-Pernaute, O.; Prieto-Potin, I.; Lamuedra, A.; Herrero-Beaumont, G.; Largo, R. Chondrocyte enlargement is a marker of osteoarthritis severity. Osteoarthr. Cartil. 2019, 27, 1229–1234. [Google Scholar] [CrossRef] [PubMed]
- Lauer, J.C.; Selig, M.; Hart, M.L.; Kurz, B.; Rolauffs, B. Articular chondrocyte phenotype regulation through the cytoskeleton and the signaling processes that originate from or converge on the cytoskeleton: Towards a novel understanding of the intersection between actin dynamics and chondrogenic function. Int. J. Mol. Sci. 2021, 22, 3279. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Scott, A.K.; Seelbinder, B.; Barthold, J.E.; Martin, B.M.S.; Kaonis, S.; Schneider, S.E.; Henderson, J.T.; Neu, C.P. Dedifferentiation alters chondrocyte nuclear mechanics during in vitro culture and expansion. Biophys. J. 2022, 121, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Bobick, B.E.; Chen, F.H.; Le, A.M.; Tuan, R.S. Regulation of the chondrogenic phenotype in culture. Birth Defects Res. C Embryo Today 2009, 87, 351–371. [Google Scholar] [CrossRef]
- Sasazaki, Y.; Seedhom, B.B.; Shore, R. Morphology of the bovine chondrocyte and of its cytoskeleton in isolation and in situ: Are chondrocytes ubiquitously paired through the entire layer of articular cartilage? Rheumatology 2008, 47, 1641–1646. [Google Scholar] [CrossRef]
- Shin, H.; Lee, M.N.; Choung, J.S.; Kim, S.; Choi, B.H.; Noh, M.; Shin, J.H. Focal adhesion assembly induces phenotypic changes and dedifferentiation in chondrocytes. J. Cell Physiol. 2016, 231, 1822–1831. [Google Scholar] [CrossRef]
- Dominice, J.; Levasseur, C.; Larno, S.; Ronot, X.; Adolphe, M. Age-related changes in rabbit articular chondrocytes. Mech Ageing Dev. 1986, 37, 231–240. [Google Scholar] [CrossRef]
- Duan, W.; Wei, L.; Zhang, J.; Hao, Y.; Li, C.; Li, H.; Li, Q.; Zhang, Q.; Chen, W.; Wei, X. Alteration of viscoelastic properties is associated with a change in cytoskeleton components of ageing chondrocytes from rabbit knee articular cartilage. Mol. Cell Biomech. 2011, 8, 253–274. [Google Scholar]
- Tsolis, K.C.; Bei, E.S.; Papathanasiou, I.; Kostopoulou, F.; Gkretsi, V.; Kalantzaki, K.; Malizos, K.; Zervakis, M.; Tsezou, A.; Economou, A. Comparative proteomic analysis of hypertrophic chondrocytes in osteoarthritis. Clin. Proteom. 2015, 12, 12. [Google Scholar] [CrossRef]
- Shen, H.; He, Y.; Wang, N.; Fritch, M.R.; Li, X.; Lin, H.; Tuan, R.S. Enhancing the potential of aged human articular chondrocytes for high-quality cartilage regeneration. FASEB J. 2021, 35, e21410. [Google Scholar] [CrossRef] [PubMed]
- Coryell, P.R.; Diekman, B.O.; Loeser, R.F. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat. Rev. Rheumatol. 2021, 17, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Chen, X.; Chen, J.; Zheng, G.; Xie, C.; Wu, H.; Miao, Z.; Lin, Y.; Wang, X.; Gao, W.; et al. STING promotes senescence, apoptosis, and extracellular matrix degradation in osteoarthritis via the NF-κB signaling pathway. Cell Death Dis. 2021, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Rando, T.A.; Jones, D.L. Regeneration, rejuvenation, and replacement: Turning back the clock on tissue aging. Cold Spring Harb. Perspect. Biol. 2021, 13, a040907. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T.B.L. Understanding the odd science of aging. Cell 2005, 120, 437–447. [Google Scholar] [CrossRef]
- Shane Anderson, A.; Loeser, R.F. Why is osteoarthri.itis an age-related disease? Best Pract. Res. Clin. Rheumatol. 2010, 24, 15–26. [Google Scholar] [CrossRef]
- Lawrence, R.C.; Felson, D.T.; Helmick, C.G.; Arnold, L.M.; Choi, H.; Deyo, R.A.; Gabriel, S.; Hirsch, R.; Hochberg, M.C.; Hunder, G.G.; et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008, 58, 26–35. [Google Scholar] [CrossRef]
- Dillon, C.F.; Rasch, E.K.; Gu, Q.; Hirsch, R. Prevalence of knee osteoarthritis in the United States: Arthritis data from the Third National Health and Nutrition Examination Survey 1991–94. J. Rheumatol. 2006, 33, 2271–2279. [Google Scholar]
- Ho-Pham, L.T.; Lai, T.Q.; Mai, L.D.; Doan, M.C.; Pham, H.N.; Nguyen, T.V. Prevalence of radiographic osteoarthritis of the knee and its relationship to self-reported pain. PLoS ONE 2014, 9, e94563. [Google Scholar] [CrossRef]
- Robinson, W.H.; Lepus, C.M.; Wang, Q.; Raghu, H.; Mao, R.; Lindstrom, T.M.; Sokolove, J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 580–592. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Livshits, G.; Zhai, G.; Hart, D.J.; Kato, B.S.; Wang, H.; Williams, F.M.; Spector, T.D. Interleukin-6 is a significant predictor of radiographic knee osteoarthritis: The Chingford Study. Arthritis Rheum. 2009, 60, 2037–2045. [Google Scholar] [CrossRef] [PubMed]
- Spector, T.D.; Hart, D.J.; Nandra, D.; Doyle, D.V.; Mackillop, N.; Gallimore, J.R.; Pepys, M.B. Low-level increases in serum C-reactive protein are present in early osteoarthritis of the knee and predict progressive disease. Arthritis Rheum. 1997, 40, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Stannus, O.P.; Jones, G.; Blizzard, L.; Cicuttini, F.M.; Ding, C. Associations between serum levels of inflammatory markers and change in knee pain over 5 years in older adults: A prospective cohort study. Ann. Rheum. Dis. 2013, 72, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.P.; Martel-Pelletier, J.; Abramson, S.B. Osteoarthritis, an inflammatory disease: Potential implication for the selection of new therapeutic targets. Arthritis Rheum. 2001, 44, 1237–1247. [Google Scholar] [CrossRef]
- Greene, M.A.; Loeser, R.F. Aging-related inflammation in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1966–1971. [Google Scholar] [CrossRef]
- Scanzello, C.R.; McKeon, B.; Swaim, B.H.; DiCarlo, E.; Asomugha, E.U.; Kanda, V.; Nair, A.; Lee, D.M.; Richmond, J.C.; Katz, J.N.; et al. Synovial inflammation in patients undergoing arthroscopic meniscectomy: Molecular characterization and relationship to symptoms. Arthritis Rheum. 2011, 63, 391–400. [Google Scholar] [CrossRef]
- Sellam, J.; Berenbaum, F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat. Rev. Rheumatol. 2010, 6, 625–635. [Google Scholar] [CrossRef]
- Roemer, F.W.; Guermazi, A.; Felson, D.T.; Niu, J.; Nevitt, M.C.; Crema, M.D.; Lynch, J.A.; Lewis, C.E.; Torner, J.; Zhang, Y. Presence of MRI-detected joint effusion and synovitis increases the risk of cartilage loss in knees without osteoarthritis at 30-month follow-up: The MOST study. Ann. Rheum. Dis. 2011, 70, 1804–1809. [Google Scholar] [CrossRef]
- Hill, C.L.; Hunter, D.J.; Niu, J.; Clancy, M.; Guermazi, A.; Genant, H.; Gale, D.; Grainger, A.; Conaghan, P.; Felson, D.T. Synovitis detected on magnetic resonance imaging and its relation to pain and cartilage loss in knee osteoarthritis. Ann. Rheum. Dis. 2007, 66, 1599–1603. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Dodig, S.; Čepelak, I.; Pavić, I. Hallmarks of senescence and aging. Biochem. Med. 2019, 29, 030501. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of cellular senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Denoth-Lippuner, A.; Jessberger, S. Mechanisms of cellular rejuvenation. FEBS Lett. 2019, 593, 3381–3392. [Google Scholar] [CrossRef]
- Zhang, W.; Qu, J.; Liu, G.H.; Belmonte, J.C.I. The ageing epigenome and its rejuvenation. Nat. Rev. Mol. Cell Biol. 2020, 21, 137–150. [Google Scholar] [CrossRef]
- Blackburn, E.H. The molecular structure of centromeres and telomeres. Annu. Rev. Biochem. 1984, 53, 163–194. [Google Scholar] [CrossRef]
- Fragkiadaki, P.; Nikitovic, D.; Kalliantasi, K.; Sarandi, E.; Thanasoula, M.; Stivaktakis, P.D.; Nepka, C.; Spandidos, D.A.; Tosounidis, T.; Tsatsakis, A. Telomere length and telomerase activity in osteoporosis and osteoarthritis. Exp. Ther. Med. 2020, 19, 1626–1632. [Google Scholar] [CrossRef]
- Xin, H.; Liu, D.; Songyang, Z. The telosome/shelterin complex and its functions. Genome Biol. 2008, 9, 232. [Google Scholar] [CrossRef]
- Ain, Q.; Schmeer, C.; Penndorf, D.; Fischer, M.; Bondeva, T.; Förster, M.; Haenold, R.; Witte, O.W.; Kretz, A. Cell cycle-dependent and -independent telomere shortening accompanies murine brain aging. Aging 2018, 10, 3397–3420. [Google Scholar] [CrossRef]
- Saretzki, G. Telomeres, telomerase and ageing. Subcell Biochem. 2018, 90, 221–308. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.; Novakofski, K.D.; Donocoff, R.S.; Liang, Y.X.; Fortier, L.A. Telomerase activity in articular chondrocytes is lost after puberty. Cartilage 2014, 5, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Price, J.S.; Waters, J.G.; Darrah, C.; Pennington, C.; Edwards, D.R.; Donell, S.T.; Clark, I.M. The role of chondrocyte senescence in osteoarthritis. Aging Cell 2002, 1, 57–65. [Google Scholar] [CrossRef]
- Harbo, M.; Bendix, L.; Bay-Jensen, A.C.; Graakjaer, J.; Søe, K.; Andersen, T.L.; Kjaersgaard-Andersen, P.; Koelvraa, S.; Delaisse, J.M. The distribution pattern of critically short telomeres in human osteoarthritic knees. Arthritis Res. Ther. 2012, 14, R12. [Google Scholar] [CrossRef]
- Manoy, P.; Yuktanandana, P.; Tanavalee, A.; Tanpowpong, T.; Ittipanichpong, T.; Honsawek, S. Telomere shortening is associated with poor physical performance in knee osteoarthritis. Biomed. Rep. 2020, 13, 27. [Google Scholar] [CrossRef]
- Yang, J.; Xu, H.; Cai, B.; Wei, J.; Sun, L.; Li, Y.; Wang, T.; Li, Y. Genetically predicted longer telomere length may reduce risk of hip osteoarthritis. Front. Genet. 2021, 12, 718890. [Google Scholar] [CrossRef]
- Martin, J.A.; Buckwalter, J.A. Telomere erosion and senescence in human articular cartilage chondrocytes. J. Gerontol. A Biol. Sci. Med. Sci. 2001, 56, B172–B179. [Google Scholar] [CrossRef]
- Nagai, K.; Matsushita, T.; Matsuzaki, T.; Takayama, K.; Matsumoto, T.; Kuroda, R.; Kurosaka, M. Depletion of SIRT6 causes cellular senescence, DNA damage, and telomere dysfunction in human chondrocytes. Osteoarthr. Cartil. 2015, 23, 1412–1420. [Google Scholar] [CrossRef]
- Wang, Y.; Yuan, Q.; Xie, L. Histone Modifications in Aging: The Underlying Mechanisms and Implications. Curr. Stem Cell Res. Ther. 2018, 13, 125–135. [Google Scholar] [CrossRef]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, J. Epigenetics and Osteoarthritis. Genes Dis. 2015, 2, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.; Salazar, L.A. Autophagy and polyphenols in osteoarthritis: A focus on epigenetic regulation. Int. J. Mol. Sci. 2021, 23, 421. [Google Scholar] [CrossRef]
- Shen, J.; Abu-Amer, Y.; O’Keefe, R.J.; McAlinden, A. Inflammation and epigenetic regulation in osteoarthritis. Connect. Tissue Res. 2017, 58, 49–63. [Google Scholar] [CrossRef]
- Simon, T.C.; Jeffries, M.A. The epigenomic landscape in osteoarthritis. Curr. Rheumatol. Rep. 2017, 19, 30. [Google Scholar] [CrossRef]
- Reynard, L.N. Analysis of genetics and DNA methylation in osteoarthritis: What have we learnt about the disease? Semin. Cell Dev. Biol. 2017, 62, 57–66. [Google Scholar] [CrossRef]
- Rice, S.J.; Roberts, J.B.; Tselepi, M.; Brumwell, A.; Falk, J.; Steven, C.; Loughlin, J. Genetic and epigenetic fine-tuning of TGFB1 expression within the human osteoarthritic joint. Arthritis Rheumatol. 2021, 73, 1866–1877. [Google Scholar] [CrossRef]
- Ma, P.; Schultz, R.M. HDAC1 and HDAC2 in mouse oocytes and preimplantation embryos: Specificity versus compensation. Cell Death Differ. 2016, 23, 1119–1127. [Google Scholar] [CrossRef]
- Coutinho de Almeida, R.; Ramos, Y.F.M.; Mahfouz, A.; den Hollander, W.; Lakenberg, N.; Houtman, E.; van Hoolwerff, M.; Suchiman, H.E.D.; Rodríguez Ruiz, A.; Slagboom, P.E.; et al. RNA sequencing data integration reveals an miRNA interactome of osteoarthritis cartilage. Ann. Rheum. Dis. 2019, 78, 270–277. [Google Scholar] [CrossRef]
- Koch, R.E.; Josefson, C.C.; Hill, G.E. Mitochondrial function, ornamentation, and immunocompetence. Biol. Rev. Camb. Philos Soc. 2017, 92, 1459–1474. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Menzies, K.J.; Auwerx, J. The role of mitochondria in stem cell fate and aging. Development 2018, 145, dev143420. [Google Scholar] [CrossRef] [PubMed]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.G. Mammalian mitochondria and aging: An update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.Y.; Blum, A.; Liu, J.; Finkel, T. The role of mitochondria in aging. J. Clin. Investig. 2018, 128, 3662–3670. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Youle, R.J.; Finkel, T. The mitochondrial basis of aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef]
- Zhang, T.; Xi, Q.; Wang, D.; Li, J.; Wang, M.; Li, D.; Zhu, L.; Jin, L. Mitochondrial dysfunction and endoplasmic reticulum stress involved in oocyte aging: An analysis using single-cell RNA-sequencing of mouse oocytes. J. Ovarian Res. 2019, 12, 53. [Google Scholar] [CrossRef]
- Ruan, L.; Wang, Y.; Zhang, X.; Tomaszewski, A.; McNamara, J.T.; Li, R. Mitochondria-associated proteostasis. Annu. Rev. Biophys. 2020, 49, 41–67. [Google Scholar] [CrossRef]
- Deshwal, S.; Fiedler, K.U.; Langer, T. Mitochondrial proteases: Multifaceted regulators of mitochondrial plasticity. Annu. Rev. Biochem. 2020, 89, 501–528. [Google Scholar] [CrossRef]
- Sun, K.; Jing, X.; Guo, J.; Yao, X.; Guo, F. Mitophagy in degenerative joint diseases. Autophagy 2021, 17, 2082–2092. [Google Scholar] [CrossRef]
- Kan, S.; Duan, M.; Liu, Y.; Wang, C.; Xie, J. Role of mitochondria in physiology of chondrocytes and diseases of osteoarthritis and rheumatoid arthritis. Cartilage 2021, 13, 1102s–1121s. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. Embo J. 2018, 37, e99238. [Google Scholar] [CrossRef] [PubMed]
- Gadicherla, A.K.; Wang, N.; Bulic, M.; Agullo-Pascual, E.; Lissoni, A.; De Smet, M.; Delmar, M.; Bultynck, G.; Krysko, D.V.; Camara, A.; et al. Mitochondrial Cx43 hemichannels contribute to mitochondrial calcium entry and cell death in the heart. Basic Res. Cardiol. 2017, 112, 27. [Google Scholar] [CrossRef] [PubMed]
- Amorim, J.A.; Coppotelli, G.; Rolo, A.P.; Palmeira, C.M.; Ross, J.M.; Sinclair, D.A. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat. Rev. Endocrinol. 2022, 18, 243–258. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Makarczyk, M.J.; Lin, H. Role of mitochondria in mediating chondrocyte response to mechanical stimuli. Life Sci. 2020, 263, 118602. [Google Scholar] [CrossRef]
- Yao, X.; Zhang, J.; Jing, X.; Ye, Y.; Guo, J.; Sun, K.; Guo, F. Fibroblast growth factor 18 exerts anti-osteoarthritic effects through PI3K-AKT signaling and mitochondrial fusion and fission. Pharmacol. Res. 2019, 139, 314–324. [Google Scholar] [CrossRef]
- Zhang, J.; Hao, X.; Chi, R.; Qi, J.; Xu, T. Moderate mechanical stress suppresses the IL-1β-induced chondrocyte apoptosis by regulating mitochondrial dynamics. J. Cell Physiol. 2021, 236, 7504–7515. [Google Scholar] [CrossRef]
- Rego-Pérez, I.; Durán-Sotuela, A.; Ramos-Louro, P.; Blanco, F.J. Mitochondrial genetics and epigenetics in osteoarthritis. Front. Genet. 2019, 10, 1335. [Google Scholar] [CrossRef]
- He, Y.; Yocum, L.; Alexander, P.G.; Jurczak, M.J.; Lin, H. Urolithin A protects chondrocytes from mechanical overloading-induced injuries. Front. Pharmacol. 2021, 12, 703847. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhang, Z.; Sheng, P.; Mobasheri, A. The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res. Rev. 2021, 66, 101249. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Santra, M.; Dill, K.A.; de Graff, A.M.R. Proteostasis collapse is a driver of cell aging and death. Proc. Natl. Acad. Sci. USA 2019, 116, 22173–22178. [Google Scholar] [CrossRef] [PubMed]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- Fernando, R.; Drescher, C.; Nowotny, K.; Grune, T.; Castro, J.P. Impaired proteostasis during skeletal muscle aging. Free Radic. Biol. Med. 2019, 132, 58–66. [Google Scholar] [CrossRef]
- Korovila, I.; Hugo, M.; Castro, J.P.; Weber, D.; Höhn, A.; Grune, T.; Jung, T. Proteostasis, oxidative stress and aging. Redox Biol. 2017, 13, 550–567. [Google Scholar] [CrossRef]
- Serrano, R.L.; Chen, L.Y.; Lotz, M.K.; Liu-Bryan, R.; Terkeltaub, R. Impaired proteasomal function in human osteoarthritic chondrocytes can contribute to decreased levels of SOX9 and Aggrecan. Arthritis Rheumatol. 2018, 70, 1030–1041. [Google Scholar] [CrossRef]
- Tan, L.; Register, T.C.; Yammani, R.R. Age-related decline in expression of molecular chaperones induces endoplasmic reticulum stress and chondrocyte apoptosis in articular cartilage. Aging Dis. 2020, 11, 1091–1102. [Google Scholar] [CrossRef]
- Rellmann, Y.; Gronau, I.; Hansen, U.; Dreier, R. 4-phenylbutyric acid reduces endoplasmic reticulum stress in chondrocytes that is caused by loss of the protein disulfide isomerase ERp57. Oxid. Med. Cell Longev. 2019, 2019, 6404035. [Google Scholar] [CrossRef]
- Lan, C.N.; Cai, W.J.; Shi, J.; Yi, Z.J. MAPK inhibitors protect against early-stage osteoarthritis by activating autophagy. Mol. Med. Rep. 2021, 24, 829. [Google Scholar] [CrossRef] [PubMed]
- Dzięgielewska-Gęsiak, S.; Stołtny, D.; Brożek, A.; Muc-Wierzgoń, M.; Wysocka, E. Are insulin-resistance and oxidative stress cause or consequence of aging. Exp. Biol. Med. 2020, 245, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Rowan, S.; Bejarano, E.; Taylor, A. Mechanistic targeting of advanced glycation end-products in age-related diseases. Biochim Biophys. Acta Mol. Basis Dis. 2018, 1864, 3631–3643. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Collins, J.A.; Diekman, B.O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Archer, C.W.; Francis-West, P. The chondrocyte. Int. J. Biochem. Cell Biol. 2003, 35, 401–404. [Google Scholar] [CrossRef]
- Lepetsos, P.; Papavassiliou, A.G. ROS/oxidative stress signaling in osteoarthritis. Biochim. Biophys. Acta 2016, 1862, 576–591. [Google Scholar] [CrossRef]
- Choi, W.S.; Lee, G.; Song, W.H.; Koh, J.T.; Yang, J.; Kwak, J.S.; Kim, H.E.; Kim, S.K.; Son, Y.O.; Nam, H.; et al. The CH25H-CYP7B1-RORα axis of cholesterol metabolism regulates osteoarthritis. Nature 2019, 566, 254–258. [Google Scholar] [CrossRef]
- Loef, M.; Schoones, J.W.; Kloppenburg, M.; Ioan-Facsinay, A. Fatty acids and osteoarthritis: Different types, different effects. Jt. Bone Spine 2019, 86, 451–458. [Google Scholar] [CrossRef]
- Arra, M.; Swarnkar, G.; Ke, K.; Otero, J.E.; Ying, J.; Duan, X.; Maruyama, T.; Rai, M.F.; O’Keefe, R.J.; Mbalaviele, G.; et al. LDHA-mediated ROS generation in chondrocytes is a potential therapeutic target for osteoarthritis. Nat. Commun. 2020, 11, 3427. [Google Scholar] [CrossRef]
- Wei, F.Y.; Lee, J.K.; Wei, L.; Qu, F.; Zhang, J.Z. Correlation of insulin-like growth factor 1 and osteoarthritic cartilage degradation: A spontaneous osteoarthritis in guinea-pig. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4493–4500. [Google Scholar]
- Wen, C.; Xu, L.; Xu, X.; Wang, D.; Liang, Y.; Duan, L. Insulin-like growth factor-1 in articular cartilage repair for osteoarthritis treatment. Arthritis Res. Ther. 2021, 23, 277. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhao, L.; Chen, D. Growth factor signalling in osteoarthritis. Growth Factors 2018, 36, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Cherifi, C.; Monteagudo, S.; Lories, R.J. Promising targets for therapy of osteoarthritis: A review on the Wnt and TGF-β signalling pathways. Ther. Adv. Musculoskelet. Dis. 2021, 13, 1759720x211006959. [Google Scholar] [CrossRef] [PubMed]
- Blaney Davidson, E.N.; Scharstuhl, A.; Vitters, E.L.; van der Kraan, P.M.; van den Berg, W.B. Reduced transforming growth factor-beta signaling in cartilage of old mice: Role in impaired repair capacity. Arthritis Res. Ther. 2005, 7, R1338–R1347. [Google Scholar] [CrossRef] [PubMed]
- Baugé, C.; Girard, N.; Lhuissier, E.; Bazille, C.; Boumediene, K. Regulation and role of TGFβ signaling pathway in aging and osteoarthritis joints. Aging Dis. 2013, 5, 394–405. [Google Scholar] [CrossRef]
- van der Kraan, P.M.; van den Berg, W.B. Chondrocyte hypertrophy and osteoarthritis: Role in initiation and progression of cartilage degeneration? Osteoarthr. Cartil. 2012, 20, 223–232. [Google Scholar] [CrossRef]
- Baugé, C.; Duval, E.; Ollitrault, D.; Girard, N.; Leclercq, S.; Galéra, P.; Boumédiene, K. Type II TGFβ receptor modulates chondrocyte phenotype. Age 2013, 35, 1105–1116. [Google Scholar] [CrossRef]
- Loeser, R.F.; Gandhi, U.; Long, D.L.; Yin, W.; Chubinskaya, S. Aging and oxidative stress reduce the response of human articular chondrocytes to insulin-like growth factor 1 and osteogenic protein 1. Arthritis Rheumatol. 2014, 66, 2201–2209. [Google Scholar] [CrossRef]
- Vijg, J.; Suh, Y. Genome instability and aging. Annu. Rev. Physiol. 2013, 75, 645–668. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Gurkar, A.U.; Wang, Y.; Vijg, J.; Hoeijmakers, J.H.J.; Robbins, P.D. Nuclear Genomic Instability and Aging. Annu. Rev. Biochem. 2018, 87, 295–322. [Google Scholar] [CrossRef]
- Fakouri, N.B.; Hou, Y.; Demarest, T.G.; Christiansen, L.S.; Okur, M.N.; Mohanty, J.G.; Croteau, D.L.; Bohr, V.A. Toward understanding genomic instability, mitochondrial dysfunction and aging. FEBS J. 2019, 286, 1058–1073. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Wilson, D.M., 3rd. DNA Damage and Associated DNA Repair Defects in Disease and Premature Aging. Am. J. Hum. Genet. 2019, 105, 237–257. [Google Scholar] [CrossRef] [PubMed]
- Neri, S.; Guidotti, S.; Bini, C.; Pelotti, S.; D’Adamo, S.; Minguzzi, M.; Platano, D.; Santi, S.; Mariani, E.; Cattini, L.; et al. Oxidative stress-induced DNA damage and repair in primary human osteoarthritis chondrocytes: Focus on IKKα and the DNA Mismatch Repair System. Free Radic. Biol. Med. 2021, 166, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Yudoh, K.; Nguyen v, T.; Nakamura, H.; Hongo-Masuko, K.; Kato, T.; Nishioka, K. Potential involvement of oxidative stress in cartilage senescence and development of osteoarthritis: Oxidative stress induces chondrocyte telomere instability and downregulation of chondrocyte function. Arthritis Res. Ther. 2005, 7, R380–R391. [Google Scholar] [CrossRef]
- Teerawattanapong, N.; Udomsinprasert, W.; Ngarmukos, S.; Tanavalee, A.; Honsawek, S. Blood leukocyte LINE-1 hypomethylation and oxidative stress in knee osteoarthritis. Heliyon 2019, 5, e01774. [Google Scholar] [CrossRef]
- Jeong, S.; Lee, K.; Wen, X.; Kim, Y.; Cho, N.Y.; Jang, J.J.; Kang, G.H. Tumoral LINE-1 hypomethylation is associated with poor survival of patients with intrahepatic cholangiocarcinoma. BMC Cancer 2017, 17, 588. [Google Scholar] [CrossRef]
- Karouzakis, E.; Gay, R.E.; Gay, S.; Neidhart, M. Increased recycling of polyamines is associated with global DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2012, 64, 1809–1817. [Google Scholar] [CrossRef]
- Neri, S.; Guidotti, S.; Lilli, N.L.; Cattini, L.; Mariani, E. Infrapatellar fat pad-derived mesenchymal stromal cells from osteoarthritis patients: In vitro genetic stability and replicative senescence. J. Orthop. Res. 2017, 35, 1029–1037. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Caglar, H.O.; Biray Avci, C. Alterations of cell cycle genes in cancer: Unmasking the role of cancer stem cells. Mol. Biol. Rep. 2020, 47, 3065–3076. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol. 2017, 27, 2652–2660. [Google Scholar] [CrossRef] [PubMed]
- Petrova, N.V.; Velichko, A.K.; Razin, S.V.; Kantidze, O.L. Small molecule compounds that induce cellular senescence. Aging Cell 2016, 15, 999–1017. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Ramasamy, T.S.; Yee, Y.M.; Khan, I.M. Chondrocyte ag.ging: The molecular determinants and therapeutic opportunities. Front. Cell Dev. Biol. 2021, 9, 625497. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Gao, S.G.; Zeng, C.; Li, L.J.; Luo, W.; Zhang, F.J.; Tian, J.; Cheng, C.; Tu, M.; Xiong, Y.L.; Jiang, W.; et al. Correlation between senescence-associated beta-galactosidase expression in articular cartilage and disease severity of patients with knee osteoarthritis. Int. J. Rheum. Dis. 2016, 19, 226–232. [Google Scholar] [CrossRef]
- Vinatier, C.; Domínguez, E.; Guicheux, J.; Caramés, B. Role of the inflammation-autophagy-senescence integrative network in osteoarthritis. Front. Physiol. 2018, 9, 706. [Google Scholar] [CrossRef]
- Ohno-Iwashita, Y.; Shimada, Y.; Hayashi, M.; Inomata, M. Plasma membrane microdomains in aging and disease. Geriatr. Gerontol. Int. 2010, 10 (Suppl 1), S41–S52. [Google Scholar] [CrossRef]
- Xie, J.; Wang, Y.; Lu, L.; Liu, L.; Yu, X.; Pei, F. Cellular senescence in knee osteoarthritis: Molecular mechanisms and therapeutic implications. Ageing Res. Rev. 2021, 70, 101413. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhang, X.; Rothrauff, B.B.; Fritch, M.R.; Chang, A.; He, Y.; Yeung, M.; Liu, S.; Lipa, K.E.; Lei, G.; et al. Novel role of estrogen receptor-α on regulating chondrocyte phenotype and response to mechanical loading. Osteoarthr. Cartil. 2022, 30, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Pan, J.; Li, J.; Zeng, C.; Qi, W.; Shao, Y.; Liu, X.; Liu, L.; Xiao, G.; Zhang, H.; et al. Metformin attenuates cartilage degeneration in an experimental osteoarthritis model by regulating AMPK/mTOR. Aging 2020, 12, 1087–1103. [Google Scholar] [CrossRef] [PubMed]
- Si, H.B.; Yang, T.M.; Li, L.; Tian, M.; Zhou, L.; Li, D.P.; Huang, Q.; Kang, P.D.; Yang, J.; Zhou, Z.K.; et al. miR-140 attenuates the progression of early-stage osteoarthritis by retarding chondrocyte senescence. Mol. Ther. Nucleic Acids 2020, 19, 15–30. [Google Scholar] [CrossRef]
- Chagin, A.S.; Vuppalapati, K.K.; Kobayashi, T.; Guo, J.; Hirai, T.; Chen, M.; Offermanns, S.; Weinstein, L.S.; Kronenberg, H.M. G-protein stimulatory subunit alpha and Gq/11α G-proteins are both required to maintain quiescent stem-like chondrocytes. Nat. Commun. 2014, 5, 3673. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Ferreira-Gonzalez, S.; Lu, W.Y.; Raven, A.; Dwyer, B.; Man, T.Y.; O’Duibhir, E.; Lewis, P.J.S.; Campana, L.; Kendall, T.J.; Bird, T.G.; et al. Paracrine cellular senescence exacerbates biliary injury and impairs regeneration. Nat. Commun. 2018, 9, 1020. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367. [Google Scholar] [CrossRef]
- Asghar, S.; Litherland, G.J.; Lockhart, J.C.; Goodyear, C.S.; Crilly, A. Exosomes in intercellular communication and implications for osteoarthritis. Rheumatology 2020, 59, 57–68. [Google Scholar] [CrossRef]
- Zheng, L.; Wang, Y.; Qiu, P.; Xia, C.; Fang, Y.; Mei, S.; Fang, C.; Shi, Y.; Wu, K.; Chen, Z.; et al. Primary chondrocyte exosomes mediate osteoarthritis progression by regulating mitochondrion and immune reactivity. Nanomedicine 2019, 14, 3193–3212. [Google Scholar] [CrossRef]
- Ni, Z.; Kuang, L.; Chen, H.; Xie, Y.; Zhang, B.; Ouyang, J.; Wu, J.; Zhou, S.; Chen, L.; Su, N.; et al. The exosome-like vesicles from osteoarthritic chondrocyte enhanced mature IL-1β production of macrophages and aggravated synovitis in osteoarthritis. Cell Death Dis. 2019, 10, 522. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lin, L.; Zou, R.; Wen, C.; Wang, Z.; Lin, F. MSC-derived exosomes promote proliferation and inhibit apoptosis of chondrocytes via lncRNA-KLF3-AS1/miR-206/GIT1 axis in osteoarthritis. Cell Cycle 2018, 17, 2411–2422. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, J.A.; Collins, J.A.; Loeser, R.F. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic. Biol. Med. 2019, 132, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Qi, W.; Zhan, J.; Lin, Z.; Lin, J.; Xue, X.; Pan, X.; . Zhou, Y. Activating Nrf2 signalling alleviates osteoarthritis development by inhibiting inflammasome activation. J. Cell Mol. Med. 2020, 24, 13046–13057. [Google Scholar] [CrossRef]
- He, Y.; Gao, M.; Cao, Y.; Tang, H.; Liu, S.; Tao, Y. Nuclear localization of metabolic enzymes in immunity and metastasis. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 359–371. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef]
- Guan, Y.; Wang, S.R.; Huang, X.Z.; Xie, Q.H.; Xu, Y.Y.; Shang, D.; Hao, C.M. Nicotinamide Mononucleotide, an NAD(+) Precursor, Rescues Age-Associated Susceptibility to AKI in a Sirtuin 1-Dependent Manner. J. Am. Soc. Nephrol. 2017, 28, 2337–2352. [Google Scholar] [CrossRef]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD(+) supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef]
- Trammell, S.A.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef]
- Dellinger, R.W.; Santos, S.R.; Morris, M.; Evans, M.; Alminana, D.; Guarente, L.; Marcotulli, E. Repeat dose NRPT (nicotinamide riboside and pterostilbene) increases NAD(+) levels in humans safely and sustainably: A randomized, double-blind, placebo-controlled study. NPJ Aging Mech. Dis. 2017, 3, 17. [Google Scholar] [CrossRef]
- Goiran, T.; Duplan, E.; Rouland, L.; El Manaa, W.; Lauritzen, I.; Dunys, J.; You, H.; Checler, F.; Alves da Costa, C. Nuclear p53-mediated repression of autophagy involves PINK1 transcriptional down-regulation. Cell Death Differ. 2018, 25, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Zhao, F.; Wu, R.; Qin, S.; Nowsheen, S.; Huang, J.; Zhou, Q.; Chen, Y.; Deng, M.; Guo, G.; et al. ZFP161 regulates replication fork stability and maintenance of genomic stability by recruiting the ATR/ATRIP complex. Nat. Commun. 2019, 10, 5304. [Google Scholar] [CrossRef]
- Khan, N.M.; Ahmad, I.; Ansari, M.Y.; Haqqi, T.M. Wogonin, a natural flavonoid, intercalates with genomic DNA and exhibits protective effects in IL-1β stimulated osteoarthritis chondrocytes. Chem. Biol. Interact. 2017, 274, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Jäger, K.; Walter, M. Therapeutic Targeting of Telomerase. Genes 2016, 7, 39. [Google Scholar] [CrossRef]
- Mendelsohn, A.R.; Larrick, J.W. Ectopic expression of telomerase safely increases health span and life span. Rejuvenation Res. 2012, 15, 435–438. [Google Scholar] [CrossRef]
- Harley, C.B.; Liu, W.; Blasco, M.; Vera, E.; Andrews, W.H.; Briggs, L.A.; Raffaele, J.M. A natural product telomerase activator as part of a health maintenance program. Rejuvenation Res. 2011, 14, 45–56. [Google Scholar] [CrossRef]
- Bernardes de Jesus, B.; Schneeberger, K.; Vera, E.; Tejera, A.; Harley, C.B.; Blasco, M.A. The telomerase activator TA-65 elongates short telomeres and increases health span of adult/old mice without increasing cancer incidence. Aging Cell 2011, 10, 604–621. [Google Scholar] [CrossRef]
- Dock, J.N.; Effros, R.B. Role of CD8 T cell replicative senescence in human aging and in HIV-mediated immunosenescence. Aging Dis. 2011, 2, 382–397. [Google Scholar]
- Dow, C.T.; Harley, C.B. Evaluation of an oral telomerase activator for early age-related macular degeneration-a pilot study. Clin. Ophthalmol. 2016, 10, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Sprouse, A.A.; Steding, C.E.; Herbert, B.S. Pharmaceutical regulation of telomerase and its clinical potential. J. Cell Mol. Med. 2012, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Eitan, E.; Tichon, A.; Gazit, A.; Gitler, D.; Slavin, S.; Priel, E. Novel telomerase-increasing compound in mouse brain delays the onset of amyotrophic lateral sclerosis. EMBO Mol. Med. 2012, 4, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Haendeler, J.; Hoffmann, J.; Diehl, J.F.; Vasa, M.; Spyridopoulos, I.; Zeiher, A.M.; Dimmeler, S. Antioxidants inhibit nuclear export of telomerase reverse transcriptase and delay replicative senescence of endothelial cells. Circ. Res. 2004, 94, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Moritoh, Y.; Miwa, N. Age-dependent telomere-shortening is repressed by phosphorylated alpha-tocopherol together with cellular longevity and intracellular oxidative-stress reduction in human brain microvascular endotheliocytes. J. Cell Biochem. 2007, 102, 689–703. [Google Scholar] [CrossRef]
- Dong, X.X.; Hui, Z.J.; Xiang, W.X.; Rong, Z.F.; Jian, S.; Zhu, C.J. Ginkgo biloba extract reduces endothelial progenitor-cell senescence through augmentation of telomerase activity. J. Cardiovasc. Pharmacol. 2007, 49, 111–115. [Google Scholar] [CrossRef]
- Shim, J.H.; Lee, T.R.; Shin, D.W. Novel in vitro culture condition improves the stemness of human dermal stem/progenitor cells. Mol. Cells 2013, 36, 556–563. [Google Scholar] [CrossRef]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef]
- Harari, Y.; Zadok-Laviel, S.; Kupiec, M. Long Telomeres Do Not Affect Cellular Fitness in Yeast. MBio 2017, 8, e01314-17. [Google Scholar] [CrossRef]
- Brunet, A.; Berger, S.L. Epigenetics of aging and aging-related disease. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl 1), S17–S20. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech. Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Lee, J.H.; Lee, H.Y.; Min, K.J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, C.; Vassilopoulos, A. Sirtuins at the crossroads of stemness, aging, and cancer. Aging Cell 2017, 16, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Sae-Lee, C.; Corsi, S.; Barrow, T.M.; Kuhnle, G.G.C.; Bollati, V.; Mathers, J.C.; Byun, H.M. Dietary Intervention Modifies DNA Methylation Age Assessed by the Epigenetic Clock. Mol. Nutr. Food Res. 2018, 62, e1800092. [Google Scholar] [CrossRef]
- Rhoads, T.W.; Burhans, M.S.; Chen, V.B.; Hutchins, P.D.; Rush, M.J.P.; Clark, J.P.; Stark, J.L.; McIlwain, S.J.; Eghbalnia, H.R.; Pavelec, D.M.; et al. Caloric Restriction Engages Hepatic RNA Processing Mechanisms in Rhesus Monkeys. Cell Metab. 2018, 27, 677–688.e675. [Google Scholar] [CrossRef]
- Hahn, O.; Grönke, S.; Stubbs, T.M.; Ficz, G.; Hendrich, O.; Krueger, F.; Andrews, S.; Zhang, Q.; Wakelam, M.J.; Beyer, A.; et al. Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biol. 2017, 18, 56. [Google Scholar] [CrossRef]
- Madry, H.; Cucchiarini, M. Gene therapy for human osteoarthritis: Principles and clinical translation. Expert Opin. Biol. Ther. 2016, 16, 331–346. [Google Scholar] [CrossRef]
- Arden, N.K.; Perry, T.A.; Bannuru, R.R.; Bruyère, O.; Cooper, C.; Haugen, I.K.; Hochberg, M.C.; McAlindon, T.E.; Mobasheri, A.; Reginster, J.Y. Non-surgical management of knee osteoarthritis: Comparison of ESCEO and OARSI 2019 guidelines. Nat. Rev. Rheumatol. 2021, 17, 59–66. [Google Scholar] [CrossRef]
- Radtke, S.; Kiem, H.P. Bringing gene therapy to where it’s needed. Trends Mol. Med. 2022, 28, 171–172. [Google Scholar] [CrossRef]
- Tao, K.; Rey-Rico, A.; Frisch, J.; Venkatesan, J.K.; Schmitt, G.; Madry, H.; Lin, J.; Cucchiarini, M. rAAV-mediated combined gene transfer and overexpression of TGF-β and SOX9 remodels human osteoarthritic articular cartilage. J. Orthop. Res. 2016, 34, 2181–2190. [Google Scholar] [CrossRef]
- Maihöfer, J.; Madry, H.; Rey-Rico, A.; Venkatesan, J.K.; Goebel, L.; Schmitt, G.; Speicher-Mentges, S.; Cai, X.; Meng, W.; Zurakowski, D.; et al. Hydrogel-guided, rAAV-mediated IGF-I overexpression enables long-term cartilage repair and protection against perifocal osteoarthritis in a large-animal full-thickness chondral defect model at one year in vivo. Adv. Mater. 2021, 33, e2008451. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Madry, H.; Venkatesan, J.K.; Schmitt, G.; Speicher-Mentges, S.; Zurakowski, D.; Menger, M.D.; Laschke, M.W.; Cucchiarini, M. rAAV-mediated sox9 overexpression improves the repair of osteochondral defects in a clinically relevant large animal model over time in vivo and reduces perifocal osteoarthritic changes. Am. J. Sports Med. 2021, 49, 3696–3707. [Google Scholar] [CrossRef] [PubMed]
- Urich, J.; Cucchiarini, M.; Rey-Rico, A. Therapeutic delivery of rAAV sox9 via polymeric micelles counteracts the effects of osteoarthritis-associated inflammatory cytokines in human articular chondrocytes. Nanomaterials 2020, 10, 1238. [Google Scholar] [CrossRef] [PubMed]
- Rey-Rico, A.; Venkatesan, J.K.; Schmitt, G.; Concheiro, A.; Madry, H.; Alvarez-Lorenzo, C.; Cucchiarini, M. rAAV-mediated overexpression of TGF-β via vector delivery in polymeric micelles stimulates the biological and reparative activities of human articular chondrocytes in vitro and in a human osteochondral defect model. Int. J. Nanomed. 2017, 12, 6985–6996. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Shen, J.; Hui, T. Epigenetic and microRNA regulation during osteoarthritis development. F1000Research 2015, 4. [Google Scholar] [CrossRef]
- Song, J.; Jin, E.H.; Kim, D.; Kim, K.Y.; Chun, C.H.; Jin, E.J. MicroRNA-222 regulates MMP-13 via targeting HDAC-4 during osteoarthritis pathogenesis. BBA Clin. 2015, 3, 79–89. [Google Scholar] [CrossRef]
- Bai, Y.; Chen, K.; Zhan, J.; Wu, M. miR-122/SIRT1 axis regulates chondrocyte extracellular matrix degradation in osteoarthritis. Biosci. Rep. 2020, 40, BSR20191908. [Google Scholar] [CrossRef]
- Wang, J.H.; Shih, K.S.; Wu, Y.W.; Wang, A.W.; Yang, C.R. Histone deacetylase inhibitors increase microRNA-146a expression and enhance negative regulation of interleukin-1β signaling in osteoarthritis fibroblast-like synoviocytes. Osteoarthr. Cartil. 2013, 21, 1987–1996. [Google Scholar] [CrossRef]
- Ben-Meir, A.; Burstein, E.; Borrego-Alvarez, A.; Chong, J.; Wong, E.; Yavorska, T.; Naranian, T.; Chi, M.; Wang, Y.; Bentov, Y.; et al. Coenzyme Q10 restores oocyte mitochondrial function and fertility during reproductive aging. Aging Cell 2015, 14, 887–895. [Google Scholar] [CrossRef]
- Foote, K.; Reinhold, J.; Yu, E.P.K.; Figg, N.L.; Finigan, A.; Murphy, M.P.; Bennett, M.R. Restoring mitochondrial DNA copy number preserves mitochondrial function and delays vascular aging in mice. Aging Cell 2018, 17, e12773. [Google Scholar] [CrossRef]
- Wang, J.; Li, S.; Wang, J.; Wu, F.; Chen, Y.; Zhang, H.; Guo, Y.; Lin, Y.; Li, L.; Yu, X.; et al. Spermidine alleviates cardiac aging by improving mitochondrial biogenesis and function. Aging 2020, 12, 650–671. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, M.; Li, L.; Xu, S.; Huang, D.; Ju, M.; Huang, J.; Chen, K.; Gu, H. Trehalose, sucrose and raffinose are novel activators of autophagy in human keratinocytes through an mTOR-independent pathway. Sci. Rep. 2016, 6, 28423. [Google Scholar] [CrossRef]
- Wang, Q.; Ren, J. mTOR-independent autophagy inducer trehalose rescues against insulin resistance-induced myocardial contractile anomalies: Role of p38 MAPK and Foxo1. Pharmacol. Res. 2016, 111, 357–373. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Zheng, G.; Feng, Z.; Chen, Y.; Lou, Y.; Wang, C.; Zhang, X.; Zhang, Y.; Xu, H.; Shang, P.; et al. Trehalose ameliorates oxidative stress-mediated mitochondrial dysfunction and ER stress via selective autophagy stimulation and autophagic flux restoration in osteoarthritis development. Cell Death Dis. 2017, 8, e3081. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wu, Z.; Xu, L.; Xu, K.; Chen, Z.; Ran, J.; Wu, L. The role of SIRT3-mediated mitochondrial homeostasis in osteoarthritis. Cell Mol. Life Sci. 2020, 77, 3729–3743. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.; Wang, Y.; Terkeltaub, R.; Liu-Bryan, R. Activation of AMPK-SIRT3 signaling is chondroprotective by preserving mitochondrial DNA integrity and function. Osteoarthr. Cartil. 2018, 26, 1539–1550. [Google Scholar] [CrossRef]
- Castro, C.M.; Corciulo, C.; Solesio, M.E.; Liang, F.; Pavlov, E.V.; Cronstein, B.N. Adenosine A2A receptor (A2AR) stimulation enhances mitochondrial metabolism and mitigates reactive oxygen species-mediated mitochondrial injury. FASEB J. 2020, 34, 5027–5045. [Google Scholar] [CrossRef]
- Hu, S.; Zhang, C.; Ni, L.; Huang, C.; Chen, D.; Shi, K.; Jin, H.; Zhang, K.; Li, Y.; Xie, L.; et al. Stabilization of HIF-1α alleviates osteoarthritis via enhancing mitophagy. Cell Death Dis. 2020, 11, 481. [Google Scholar] [CrossRef]
- Ansari, M.Y.; Ahmad, N.; Voleti, S.; Wase, S.J.; Novak, K.; Haqqi, T.M. Mitochondrial dysfunction triggers a catabolic response in chondrocytes via ROS-mediated activation of the JNK/AP1 pathway. J. Cell Sci. 2020, 133, jcs247353. [Google Scholar] [CrossRef]
- Basisty, N.; Meyer, J.G.; Schilling, B. Protein turnover in aging and longevity. Proteomics 2018, 18, e1700108. [Google Scholar] [CrossRef] [PubMed]
- Puglielli, L. Aging of the brain, neurotrophin signaling, and Alzheimer’s disease: Is IGF1-R the common culprit? Neurobiol. Aging 2008, 29, 795–811. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, A.S.; Alexandrov, A.I.; Makarova, N.E.; Gladyshev, V.N.; Dmitriev, S.E. Protein synthesis and quality control in aging. Aging 2018, 10, 4269–4288. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Brielmann, R.M.; Neto, M.F.; Lin, Y.F.; Haynes, C.M.; Morimoto, R.I. Mitochondrial stress restores the heat shock response and prevents proteostasis collapse during aging. Cell Rep. 2017, 21, 1481–1494. [Google Scholar] [CrossRef]
- Pellegrino, M.W.; Haynes, C.M. Mitophagy and the mitochondrial unfolded protein response in neurodegeneration and bacterial infection. BMC Biol. 2015, 13, 22. [Google Scholar] [CrossRef]
- Moehle, E.A.; Shen, K.; Dillin, A. Mitochondrial proteostasis in the context of cellular and organismal health and aging. J. Biol. Chem. 2019, 294, 5396–5407. [Google Scholar] [CrossRef]
- Kumsta, C.; Chang, J.T.; Lee, R.; Tan, E.P.; Yang, Y.; Loureiro, R.; Choy, E.H.; Lim, S.H.Y.; Saez, I.; Springhorn, A.; et al. The autophagy receptor p62/SQST-1 promotes proteostasis and longevity in C. elegans by inducing autophagy. Nat. Commun. 2019, 10, 5648. [Google Scholar] [CrossRef]
- Merry, B.J. Calorie restriction and age-related ox.xidative stress. Ann. N. Y. Acad. Sci. 2000, 908, 180–198. [Google Scholar] [CrossRef]
- Radakovich, L.B.; Marolf, A.J.; Culver, L.A.; Santangelo, K.S. Calorie restriction with regular chow, but not a high-fat diet, delays onset of spontaneous osteoarthritis in the Hartley guinea pig model. Arthritis Res. Ther. 2019, 21, 145. [Google Scholar] [CrossRef]
- Gabandé-Rodríguez, E.; Gómez de Las Heras, M.M.; Mittelbrunn, M. Control of Inflammation by Calorie Restriction Mimetics: On the Crossroad of Autophagy and Mitochondria. Cells 2019, 9, 82. [Google Scholar] [CrossRef]
- Hwangbo, D.S.; Lee, H.Y.; Abozaid, L.S.; Min, K.J. Mechanisms of lifespan regulation by calorie restriction and intermittent fasting in model organisms. Nutrients 2020, 12, 1194. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Mattson, M.P. Fasting: Molecular mechanisms and clinical applications. Cell Metab. 2014, 19, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriou, D.; Benetou, V.; Trichopoulou, A.; La Vecchia, C.; Bamia, C. Mediterranean diet and its components in relation to all-cause mortality: Meta-analysis. Br. J. Nutr. 2018, 120, 1081–1097. [Google Scholar] [CrossRef]
- Aiello, A.; Accardi, G.; Candore, G.; Gambino, C.M.; Mirisola, M.; Taormina, G.; Virruso, C.; Caruso, C. Nutrient sensing pathways as therapeutic targets for healthy ageing. Expert Opin. Ther. Targets 2017, 21, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Ros, M.; Carrascosa, J.M. Current nutritional and pharmacological anti-aging interventions. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165612. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a tool to target aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Snell, T.W.; Johnston, R.K.; Srinivasan, B.; Zhou, H.; Gao, M.; Skolnick, J. Repurposing FDA-approved drugs for anti-aging therapies. Biogerontology 2016, 17, 907–920. [Google Scholar] [CrossRef]
- Bettedi, L.; Foukas, L.C. Growth factor, energy and nutrient sensing signalling pathways in metabolic ageing. Biogerontology 2017, 18, 913–929. [Google Scholar] [CrossRef]
- Templeman, N.M.; Murphy, C.T. Regulation of reproduction and longevity by nutrient-sensing pathways. J. Cell Biol. 2018, 217, 93–106. [Google Scholar] [CrossRef]
- Schell, J.; Scofield, R.H.; Barrett, J.R.; Kurien, B.T.; Betts, N.; Lyons, T.J.; Zhao, Y.D.; Basu, A. Strawberries improve pain and inflammation in obese adults with radiographic evidence of knee osteoarthritis. Nutrients 2017, 9, 949. [Google Scholar] [CrossRef]
- Du, C.; Smith, A.; Avalos, M.; South, S.; Crabtree, K.; Wang, W.; Kwon, Y.H.; Vijayagopal, P.; Juma, S. Blueberries improve pain, gait performance, and inflammation in individuals with symptomatic knee osteoarthritis. Nutrients 2019, 11, 290. [Google Scholar] [CrossRef] [PubMed]
- Toh, W.S.; Brittberg, M.; Farr, J.; Foldager, C.B.; Gomoll, A.H.; Hui, J.H.; Richardson, J.B.; Roberts, S.; Spector, M. Cellular senescence in aging and osteoarthritis. Acta Orthop. 2016, 87, 6–14. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 2017, 169, 132–147.e116. [Google Scholar] [CrossRef]
- Soto-Gamez, A.; Demaria, M. Therapeutic interventions for aging: The case of cellular senescence. Drug Discov. Today 2017, 22, 786–795. [Google Scholar] [CrossRef]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]
- Batshon, G.; Elayyan, J.; Qiq, O.; Reich, E.; Ben-Aderet, L.; Kandel, L.; Haze, A.; Steinmeyer, J.; Lefebvre, V.; Zhang, H.; et al. Serum NT/CT SIRT1 ratio reflects early osteoarthritis and chondrosenescence. Ann. Rheum. Dis. 2020, 79, 1370–1380. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Wang, B.; Shi, Y.; Xie, C.; Huang, C.; Chen, B.; Zhang, H.; Zeng, G.; Liang, H.; Wu, Y.; et al. Senolytic agent Quercetin ameliorates intervertebral disc degeneration via the Nrf2/NF-κB axis. Osteoarthr. Cartil. 2021, 29, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Lagoumtzi, S.M.; Chondrogianni, N. Senolytics and senomorphics: Natural and synthetic therapeutics in the treatment of aging and chronic diseases. Free Radic. Biol. Med. 2021, 171, 169–190. [Google Scholar] [CrossRef] [PubMed]
- Nacarelli, T.; Sell, C. Targeting metabolism in cellular senescence, a role for intervention. Mol. Cell Endocrinol. 2017, 455, 83–92. [Google Scholar] [CrossRef]
- Kritsilis, M.; Rizou, S.V.; Koutsoudaki, P.N.; Evangelou, K.; Gorgoulis, V.G.; Papadopoulos, D. Ageing, cellular senescence and neurodegenerative disease. Int. J. Mol. Sci. 2018, 19, 2937. [Google Scholar] [CrossRef]
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 2016, 167, 1719–1733.e1712. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Wilkinson, J.E.; Hughes, B.; Gadela, N.; Ladiges, W.C.; Vo, N.; Niedernhofer, L.J.; Huffman, D.M.; Robbins, P.D. Heterochronic parabiosis regulates the extent of cellular senescence in multiple tissues. Geroscience 2020, 42, 951–961. [Google Scholar] [CrossRef]
- Sahu, A.; Clemens, Z.J.; Shinde, S.N.; Sivakumar, S.; Pius, A.; Bhatia, A.; Picciolini, S.; Carlomagno, C.; Gualerzi, A.; Bedoni, M.; et al. Regulation of aged skeletal muscle regeneration by circulating extracellular vesicles. Nat. Aging 2021, 1, 1148–1161. [Google Scholar] [CrossRef]
- Zhao, X.; Zhao, Y.; Sun, X.; Xing, Y.; Wang, X.; Yang, Q. Immunomodulation of MSCs and MSC-Derived Extracellular Vesicles in Osteoarthritis. Front. Bioeng. Biotechnol. 2020, 8, 575057. [Google Scholar] [CrossRef]
- Mas-Bargues, C.; Sanz-Ros, J.; Román-Domínguez, A.; Gimeno-Mallench, L.; Inglés, M.; Viña, J.; Borrás, C. Extracellular vesicles from healthy cells improves cell function and stemness in premature senescent stem cells by miR-302b and HIF-1α activation. Biomolecules 2020, 10, 957. [Google Scholar] [CrossRef]
- Planat-Benard, V.; Varin, A.; Casteilla, L. MSCs and inflammatory cells crosstalk in regenerative medicine: Concerted actions for cptimized resolution driven by energy metabolism. Front. Immunol. 2021, 12, 626755. [Google Scholar] [CrossRef] [PubMed]
- Pegtel, D.M.; Gould, S.J. Exosomes. Annu. Rev. Biochem. 2019, 88, 487–514. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, L.; Ma, C.; Wang, G.; Zhang, Y.; Sun, S. Exosomes derived from platelet-rich plasma present a novel potential in alleviating knee osteoarthritis by promoting proliferation and inhibiting apoptosis of chondrocyte via Wnt/β-catenin signaling pathway. J. Orthop. Surg. Res. 2019, 14, 470. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Zhou, S.; Li, S.; Kuang, L.; Chen, H.; Luo, X.; Ouyang, J.; He, M.; Du, X.; Chen, L. Exosomes: Roles and therapeutic potential in osteoarthritis. Bone Res. 2020, 8, 25. [Google Scholar] [CrossRef]
- Fan, Y.; Li, Z.; He, Y. Exosomes in the pathogenesis, progression, and treatment of osteoarthritis. Bioengineering 2022, 9, 99. [Google Scholar] [CrossRef]
- Millerand, M.; Berenbaum, F.; Jacques, C. Danger signals and inflammaging in osteoarthritis. Clin. Exp. Rheumatol. 2019, 37 (Suppl 120), 48–56. [Google Scholar]
- Rezuș, E.; Cardoneanu, A.; Burlui, A.; Luca, A.; Codreanu, C.; Tamba, B.I.; Stanciu, G.-D.; Dima, N.; Bădescu, C.; Rezuș, C. The link between inflammaging and degenerative joint diseases. Int. J. Mol. Sci. 2019, 20, 614. [Google Scholar] [CrossRef]
- Gago-Fuentes, R.; Carpintero-Fernandez, P.; Goldring, M.B.; Brink, P.R.; Mayan, M.D.; Blanco, F.J. Biochemical evidence for gap junctions and Cx43 expression in immortalized human chondrocyte cell line: A potential model in the study of cell communication in human chondrocytes. Osteoarthr. Cartil. 2014, 22, 586–590. [Google Scholar] [CrossRef]
- Mayan, M.D.; Gago-Fuentes, R.; Carpintero-Fernandez, P.; Fernandez-Puente, P.; Filgueira-Fernandez, P.; Goyanes, N.; Valiunas, V.; Brink, P.R.; Goldberg, G.S.; Blanco, F.J. Articular chondrocyte network mediated by gap junctions: Role in metabolic cartilage homeostasis. Ann. Rheum. Dis. 2015, 74, 275–284. [Google Scholar] [CrossRef]
- Carpintero-Fernandez, P.; Gago-Fuentes, R.; Wang, H.Z.; Fonseca, E.; Caeiro, J.R.; Valiunas, V.; Brink, P.R.; Mayan, M.D. Intercellular communication via gap junction channels between chondrocytes and bone cells. Biochim. Biophys. Acta Biomembr. 2018, 1860, 2499–2505. [Google Scholar] [CrossRef]
- Marino, A.A.; Waddell, D.D.; Kolomytkin, O.V.; Meek, W.D.; Wolf, R.; Sadasivan, K.K.; Albright, J.A. Increased intercellular communication through gap junctions may contribute to progression of osteoarthritis. Clin. Orthop. Relat. Res. 2004, 422, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Niger, C.; Buo, A.M.; Eidelman, E.R.; Chen, R.J.; Stains, J.P. Connexin43 enhances the expression of osteoarthritis-associated genes in synovial fibroblasts in culture. BMC Musculoskelet. Disord. 2014, 15, 425. [Google Scholar] [CrossRef] [PubMed]
- Mayan, M.D.; Carpintero-Fernandez, P.; Gago-Fuentes, R.; Martinez-de-Ilarduya, O.; Wang, H.Z.; Valiunas, V.; Brink, P.; Blanco, F.J. Human articular chondrocytes express multiple gap junction proteins: Differential expression of connexins in normal and osteoarthritic cartilage. Am. J. Pathol. 2013, 182, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Varela-Eirín, M.; Varela-Vázquez, A.; Guitián-Caamaño, A.; Paíno, C.L.; Mato, V.; Largo, R.; Aasen, T.; Tabernero, A.; Fonseca, E.; Kandouz, M.; et al. Targeting of chondrocyte plasticity via connexin43 modulation attenuates cellular senescence and fosters a pro-regenerative environment in osteoarthritis. Cell Death Dis. 2018, 9, 1166. [Google Scholar] [CrossRef] [PubMed]
- Gago-Fuentes, R.; Fernández-Puente, P.; Megias, D.; Carpintero-Fernández, P.; Mateos, J.; Acea, B.; Fonseca, E.; Blanco, F.J.; Mayan, M.D. Proteomic analysis of connexin 43 reveals novel interactors related to osteoarthritis. Mol. Cell Proteom. 2015, 14, 1831–1845. [Google Scholar] [CrossRef]
- Jallali, N.; Ridha, H.; Thrasivoulou, C.; Underwood, C.; Butler, P.E.; Cowen, T. Vulnerability to ROS-induced cell death in ageing articular cartilage: The role of antioxidant enzyme activity. Osteoarthr. Cartil. 2005, 13, 614–622. [Google Scholar] [CrossRef]
- Dozin, B.; Malpeli, M.; Camardella, L.; Cancedda, R.; Pietrangelo, A. Response of young, aged and osteoarthritic human articular chondrocytes to inflammatory cytokines: Molecular and cellular aspects. Matrix Biol. 2002, 21, 449–459. [Google Scholar] [CrossRef]
- Mobasheri, A.; Matta, C.; Zákány, R.; Musumeci, G. Chondrosenescence: Definition, hallmarks and potential role in the pathogenesis of osteoarthritis. Maturitas 2015, 80, 237–244. [Google Scholar] [CrossRef]
- Huang, Y.; He, Y.; Makarcyzk, M.J.; Lin, H. Senolytic peptide FOXO4-DRI selectively removes senescent cells from in vitro expanded human chondrocytes. Front. Bioeng. Biotechnol. 2021, 9, 677576. [Google Scholar] [CrossRef]
- Rim, Y.A.; Nam, Y.; Ju, J.H. The role of chondrocyte hypertrophy and senescence in osteoarthritis initiation and progression. Int. J. Mol. Sci. 2020, 21, 2358. [Google Scholar] [CrossRef]
- Loeser, R.F. Aging and osteoarthritis. Curr. Opin. Rheumatol. 2011, 23, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.S.; Xiao, W.F.; Luo, W. Cellular aging towards osteoarthritis. Mech. Ageing Dev. 2017, 162, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.A.; Diekman, B.O.; Loeser, R.F. Targeting aging for disease modification in osteoarthritis. Curr. Opin. Rheumatol. 2018, 30, 101–107. [Google Scholar] [CrossRef]
- Goh, S.L.; Persson, M.S.M.; Stocks, J.; Hou, Y.; Lin, J.; Hall, M.C.; Doherty, M.; Zhang, W. Efficacy and potential determinants of exercise therapy in knee and hip osteoarthritis: A systematic review and meta-analysis. Ann. Phys. Rehabil. Med. 2019, 62, 356–365. [Google Scholar] [CrossRef]
- Kan, H.S.; Chan, P.K.; Chiu, K.Y.; Yan, C.H.; Yeung, S.S.; Ng, Y.L.; Shiu, K.W.; Ho, T. Non-surgical treatment of knee osteoarthritis. Hong Kong Med. J. 2019, 25, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; He, Y.; Liu, S.; Makarcyzk, M.J.; Lei, G.; Chang, A.; Alexander, P.G.; Hao, T.; Padget, A.M.; de Pedro, N.; et al. Engineering osteoarthritic cartilage model through differentiating senescent human mesenchymal stem cells for testing disease-modifying drugs. Sci. China Life Sci. 2022, 65, 309–327. [Google Scholar] [CrossRef]
- Athanasopoulos, T.; Munye, M.M.; Yáñez-Muñoz, R.J. Nonintegrating gene therapy vectors. Hematol. Oncol. Clin. N. Am. 2017, 31, 753–770. [Google Scholar] [CrossRef]
- Senís, E.; Mosteiro, L.; Wilkening, S.; Wiedtke, E.; Nowrouzi, A.; Afzal, S.; Fronza, R.; Landerer, H.; Abad, M.; Niopek, D.; et al. AAVvector-mediated in vivo reprogramming into pluripotency. Nat. Commun. 2018, 9, 2651. [Google Scholar] [CrossRef]
- Alle, Q.; Le Borgne, E.; Milhavet, O.; Lemaitre, J.M. Reprogramming: Emerging strategies to rejuvenate aging cells and tissues. Int. J. Mol. Sci. 2021, 22, 3990. [Google Scholar] [CrossRef]
- Lin, Z.; Li, Z.; Li, E.N.; Li, X.; Del Duke, C.J.; Shen, H.; Hao, T.; O’Donnell, B.; Bunnell, B.A.; Goodman, S.B.; et al. Osteochondral tissue chip derived from iPSCs: Modeling OA pathologies and testing drugs. Front. Bioeng. Biotechnol. 2019, 7, 411. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Lipa, K.E.; Alexander, P.G.; Clark, K.L.; Lin, H. Potential Methods of Targeting Cellular Aging Hallmarks to Reverse Osteoarthritic Phenotype of Chondrocytes. Biology 2022, 11, 996. https://doi.org/10.3390/biology11070996
He Y, Lipa KE, Alexander PG, Clark KL, Lin H. Potential Methods of Targeting Cellular Aging Hallmarks to Reverse Osteoarthritic Phenotype of Chondrocytes. Biology. 2022; 11(7):996. https://doi.org/10.3390/biology11070996
Chicago/Turabian StyleHe, Yuchen, Katelyn E. Lipa, Peter G. Alexander, Karen L. Clark, and Hang Lin. 2022. "Potential Methods of Targeting Cellular Aging Hallmarks to Reverse Osteoarthritic Phenotype of Chondrocytes" Biology 11, no. 7: 996. https://doi.org/10.3390/biology11070996
APA StyleHe, Y., Lipa, K. E., Alexander, P. G., Clark, K. L., & Lin, H. (2022). Potential Methods of Targeting Cellular Aging Hallmarks to Reverse Osteoarthritic Phenotype of Chondrocytes. Biology, 11(7), 996. https://doi.org/10.3390/biology11070996