
When Nothing Turns Itself Inside out and Becomes Something: Coating Poly(Lactic-Co-Glycolic Acid) Spheres with Hydroxyapatite Nanoparticles vs. the Other Way Around



Abstract
1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. Characterization
2.3. Drug Release
2.4. Cell Culture
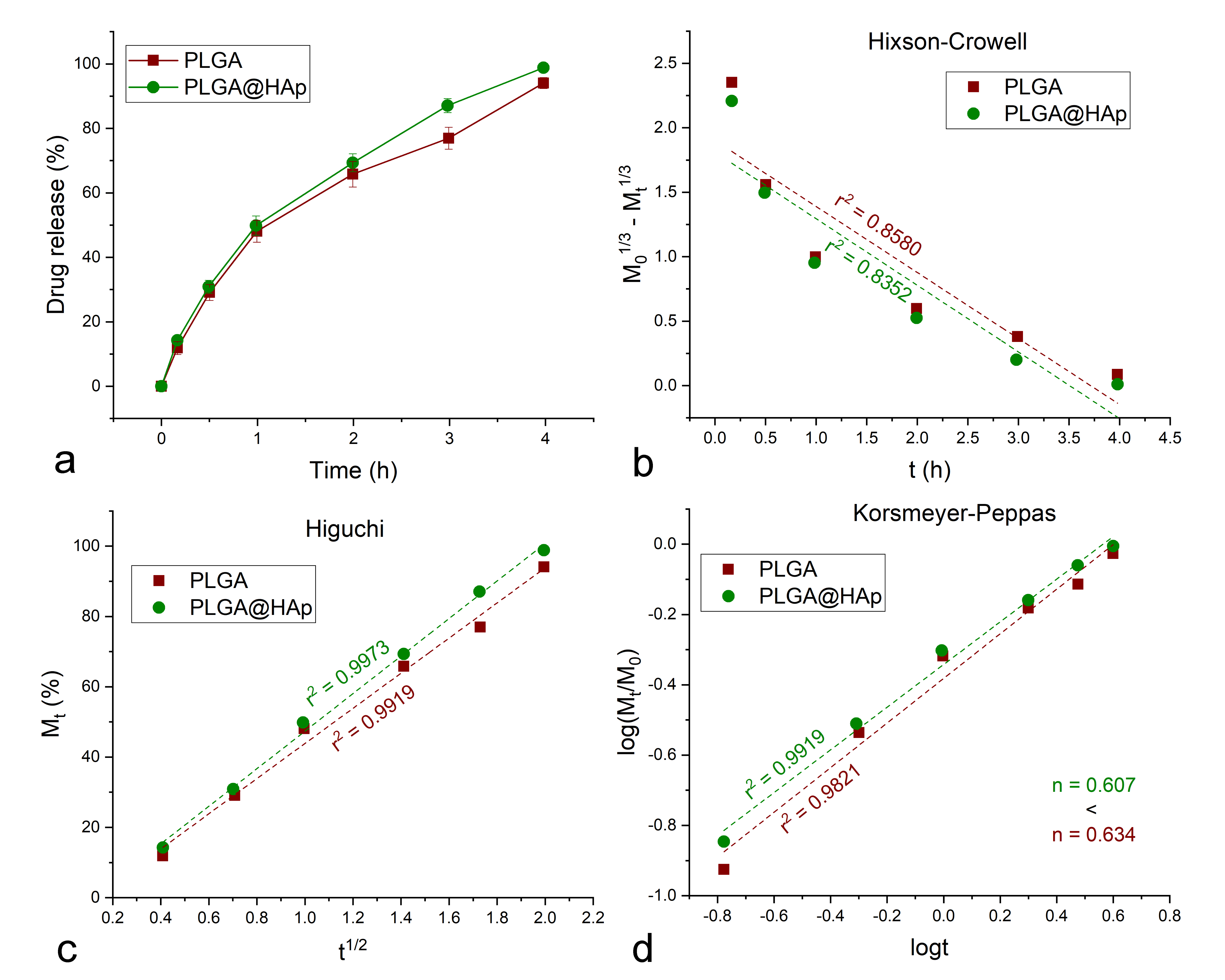
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uskoković, V. When 1 + 1 > 2: Nanostructured Composite Materials for Hard Tissue Engineering Applications. Mat. Sci. Eng. C 2015, 57, 434–451. [Google Scholar] [CrossRef] [PubMed]
- Uskoković, V. Nanotechnologies: What We Do Not Know. Tech. Soc. 2007, 29, 43–61. [Google Scholar] [CrossRef]
- Wu, V.M.; Uskoković, V. Waiting for Aπαταω: 250 Years Later. Found. Sci. 2019, 24, 617–640. [Google Scholar] [CrossRef] [PubMed]
- Meurman, J.H. Release of Salivary Proteins from Hydroxyapatite and Enamel by Elution by Phosphate Buffer. J. Dent. Res. 1977, 56, A62. [Google Scholar]
- Park, K.C.; Katz, S.; Stookey, G.K. Chlorhexidine Uptake and Release by Hydroxyapatite and Human Salivary Sediment. J. Dent. Res. 1980, 59, B946. [Google Scholar]
- Uskoković, V. Ion-Doped Hydroxyapatite: An Impasse or the Road to Follow? Ceram. Int. 2020, 46, 11443–11465. [Google Scholar] [CrossRef]
- Uskoković, V.; Uskoković, D.P. Nanosized Hydroxyapatite and Other Calcium Phosphates: Chemistry of Formation and Application as Drug and Gene Delivery Agents. J. Biomed. Mater. Res. B 2011, 96, 152–191. [Google Scholar] [CrossRef]
- Bernardi, G. Chromatography of Nucleic Acids on Hydroxyapatite. I. Chromatography of Native DNA. Biochim. Biophys. Acta 1969, 174, 423–434. [Google Scholar] [CrossRef]
- Graham, F.L.; van der Eb, A.J. A New Technique for the Assay of Infectivity of Human Adenovirus 5 DNA. Virology 1973, 52, 456–467. [Google Scholar] [CrossRef]
- Reible, D.D.; Lampert, D.J. Capping for Remediation of Contaminated Sediments. In Processes, Assessment and Remediation of Contaminated Sediments; Reible, D.D., Ed.; Springer: New York, NY, USA, 2014; pp. 325–364. [Google Scholar]
- Khan, M.A.; Wu, V.M.; Ghosh, S.; Uskoković, V. Gene Delivery Using Calcium Phosphate Nanoparticles: Optimization of the Transfection Process and the Effects of Citrate and Poly(L-Lysine) as Additives. J. Coll. Interface Sci. 2016, 471, 48–58. [Google Scholar] [CrossRef]
- Ghiasi, B.; Sefidbakht, Y.; Mozaffari-Jovin, S.; Gharachloo, B.; Mehraria, M.; Khodadadi, A.; Rezaei, M.; Ranaei-Siadat, S.O.; Uskoković, V. Hydroxyapatite as a Biomaterial—A Gift that Keeps on Giving. Drug Dev. Ind. Pharm. 2020, 46, 1035–1062. [Google Scholar] [CrossRef]
- Uskoković, V.; Tang, S.; Nikolić, M.G.; Marković, S.; Wu, V.M. Calcium Phosphate Nanoparticles as Intrinsic Inorganic Antimicrobials: In Search of the Key Particle Property. Biointerphases 2019, 14, 031001. [Google Scholar] [CrossRef]
- Kazemzadeh-Narbat, M.; Noordin, S.; Masri, B.A.; Garbuz, D.S.; Duncan, C.P.; Hancock, R.E.; Wang, R. Drug Release and Bone Growth Studies of Antimicrobial Peptide-Loaded Calcium Phosphate Coating on Titanium. J. Biomed. Mater. Res. B 2012, 100, 1344–1352. [Google Scholar] [CrossRef]
- Uskoković, V.; Desai, T.A. Phase Composition Control of Calcium Phosphate Nanoparticles for Tunable Drug Delivery Kinetics and Treatment of Osteomyelitis. I. Preparation and Drug Release. J. Biomed. Mater. Res. A 2013, 101, 1416–1426. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, K.; Wu, M.; Wu, Q.; Liu, J.; Yang, J.; Zhang, J. Unexpectedly High Adsorption Capacity of Esterified Hy-droxyapatite for Heavy Metal Removal. Langmuir 2019, 35, 16111–16119. [Google Scholar] [CrossRef]
- Itokazu, M.; Yang, W.; Aoki, T.; Ohara, A.; Kato, N. Synthesis of Antibiotic-Loaded Interporous Hydroxyapatite Blocks by Vacuum Method and in vitro Drug Release Testing. Biomaterials 1998, 19, 817–819. [Google Scholar] [CrossRef]
- Vukomanović, M.; Zavašnik-Bergant, T.; Bračko, I.; Škapin, S.D.; Ignjatović, N.; Radmilović, V.; Uskoković, D. Poly(d,l-Lactide-co-Glycolide)/Hydroxyapatite Core–Shell Nanospheres. Part 3: Properties of Hydroxyapatite Nano-rods and In-vestigation of a Distribution of the Drug within the Composite. Coll. Surf. B 2011, 87, 226–235. [Google Scholar] [CrossRef]
- Ignjatović, N.L.; Janković, R.; Uskoković, V.; Uskoković, D.P. Effects of Hydroxyapatite@Poly-Lactide-Co-Glycolide Nanoparticles Combined with Pb and Cd on Liver and Kidney Parenchyma after the Reconstruction of Mandibular Bone Defects. Toxicol. Res. 2019, 8, 287–296. [Google Scholar] [CrossRef]
- Ignjatović, N.L.; Penov-Gaši, K.M.; Wu, V.M.; Ajduković, J.J.; Kojić, V.V.; Vasiljević-Radović, D.; Kuzmanović, M.; Uskoković, V.; Uskoković, D.P. Selective Anticancer Activity of Lung-Targeting Hydroxyapatite/Chitosan-Poly(D,L)-Lactide-co-Glycolide Particles Loaded with an Androstane-Based Cancer Inhibitor. Coll. Surf. B 2016, 148, 629–639. [Google Scholar] [CrossRef]
- Ahmed, M.K.; Mansour, S.F.; Al-Wafi, R.; Afifi, M.; Uskoković, V. Gold as a Dopant in Selenium-Containing Carbonated Hydroxyapatite Fillers of Nanofibrous ε-Polycaprolactone Scaffolds for Tissue Engineering. Int. J. Pharm. 2020, 577, 118950. [Google Scholar] [CrossRef]
- Ahmed, M.K.; Mansour, S.F.; Al-Wafi, R.; El-dek, S.I.; Uskoković, V. Tuning the Mechanical, Microstructural and Cell Adhesion Properties of Electrospun ε-Polycaprolactone Microfibers by Doping Selenium-Containing Carbonated Hydroxyapatite as the Reinforcing Agent with Magnesium Ions. J. Mater. Sci. 2019, 54, 14524–14544. [Google Scholar] [CrossRef]
- Kim, S.S.; Park, M.S.; Jeon, O.; Choi, C.Y.; Kim, B.S. Poly(Lactide-co-Glycolide)/Hydroxyapatite Composite Scaffolds for Bone Tissue Engineering. Biomaterials 2006, 27, 1399–1409. [Google Scholar] [CrossRef]
- Tamai, H.; Yasuda, H. Preparation of Polymer Particles Coated with Hydroxyapatite. J. Coll. Interface Sci. 1999, 212, 585–588. [Google Scholar] [CrossRef]
- Kang, S.W.; Yang, H.S.; Seo, S.W.; Han, D.K.; Kim, B.S. Apatite-Coated Poly(Lactic-co-Glycolic Acid) Microspheres as an Injectable Scaffold for Bone Tissue Engineering. J. Biomed. Mater. Res. A 2008, 85, 747–756. [Google Scholar] [CrossRef]
- Xu, Q.; Czernuszka, J. Controlled Release of Amoxicillin From Hydroxyapatite-Coated Poly(lactic-Co-Glycolic Acid) Micro-spheres. J. Control. Release 2008, 127, 146–153. [Google Scholar] [CrossRef]
- Jongpaiboonkit, L.; Franklin-Ford, T.; Murphy, W.L. Growth of Hydroxyapatite Coatings on Biodegradable Polymer Micro-spheres. ACS Appl. Mater. Interfaces 2009, 1, 1504–1511. [Google Scholar] [CrossRef]
- Ghorbani, F.; Zamanian, A.; Behnamghader, A.; Joupari, M.D. Microwave-Induced Rapid Formation of Biomimetic Hydroxy-apatite Coating on Gelatin-Siloxane Hybrid Microspheres in 10X-SBF Solution. E-Polymers 2018, 18, 247–255. [Google Scholar] [CrossRef]
- Zhe, Z.; Zhang, S.; Venkatraman, S.S.; Lei, S. Growth of Hydroxyapatite Coating on Polymer Microspheres. Nanosci. Nanotech. Lett. 2011, 3, 472–476. [Google Scholar] [CrossRef][Green Version]
- Vukomanović, M.; Škapin, S.; Jančar, B.; Maksin, T.; Ignjatović, N.; Uskoković, V.; Uskoković, D. Poly(D,L-Lactide-Co-Glycolide)/Hydroxyapatite Core-Shell Nanospheres. Part 1: A Multifunctional System for Controlled Drug Delivery. Coll. Surf. B 2011, 82, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Uskoković, V.; Desai, T.A. In vitro Analysis of Nanoparticulate Hydroxyapatite/Chitosan Composites as Potential Drug Delivery Platforms for the Sustained Release of Antibiotics in the Treatment of Osteomyelitis. J. Pharm. Sci. 2014, 103, 567–579. [Google Scholar] [CrossRef]
- Wojcik-Pastuszka, D.; Krzak, J.; Macikowski, B.; Berkowski, R.; Osiński, B.; Musiał, W. Evaluation of the Release Kinetics of a Pharmacologically Active Substance from Model Intra-Articular Implants Replacing the Cruciate Ligaments of the Knee. Materials 2019, 12, 1202. [Google Scholar] [CrossRef]
- Quarles, L.D.; Yohay, D.A.; Lever, L.W.; Caton, R.; Wenstrup, R.J. Distinct Proliferative and Differentiated Stages of Murine MC3T3-E1 Cells in Culture: An in vitro Model of Osteoblast Development. J. Bone Miner. Res. 1992, 7, 683–692. [Google Scholar] [CrossRef]
- Uskoković, V. Factors Defining the Stability of Poly(Lactide-co-Glycolide) Spheres for the Sustained Release of a Cysteine Protease Inhibitor. Int. J. Pharm. 2020, 583, 119316. [Google Scholar] [CrossRef]
- Di Natale, C.; Onesto, V.; Lagreca, E.; Vecchione, R.; Netti, P.A. Tunable Release of Curcumin with an In Silico-Supported Ap-proach from Mixtures of Highly Porous PLGA Microparticles. Materials 2020, 13, 1807. [Google Scholar] [CrossRef]
- Son, J.; Lee, D.; Yoo, J.; Park, C.; Koo, H. A Comparative Study of the Effect of Drug Hydrophobicity on Nanoparticle Drug Delivery in vivo using Two Photosensitizers. Nanomedicine 2020, 24, 102151. [Google Scholar] [CrossRef]
- Witek, L.; Tian, H.; Tovar, N.; Torroni, A.; Neiva, R.; Gil, L.F.; Coelho, P.G. The Effect of Platelet-Rich Fibrin Exudate Addition to Porous Poly(Lactic-co-Glycolic Acid) Scaffold in Bone Healing: An in vivo Study. J. Biomed. Mater. Res. B 2020, 108, 1304–1310. [Google Scholar] [CrossRef]
- Abay Akar, N.; Gürel Peközer, G.; Torun Köse, G. Fibrous Bone Tissue Engineering Scaffolds Prepared by Wet Spinning of PLGA. Turk. J. Biol. 2019, 43, 235–245. [Google Scholar] [CrossRef]
- Duan, P.; Pan, Z.; Cao, L.; Gao, J.; Yao, H.; Liu, X.; Guo, R.; Liang, X.; Dong, J.; Ding, J. Restoration of Osteochondral Defects by Implanting Bilayered Poly(Lactide-co-Glycolide) Porous Scaffolds in Rabbit Joints for 12 and 24 Weeks. J. Orthop. Translat. 2019, 19, 68–80. [Google Scholar] [CrossRef]
- Mirakabad, F.S.T.; Nejati-Koshki, K.; Akbarzadeh, A.; Yamchi, M.R.; Milani, M.; Zarghami, N.; Zeighamian, V.; Rahimzadeh, A.; Alimohammadi, S.; Hanifehpour, Y.; et al. PLGA-Based Nanoparticles as Cancer Drug Delivery Systems. Asian Pac. J. Cancer Prev. 2014, 15, 517–535. [Google Scholar] [CrossRef]
- Uskoković, V.; Desai, T.A. Nanoparticulate Drug Delivery Platforms for Advancing Bone Infection Therapies. Exp. Opin. Drug Deliv. 2014, 11, 1899–1912. [Google Scholar] [CrossRef]
- Lemaire, S.; Van Bambeke, F.; Pierard, D.; Appelbaum, P.C.; Tulkens, P.M. Activity of Fusidic Acid Against Extracellular and Intracellular Staphylococcus aureus: Influence of pH and Comparison With Linezolid and Clindamycin. Clin. Infect. Dis. 2011, 52, S493–S503. [Google Scholar] [CrossRef]
- Tsuji, A.; Kaneko, Y.; Takahashi, K.; Ogawa, M.; Goto, S. The Effects of Temperature and pH on the Growth of Eight Enteric and Nine Glucose Non-Fermenting Species of Gram-Negative Rods. Microbiol. Immunol. 1982, 26, 15–24. [Google Scholar] [CrossRef]
- Fujii, S.; Okada, M.; Sawa, H.; Furuzono, T.; Nakamura, Y. Hydroxyapatite Nanoparticles as Particulate Emulsifier: Fabrication of Hydroxyapatite-Coated Biodegradable Microspheres. Langmuir 2009, 25, 9759–9766. [Google Scholar] [CrossRef]
- Uskoković, V.; Hoover, C.; Vukomanović, M.; Uskoković, D.P.; Desai, T.A. Osteogenic and Antimicrobial Nanoparticulate Calcium Phosphate and/or Poly-Lactide-Co-Glycolide Powders for the Treatment of Osteomyelitis. Mat. Sci. Eng. C 2013, 33, 3362–3373. [Google Scholar] [CrossRef]
- Uskoković, V.; Odsinada, R.; Djordjevic, S.; Habelitz, S. Dynamic Light Scattering and Zeta Potential of Colloidal Mixtures of Amelogenin and Hydroxyapatite in Calcium and Phosphate Rich Ionic Milieus. Arch. Oral Biol. 2011, 56, 521–532. [Google Scholar] [CrossRef]
- Wu, V.M.; Tang, S.; Uskoković, V. Calcium Phosphate Nanoparticles as Intrinsic Inorganic Antimicrobials: The Antibacterial Effect. ACS Appl. Mater. Interfaces 2018, 10, 34013–34028. [Google Scholar] [CrossRef]
- Gavini, E.; Chetoni, P.; Cossu, M.; Alvarez, M.G.; Saettone, M.F.; Giunchedi, P. PLGA Microspheres for the Ocular Delivery of a Peptide Drug, Vancomycin using Emulsification/Spray-Drying as the Preparation Method: In vitro/in vivo Studies. Eur. J. Pharm. Biopharm. 2004, 57, 207–212. [Google Scholar] [CrossRef]
- Yu, X.; Pan, Q.; Zheng, Z.; Chen, Y.; Chen, Y.; Weng, S.; Huang, L. pH-Responsive and Porous Vancomycin-Loaded PLGA Microspheres: Evidence of Controlled and Sustained Release for Localized Inflammation Inhibition in vitro. RSC Adv. 2018, 8, 37424–37432. [Google Scholar] [CrossRef]
- Tseng, Y.Y.; Kao, C.W.; Liu, K.S.; Tang, Y.L.; Liu, Y.W.; Liu, S.J. Treating Intracranial Abscesses in Rats with Stereotactic Injection of Biodegradable Vancomycin-Embedded Microparticles. Pharmaceutics 2020, 12, 91. [Google Scholar] [CrossRef]
- Uskoković, V. Revisiting the Fundamentals in the Design and Control of Nanoparticulate Colloids in the Frame of Soft Chemistry. Rev. J. Chem. 2013, 3, 271–303. [Google Scholar] [CrossRef][Green Version]
- Bian, C.; Lin, H.; Li, X.; Ma, J.; Jiang, P.; Qu, F. Preparation of Spherical Macroporous Poly(Lactic-co-Glycolic Acid) for Bone Tissue Regeneration. IET Nanobiotechnol. 2015, 9, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Buzzi, V.; Brudner, M.; Wagner, T.M.; Bazzo, G.C.; Pezzin, A.P.T.; Silva, D.A.K. Caboxymetylcellulose/Gelatin Blends Loaded with Piroxicam: Preparation, Characterization and Evaluation of in Vitro Release Profile. J. Encapsul. Adsorp. Sci. 2013, 3, 40543. [Google Scholar] [CrossRef]
- Okajima, L.S.; Martinez, E.F.; Pinheiro, I.F.; Fonseca Silva, A.S.; Demasi, A.P.D. Effect of Sodium Ascorbyl Phosphate on Osteoblast Viability and Differentiation. J. Periodontal Res. 2020, 55, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Uskoković, V.; Desai, T.A. Phase Composition Control of Calcium Phosphate Nanoparticles for Tunable Drug Delivery Kinetics and Treatment of Osteomyelitis. II. Antibacterial and Osteoblastic Response. J. Biomed. Mater. Res. A 2013, 101, 1427–1436. [Google Scholar] [CrossRef]
- Uskoković, V.; Janković-Častvan, I.; Wu, V.M. Bone Mineral Crystallinity Governs the Orchestration of Ossification and Resorption during Bone Remodeling. ACS Biomat. Sci. Eng. 2019, 5, 3483–3498. [Google Scholar] [CrossRef]
- Uskoković, V. On the Relational Character of Mind and Nature. Res. Cog. J. Phil. 2009, 6, 286–400. [Google Scholar]
- von Foerster, H. Observing Systems; Intersystems Publications: Seaside, FL, USA, 1981. [Google Scholar]
- Bateson, G. Steps to an Ecology of Mind; University of Chicago Press: Chicago, IL, USA, 1972. [Google Scholar]
- Uskoković, V.; Desai, T.A. Does Translational Symmetry Matter on the Micro Scale? Fibroblastic and Osteoblastic Interactions with the Topographically Distinct Poly(ε-Caprolactone)/Hydroxyapatite Thin Films. ACS Appl. Mater. Interfaces 2014, 6, 13209–13220. [Google Scholar] [CrossRef]
- Wang, D.X.; He, Y.; Bi, L.; Qu, Z.H.; Zou, J.W.; Pan, Z.; Fan, J.J.; Chen, L.; Dong, X.; Liu, X.N.; et al. Enhancing the Bioactivity of Poly(Lactic-co-Glycolic Acid) Scaffold with a Nano-Hydroxyapatite Coating for the Treatment of Segmental Bone Defect in a Rabbit Model. Int. J. Nanomed. 2013, 8, 1855–1865. [Google Scholar] [CrossRef]
- Nie, W.; Gao, Y.; McCoul, D.J.; Gillispie, G.J.; Zhang, Y.; Liang, L.; He, C. Rapid Mineralization of Hierarchical Poly(l-Lactic Acid)/Poly(ε-Caprolactone) Nanofibrous Scaffolds by Electrodeposition for Bone Regeneration. Int. J. Nanomed. 2019, 14, 3929–3941. [Google Scholar] [CrossRef]
- Lee, J.H.; Jang, H.L.; Lee, K.M.; Baek, H.R.; Jin, K.; Noh, J.H. Cold-Spray Coating of Hydroxyapatite on a Three-Dimensional Polyetheretherketone Implant and its Biocompatibility Evaluated by in vitro and in vivo Minipig Model. J. Biomed. Mater. Res. B 2017, 105, 647–657. [Google Scholar] [CrossRef]
- Johansson, P.; Jimbo, R.; Naito, Y.; Kjellin, P.; Currie, F.; Wennerberg, A. Polyether Ether Ketone Implants Achieve Increased Bone Fusion when Coated with Nano-Sized Hydroxyapatite: A Histomorphometric Study in Rabbit Bone. Int. J. Nanomed. 2016, 11, 1435–1442. [Google Scholar] [CrossRef]
- Huang, D.; Niu, L.; Wei, Y.; Guo, M.; Zuo, Y.; Zou, Q.; Hu, Y.; Chen, W.; Li, Y. Interfacial and Biological Properties of the Gradient Coating on Polyamide Substrate for Bone Substitute. J. R. Soc. Interface 2014, 11, 99. [Google Scholar] [CrossRef]
- Riau, A.K.; Lwin, N.C.; Gelfand, L.; Hu, H.; Liedberg, B.; Chodosh, J.; Venkatraman, S.S.; Mehta, J.S. Surface Modification of Corneal Prosthesis with Nano-Hydroxyapatite to Enhance in vivo Biointegration. Acta Biomater. 2020, 107, 299–312. [Google Scholar] [CrossRef]
- Wang, L.; Jeong, K.J.; Chiang, H.H.; Zurakowski, D.; Behlau, I.; Chodosh, J.; Dohlman, C.H.; Langer, R.; Kohane, D.S. Hydroxyapatite for Keratoprosthesis Biointegration. Invest. Ophthalmol. Vis. Sci. 2011, 52, 7392–7399. [Google Scholar] [CrossRef]
- Chanchareonsook, N.; Tideman, H.; Feinberg, S.E.; Hollister, S.J.; Jongpaiboonkit, L.; Kin, L.; Jansen, J.A. Subcutaneous Tissue Response to Titanium, Poly(ε-Caprolactone), and Carbonate-Substituted Hydroxyapatite-Coated Poly(ε-Caprolactone) Plates: A Rabbit Study. J. Biomed. Mater. Res. A 2013, 101, 2258–2266. [Google Scholar] [CrossRef]
- Høgsbro, M.; Agger, A.; Johansen, L.V. Bone Anchored Hearing Implant Surgery: 1 Year Follow-Up Data Shows No Effect of Hydroxyapatite Coating on Soft Tissue Reaction After Loading at 1 Week. Otol. Neurotol. 2017, 38, e152–e158. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward 5′-3′ Primer | Reverse 5′-3′ Primer |
|---|---|---|
| ACTB | GGCCCAGAGCAAGAGAGGTATCC | ACGCACGATTTCCCTCTCAGC |
| Col I | GCGAAGGCAACAGTCGCT | CTTGGTGGTTTTGTATTCGATGAC |
| BGLAP | CTCACAGATGCCAAGCCCA | CCAAGGTAGCGCCGGAGTCT |
| Runx2 | AAATGCCTCCGCTGTTATGAA | GCTCCGGCCCACAAATCT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uskoković, V.; Wu, V.M. When Nothing Turns Itself Inside out and Becomes Something: Coating Poly(Lactic-Co-Glycolic Acid) Spheres with Hydroxyapatite Nanoparticles vs. the Other Way Around. J. Funct. Biomater. 2022, 13, 102. https://doi.org/10.3390/jfb13030102
Uskoković V, Wu VM. When Nothing Turns Itself Inside out and Becomes Something: Coating Poly(Lactic-Co-Glycolic Acid) Spheres with Hydroxyapatite Nanoparticles vs. the Other Way Around. Journal of Functional Biomaterials. 2022; 13(3):102. https://doi.org/10.3390/jfb13030102
Chicago/Turabian StyleUskoković, Vuk, and Victoria M. Wu. 2022. "When Nothing Turns Itself Inside out and Becomes Something: Coating Poly(Lactic-Co-Glycolic Acid) Spheres with Hydroxyapatite Nanoparticles vs. the Other Way Around" Journal of Functional Biomaterials 13, no. 3: 102. https://doi.org/10.3390/jfb13030102
APA StyleUskoković, V., & Wu, V. M. (2022). When Nothing Turns Itself Inside out and Becomes Something: Coating Poly(Lactic-Co-Glycolic Acid) Spheres with Hydroxyapatite Nanoparticles vs. the Other Way Around. Journal of Functional Biomaterials, 13(3), 102. https://doi.org/10.3390/jfb13030102