
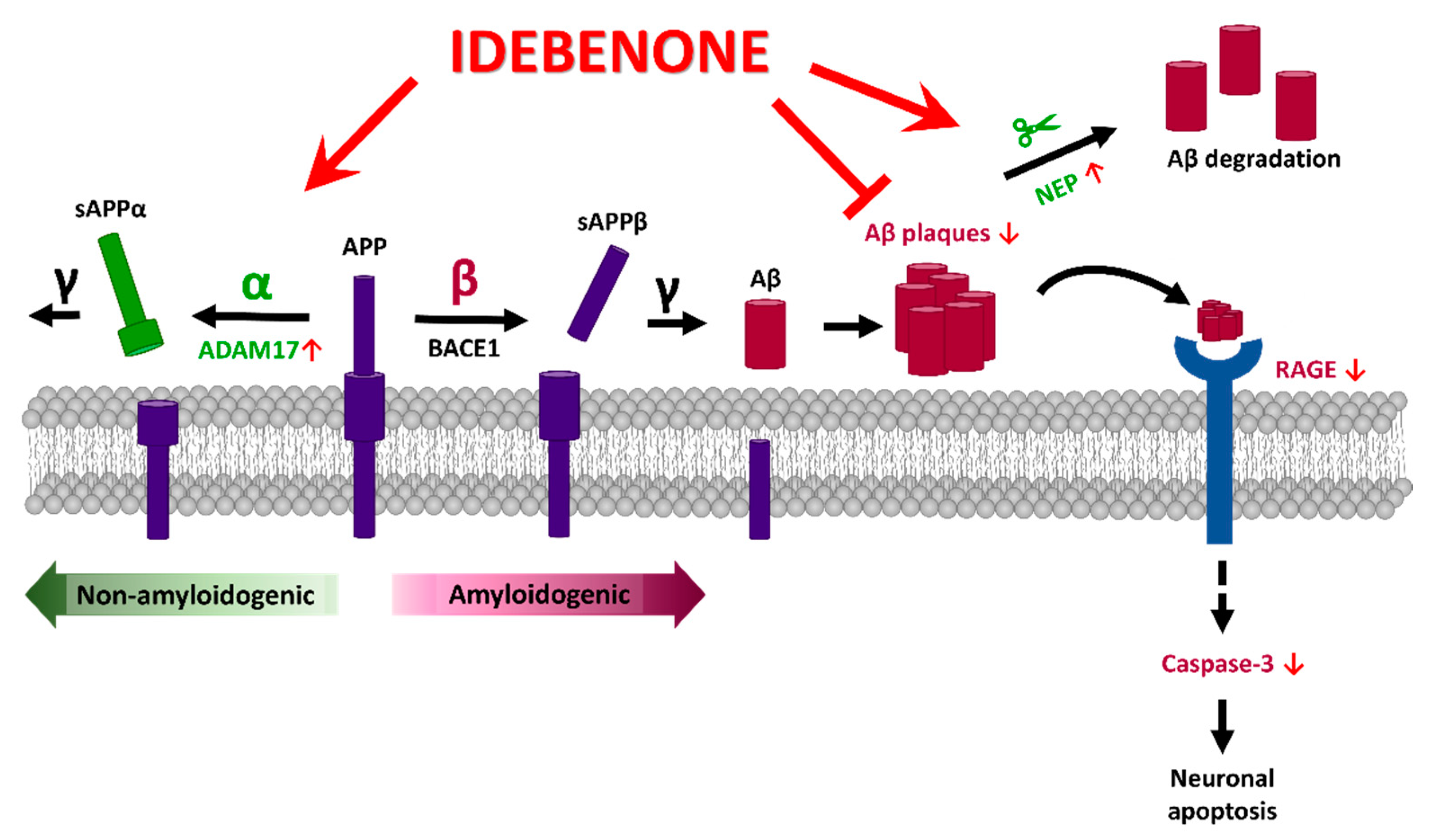
Idebenone Decreases Aβ Pathology by Modulating RAGE/Caspase-3 Signaling and the Aβ Degradation Enzyme NEP in a Mouse Model of AD

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. 5xFAD Mice
2.2. Idebenone Injection
2.3. Immunofluorescent Staining
2.4. Statistics
3. Results
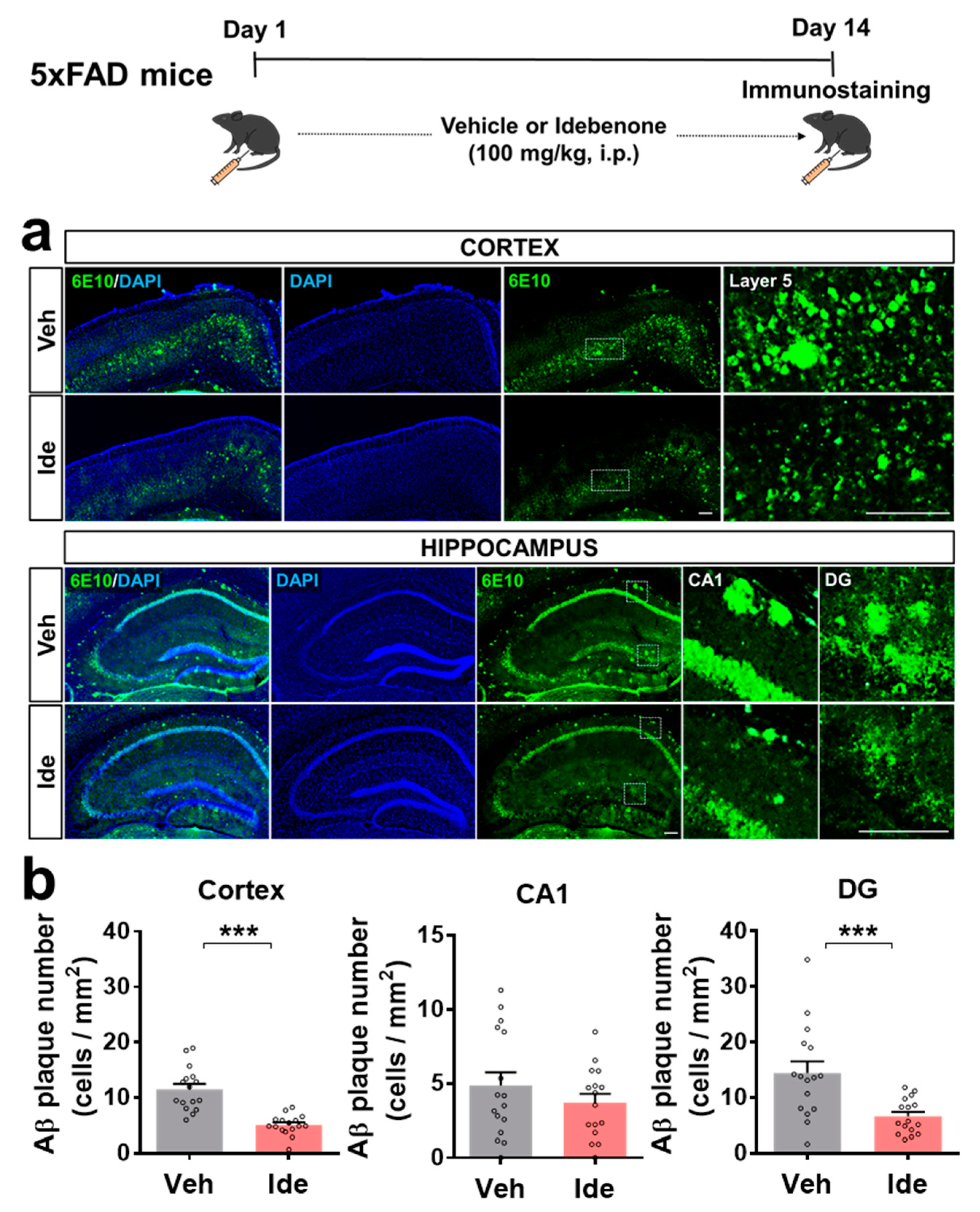
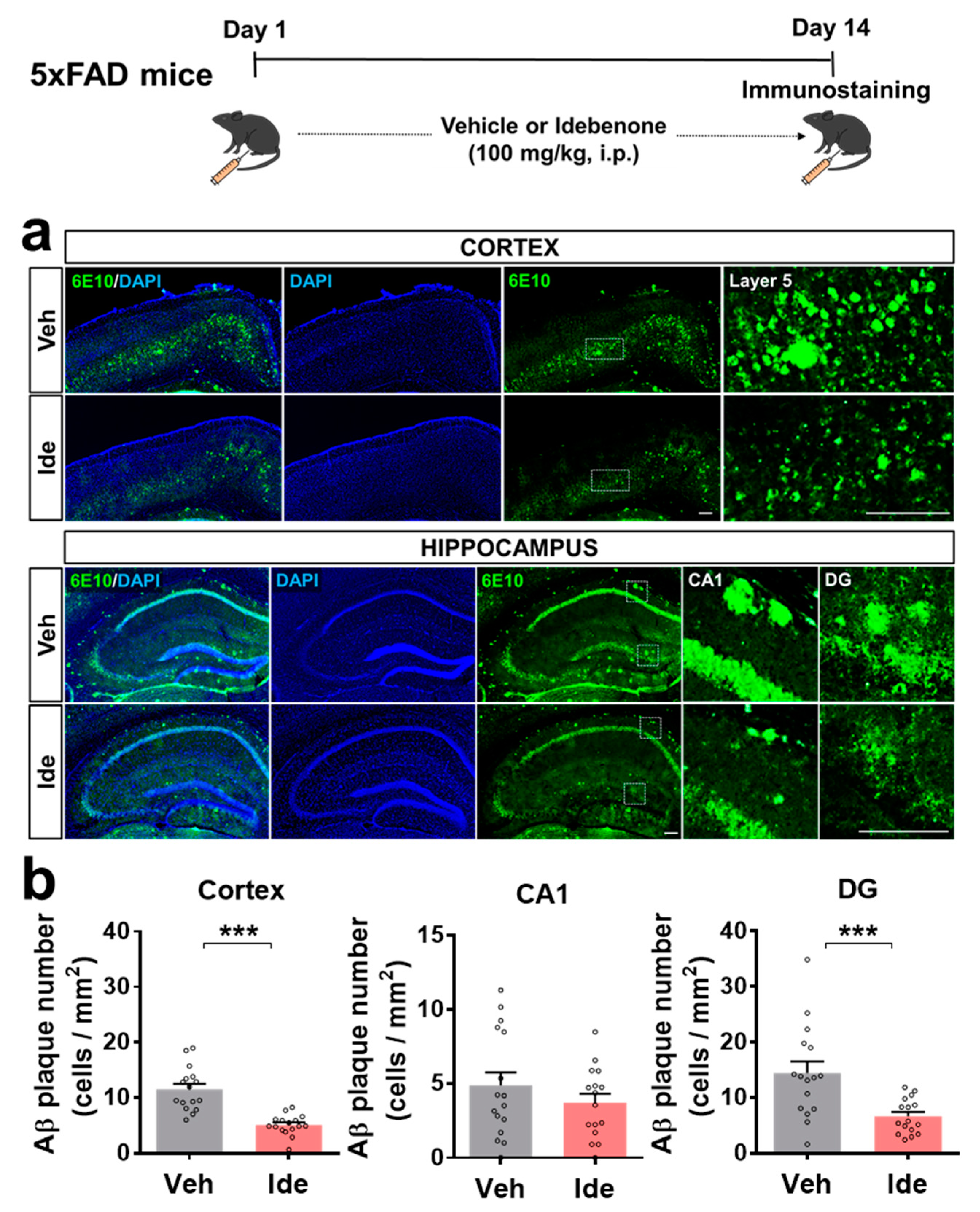
3.1. Idebenone Reduces Aβ Plaque Number in 5xFAD Mice
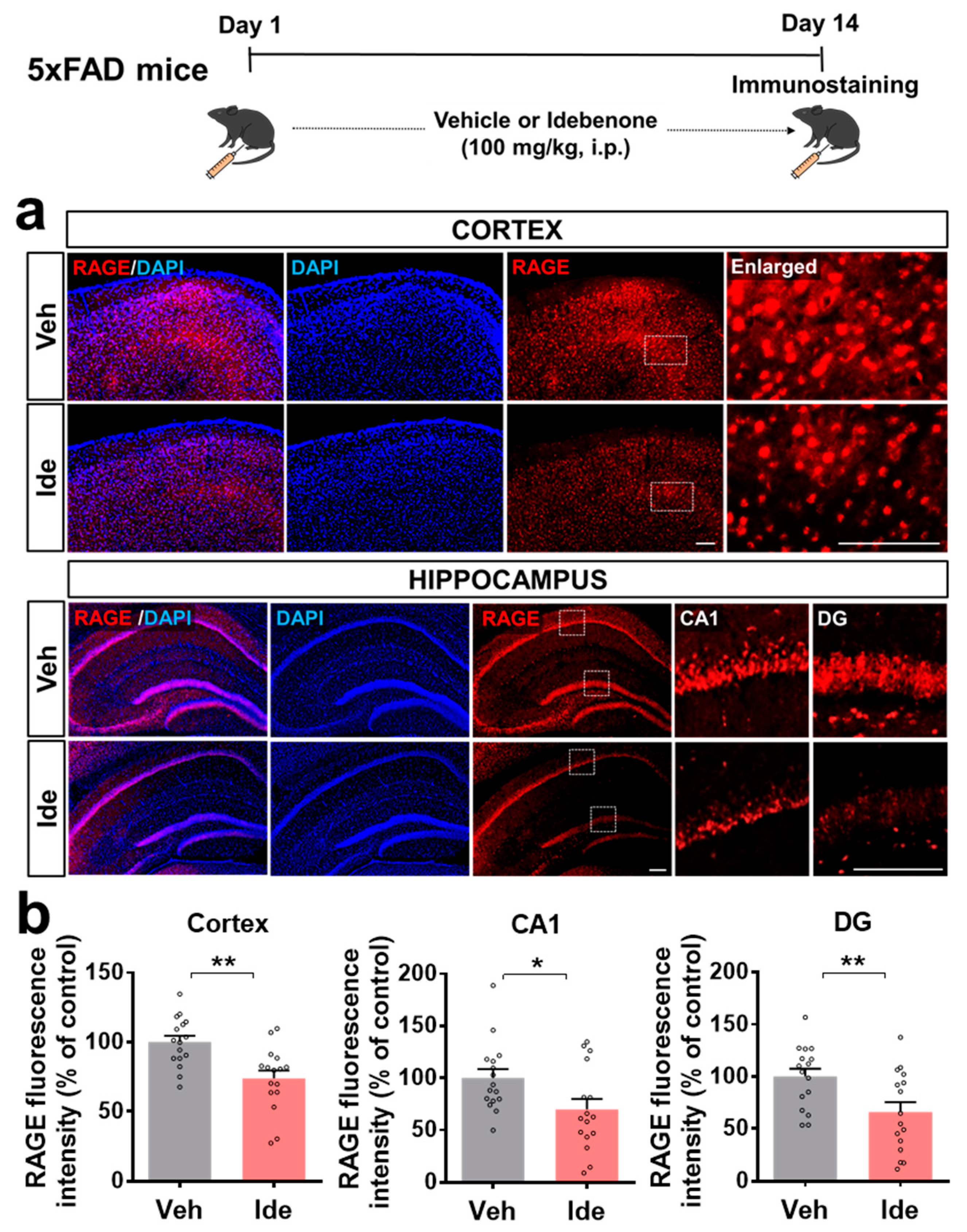
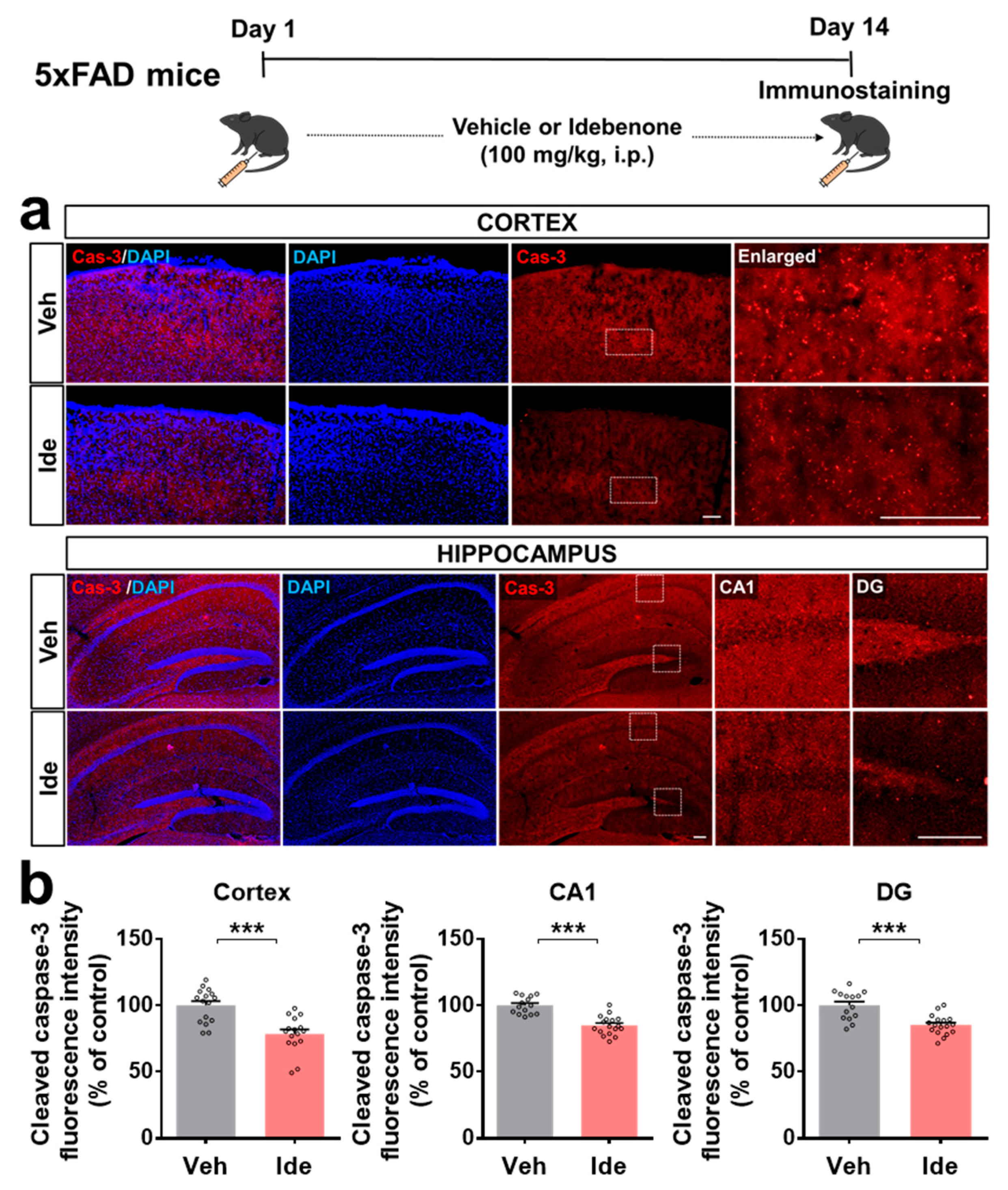
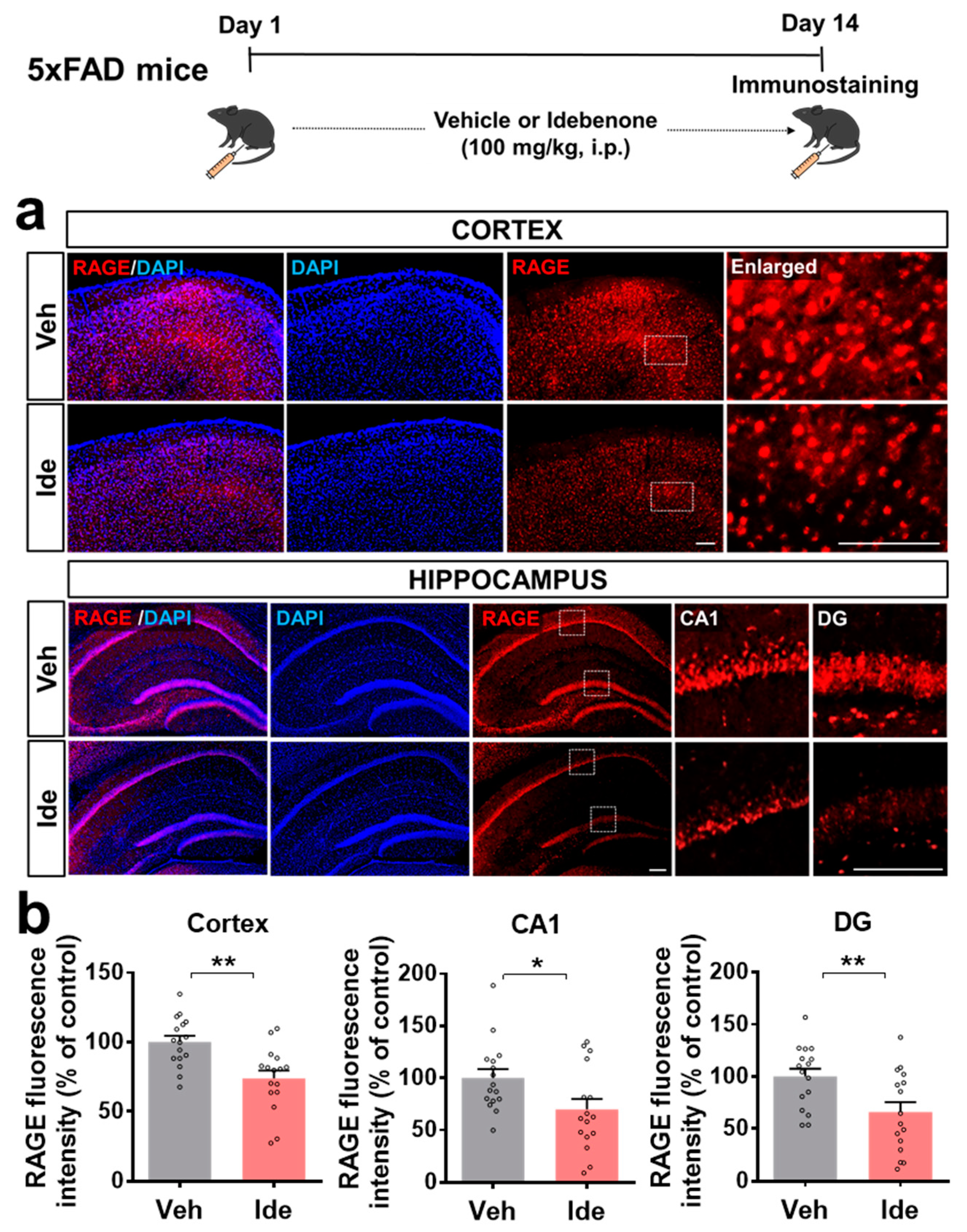
3.2. Idebenone Decreases RAGE and Caspase-3 Levels in 5xFAD Mice
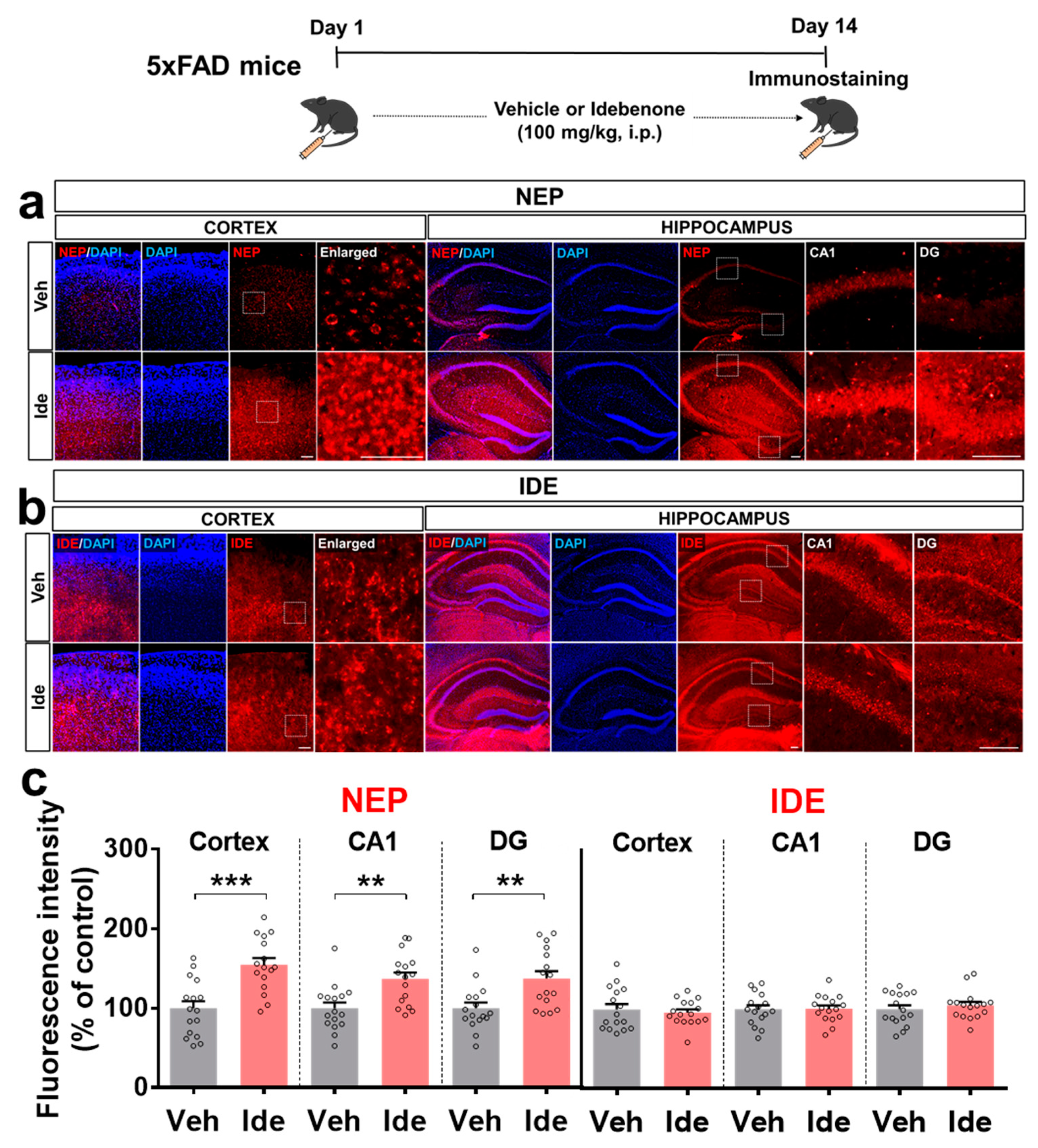
3.3. Idebenone Upregulates the Expression of the Aβ Degradation Enzyme NEP in 5xFAD Mice
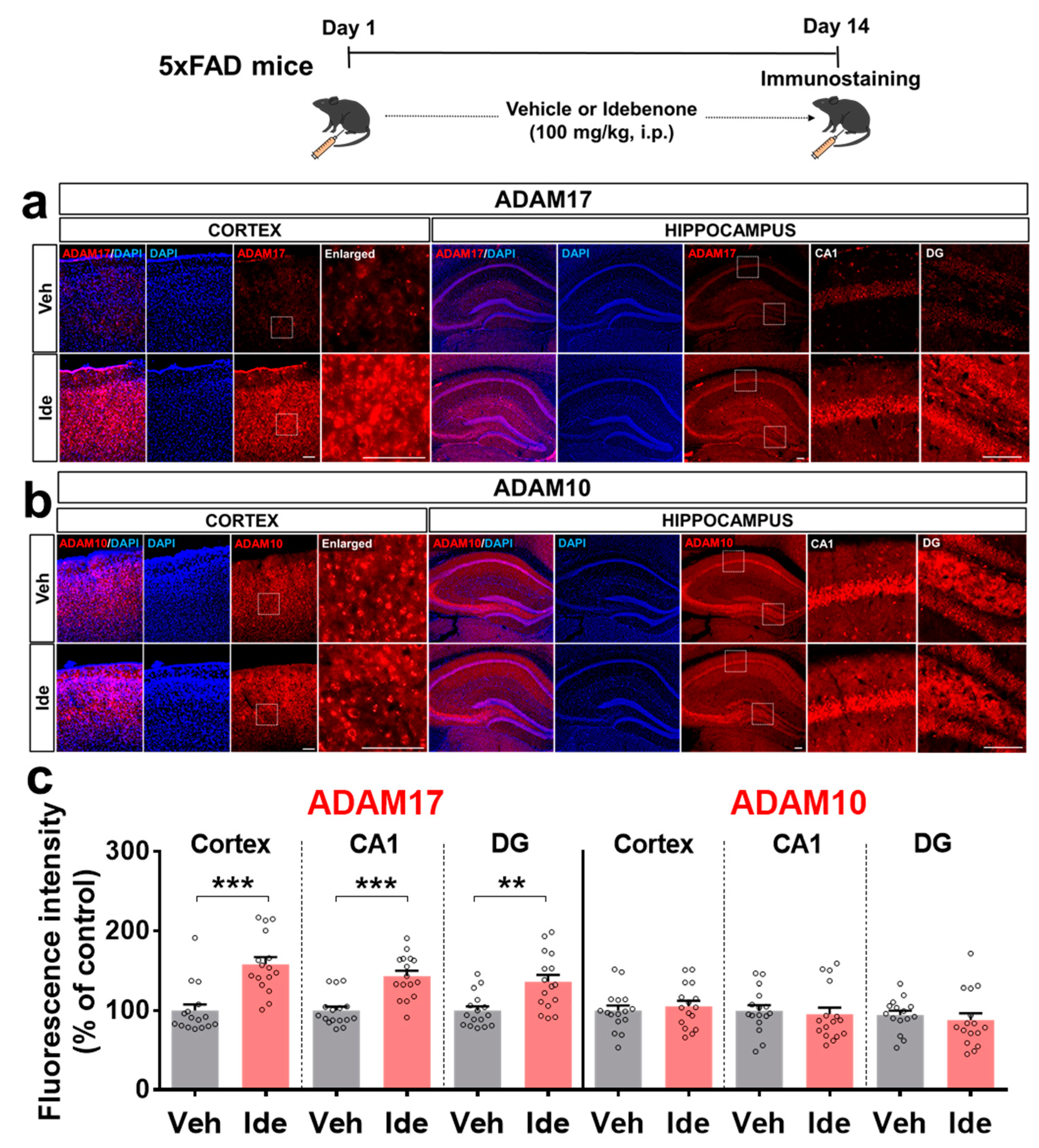
3.4. Idebenone Upregulates the Expression of the α-Secretase ADAM17 in 5xFAD Mice
3.5. Idebenone Decreases Tau Phosphorylation at Thr231 (AT180) and p-GSK3β Levels in 5xFAD Mice
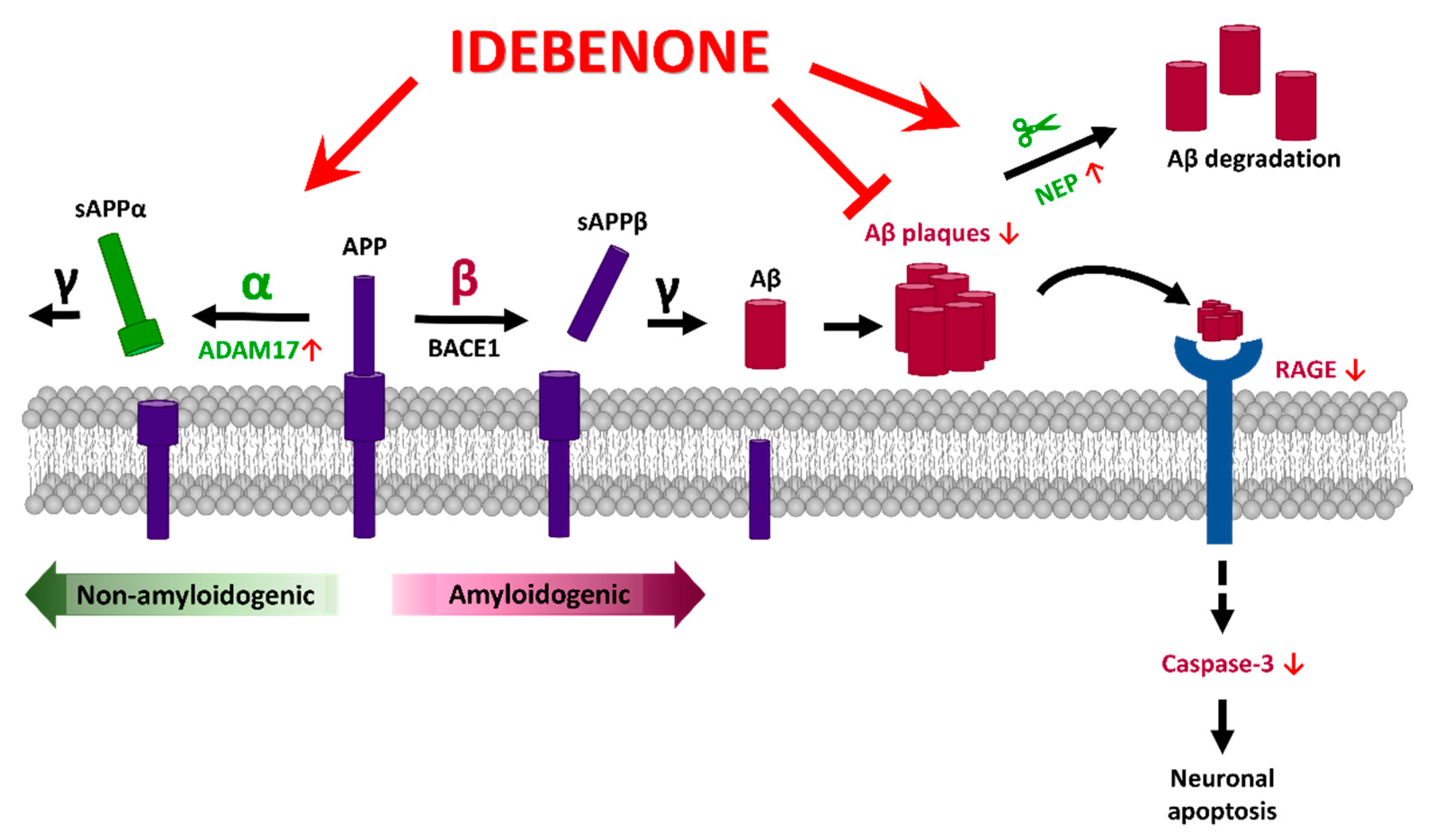
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [Green Version]
- Montagna, E.; Dorostkar, M.M.; Herms, J. The Role of APP in Structural Spine Plasticity. Front. Mol. Neurosci. 2017, 10, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, A.; Sawmiller, D.; Tan, J. Restoring Soluble Amyloid Precursor Protein alpha Functions as a Potential Treatment for Alzheimer’s Disease. J. Neurosci. Res. 2017, 95, 973–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoe, H.S.; Lee, H.K.; Pak, D.T. The upside of APP at synapses. CNS Neurosci. Ther. 2012, 18, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Klevanski, M.; Herrmann, U.; Weyer, S.W.; Fol, R.; Cartier, N.; Wolfer, D.P.; Caldwell, J.H.; Korte, M.; Muller, U.C. The APP Intracellular Domain Is Required for Normal Synaptic Morphology, Synaptic Plasticity, and Hippocampus-Dependent Behavior. J. Neurosci. 2015, 35, 16018–16033. [Google Scholar] [CrossRef]
- Kuhn, P.H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Rossner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef] [Green Version]
- Bae, N.; Byeon, S.E.; Song, J.; Lee, S.J.; Kwon, M.; Mook-Jung, I.; Cho, J.Y.; Hong, S. Knock-down of protein L-isoaspartyl O-methyltransferase increases beta-amyloid production by decreasing ADAM10 and ADAM17 levels. Acta Pharm. Sin. 2011, 32, 288–294. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Molinas, A.J.R.; Mukerjee, S.; Morgan, D.A.; Rahmouni, K.; Zsombok, A.; Lazartigues, E. Activation of ADAM17 (A Disintegrin and Metalloprotease 17) on Glutamatergic Neurons Selectively Promotes Sympathoexcitation. Hypertension 2019, 73, 1266–1274. [Google Scholar] [CrossRef]
- Gil-Bea, F.; Akterin, S.; Persson, T.; Mateos, L.; Sandebring, A.; Avila-Carino, J.; Gutierrez-Rodriguez, A.; Sundstrom, E.; Holmgren, A.; Winblad, B.; et al. Thioredoxin-80 is a product of alpha-secretase cleavage that inhibits amyloid-beta aggregation and is decreased in Alzheimer’s disease brain. EMBO Mol. Med. 2012, 4, 1097–1111. [Google Scholar] [CrossRef] [Green Version]
- Ray, B.; Maloney, B.; Sambamurti, K.; Karnati, H.K.; Nelson, P.T.; Greig, N.H.; Lahiri, D.K. Rivastigmine modifies the alpha-secretase pathway and potentially early Alzheimer’s disease. Transl. Psychiatry 2020, 10, 47. [Google Scholar] [CrossRef] [Green Version]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Yu, Q.; Arancio, O.; Chen, D.; Gore, S.S.; Yan, S.S.; Yan, S.F. RAGE mediates Abeta accumulation in a mouse model of Alzheimer’s disease via modulation of beta- and gamma-secretase activity. Hum. Mol. Genet. 2018, 27, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, L.; Hargreaves, I.P.; Georgian, A.R.; Turner, C.; Dalton, R.N.; Abbott, N.J.; Heales, S.J.R.; Preston, J.E. CoQ10 Deficient Endothelial Cell Culture Model for the Investigation of CoQ10 Blood-Brain Barrier Transport. J. Clin. Med. 2020, 9, 3236. [Google Scholar] [CrossRef]
- Wang, H.; Li, L.; Jia, K.; Wang, Q.; Sui, S.; Lin, Y.; He, Y. Idebenone protects mitochondrial function against amyloid beta toxicity in primary cultured cortical neurons. Neuroreport 2020, 31, 1104–1110. [Google Scholar] [CrossRef]
- Yamada, K.; Tanaka, T.; Han, D.; Senzaki, K.; Kameyama, T.; Nabeshima, T. Protective effects of idebenone and alpha-tocopherol on beta-amyloid-(1-42)-induced learning and memory deficits in rats: Implication of oxidative stress in beta-amyloid-induced neurotoxicity in vivo. Eur. J. Neurosci. 1999, 11, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharm. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Donahue, J.E.; Flaherty, S.L.; Johanson, C.E.; Duncan, J.A., 3rd; Silverberg, G.D.; Miller, M.C.; Tavares, R.; Yang, W.; Wu, Q.; Sabo, E.; et al. RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. 2006, 112, 405–415. [Google Scholar] [CrossRef]
- Choi, B.R.; Cho, W.H.; Kim, J.; Lee, H.J.; Chung, C.; Jeon, W.K.; Han, J.S. Increased expression of the receptor for advanced glycation end products in neurons and astrocytes in a triple transgenic mouse model of Alzheimer’s disease. Exp. Mol. Med. 2014, 46, e75. [Google Scholar] [CrossRef] [Green Version]
- Takuma, K.; Fang, F.; Zhang, W.; Yan, S.; Fukuzaki, E.; Du, H.; Sosunov, A.; McKhann, G.; Funatsu, Y.; Nakamichi, N.; et al. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2009, 106, 20021–20026. [Google Scholar] [CrossRef] [Green Version]
- Giridharan, V.V.; Thandavarayan, R.A.; Arumugam, S.; Mizuno, M.; Nawa, H.; Suzuki, K.; Ko, K.M.; Krishnamurthy, P.; Watanabe, K.; Konishi, T. Schisandrin B Ameliorates ICV-Infused Amyloid beta Induced Oxidative Stress and Neuronal Dysfunction through Inhibiting RAGE/NF-kappaB/MAPK and Up-Regulating HSP/Beclin Expression. PLoS ONE 2015, 10, e0142483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Hong, Y.S.; Jeong, J.H.; Yang, E.J.; Jhun, J.Y.; Park, M.K.; Jung, Y.O.; Min, J.K.; Kim, H.Y.; Park, S.H.; et al. Coenzyme Q10 ameliorates pain and cartilage degradation in a rat model of osteoarthritis by regulating nitric oxide and inflammatory cytokines. PLoS ONE 2013, 8, e69362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Ma, M.; Zhu, X.; Li, M.; Guo, M.; Liu, P.; He, Z.; Fu, Q. Effectiveness of idebenone nanorod formulations in the treatment of Alzheimer’s disease. J. Control. Release 2021, 336, 169–180. [Google Scholar] [CrossRef]
- Jiang, W.; Geng, H.; Lv, X.; Ma, J.; Liu, F.; Lin, P.; Yan, C. Idebenone Protects against Atherosclerosis in Apolipoprotein E-Deficient Mice Via Activation of the SIRT3-SOD2-mtROS Pathway. Cardiovasc. Drugs Ther. 2020. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.X.; Cai, W.J.; Sun, X.Y.; Dai, P.P.; Li, X.M.; Wang, Q.; Huang, X.L.; He, B.; Wang, P.P.; Wu, G.; et al. RAGE-dependent mitochondria pathway: A novel target of silibinin against apoptosis of osteoblastic cells induced by advanced glycation end products. Cell Death Dis. 2018, 9, 674. [Google Scholar] [CrossRef] [PubMed]
- Avetisyan, A.; Balasanyants, S.; Simonyan, R.; Koroev, D.; Kamynina, A.; Zinovkin, R.; Bobkova, N.; Volpina, O. Synthetic fragment (60–76) of RAGE improves brain mitochondria function in olfactory bulbectomized mice. Neurochem. Int. 2020, 140, 104799. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.S.; Jo, S.A. Mechanisms of Amyloid-beta Peptide Clearance: Potential Therapeutic Targets for Alzheimer’s Disease. Biomol. Ther. Seoul 2012, 20, 245–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miners, J.S.; Van Helmond, Z.; Chalmers, K.; Wilcock, G.; Love, S.; Kehoe, P.G. Decreased expression and activity of neprilysin in Alzheimer disease are associated with cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 2006, 65, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.M.; Mouri, A.; Kokubo, H.; Nakajima, R.; Suemoto, T.; Higuchi, M.; Staufenbiel, M.; Noda, Y.; Yamaguchi, H.; Nabeshima, T.; et al. Neprilysin-sensitive synapse-associated amyloid-beta peptide oligomers impair neuronal plasticity and cognitive function. J. Biol. Chem. 2006, 281, 17941–17951. [Google Scholar] [CrossRef] [Green Version]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Shirotani, K.; Lu, B.; Gerard, N.P.; Gerard, C.; Hama, E.; Lee, H.J.; Saido, T.C. Metabolic regulation of brain Abeta by neprilysin. Science 2001, 292, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Wang, R.; Chen, L.; Bennett, D.A.; Dickson, D.W.; Wang, D.S. Expression and functional profiling of neprilysin, insulin-degrading enzyme, and endothelin-converting enzyme in prospectively studied elderly and Alzheimer’s brain. J. Neurochem. 2010, 115, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Carty, N.; Nash, K.R.; Brownlow, M.; Cruite, D.; Wilcock, D.; Selenica, M.L.; Lee, D.C.; Gordon, M.N.; Morgan, D. Intracranial injection of AAV expressing NEP but not IDE reduces amyloid pathology in APP+PS1 transgenic mice. PLoS ONE 2013, 8, e59626. [Google Scholar] [CrossRef] [Green Version]
- Vettorazzi, J.F.; Kurauti, M.A.; Soares, G.M.; Borck, P.C.; Ferreira, S.M.; Branco, R.C.S.; Michelone, L.S.L.; Boschero, A.C.; Junior, J.M.C.; Carneiro, E.M. Bile acid TUDCA improves insulin clearance by increasing the expression of insulin-degrading enzyme in the liver of obese mice. Sci Rep. 2017, 7, 14876. [Google Scholar] [CrossRef] [Green Version]
- Del Turco, D.; Schlaudraff, J.; Bonin, M.; Deller, T. Upregulation of APP, ADAM10 and ADAM17 in the denervated mouse dentate gyrus. PLoS ONE 2014, 9, e84962. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.; May, P.; Gu, W.; Mayhaus, M.; Pichler, S.; Spaniol, C.; Glaab, E.; Bobbili, D.R.; Antony, P.; Koegelsberger, S.; et al. A rare loss-of-function variant of ADAM17 is associated with late-onset familial Alzheimer disease. Mol. Psychiatry 2020, 25, 629–639. [Google Scholar] [CrossRef]
- Sogorb-Esteve, A.; Garcia-Ayllon, M.S.; Gobom, J.; Alom, J.; Zetterberg, H.; Blennow, K.; Saez-Valero, J. Levels of ADAM10 are reduced in Alzheimer’s disease CSF. J. Neuroinflamm. 2018, 15, 213. [Google Scholar] [CrossRef] [PubMed]
- Scharfenberg, F.; Helbig, A.; Sammel, M.; Benzel, J.; Schlomann, U.; Peters, F.; Wichert, R.; Bettendorff, M.; Schmidt-Arras, D.; Rose-John, S.; et al. Degradome of soluble ADAM10 and ADAM17 metalloproteases. Cell Mol. Life Sci. 2020, 77, 331–350. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.X.; Iqbal, K. Hyperphosphorylation of microtubule-associated protein tau: A promising therapeutic target for Alzheimer disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahn, H. Memory loss in Alzheimer’s disease. Dialogues Clin. Neurosci. 2013, 15, 445–454. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immunogen | Host Species | Dilution | Manufacturer | Catalog No. |
|---|---|---|---|---|
| 6E10 | Mouse | 1:500 | BioLegend | 803002 |
| RAGE | Rabbit | 1:200 | Abcam | AB3611 |
| Caspase-3 | Rabbit | 1:100 | Cell Signaling | 9664 |
| NEP | Rabbit | 1:200 | Millipore | AB5458 |
| IDE | Rabbit | 1:200 | Abcam | AB32216 |
| ADAM17 | Rabbit | 1:100 | Abcam | AB2051 |
| ADAM10 | Rabbit | 1:100 | Abcam | AB1997 |
| AT180 | Mouse | 1:100 | Invitrogen | MN1040 |
| Tau5 | Mouse | 1:100 | Invitrogen | AHB0042 |
| p-GSK-3βY216 | Rabbit | 1:200 | Abcam | AB75745 |
| p-CDK-5 | Rabbit | 1:200 | LSBio | LS-C354604 |
| DYRK1A | Rabbit | 1:200 | Abcam | AB69811 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-j.; Jeong, H.-R.; Park, J.-H.; Hoe, H.-S. Idebenone Decreases Aβ Pathology by Modulating RAGE/Caspase-3 Signaling and the Aβ Degradation Enzyme NEP in a Mouse Model of AD. Biology 2021, 10, 938. https://doi.org/10.3390/biology10090938
Lee H-j, Jeong H-R, Park J-H, Hoe H-S. Idebenone Decreases Aβ Pathology by Modulating RAGE/Caspase-3 Signaling and the Aβ Degradation Enzyme NEP in a Mouse Model of AD. Biology. 2021; 10(9):938. https://doi.org/10.3390/biology10090938
Chicago/Turabian StyleLee, Hyun-ju, Ha-Ram Jeong, Jin-Hee Park, and Hyang-Sook Hoe. 2021. "Idebenone Decreases Aβ Pathology by Modulating RAGE/Caspase-3 Signaling and the Aβ Degradation Enzyme NEP in a Mouse Model of AD" Biology 10, no. 9: 938. https://doi.org/10.3390/biology10090938
APA StyleLee, H.-j., Jeong, H.-R., Park, J.-H., & Hoe, H.-S. (2021). Idebenone Decreases Aβ Pathology by Modulating RAGE/Caspase-3 Signaling and the Aβ Degradation Enzyme NEP in a Mouse Model of AD. Biology, 10(9), 938. https://doi.org/10.3390/biology10090938