
Density Functional Theory Study of Metal and Metal-Oxide Nucleation and Growth on the Anatase TiO2(101) Surface


Abstract
:1. Introduction
2. Methods
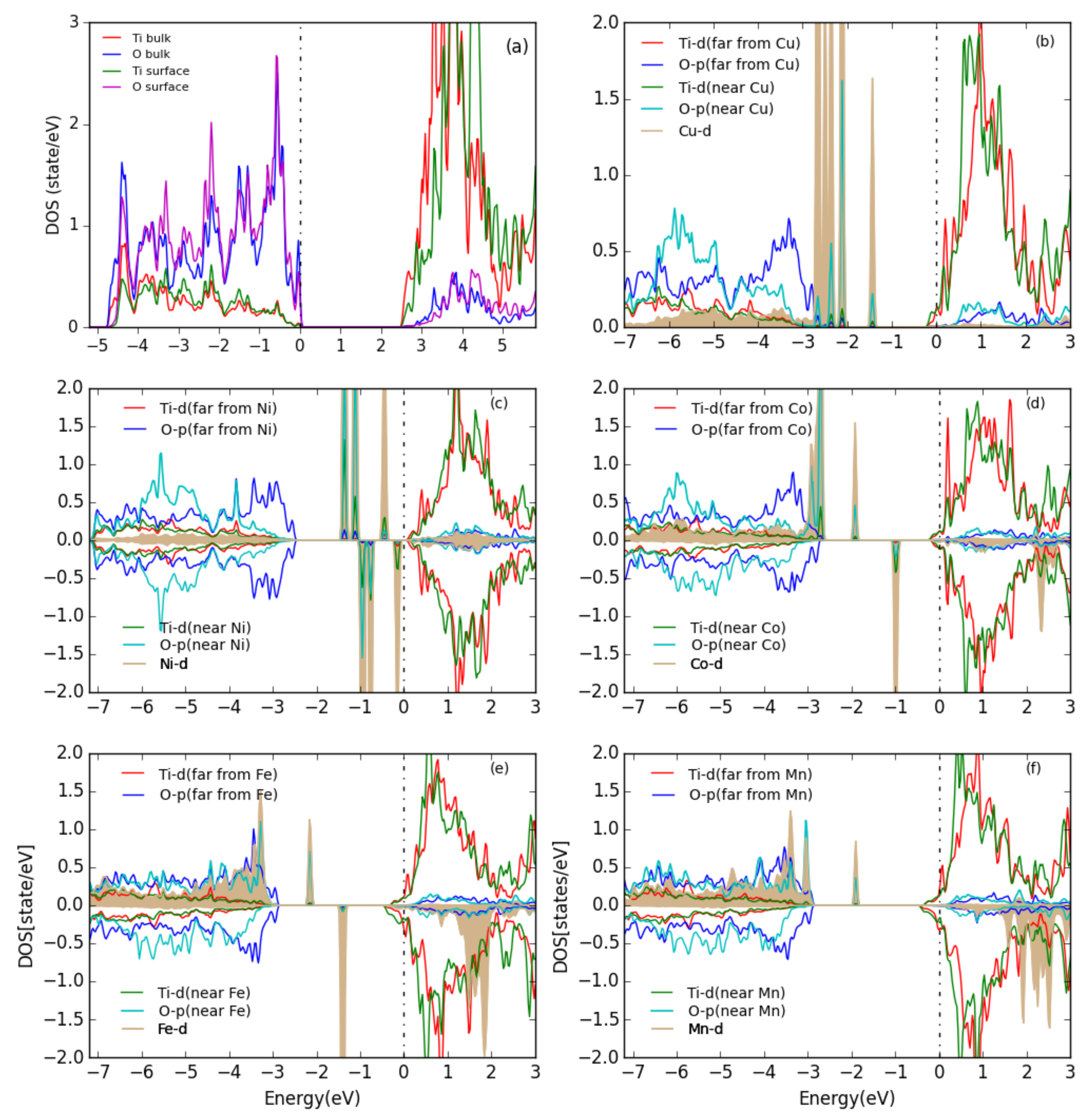
3. Results and Discussion
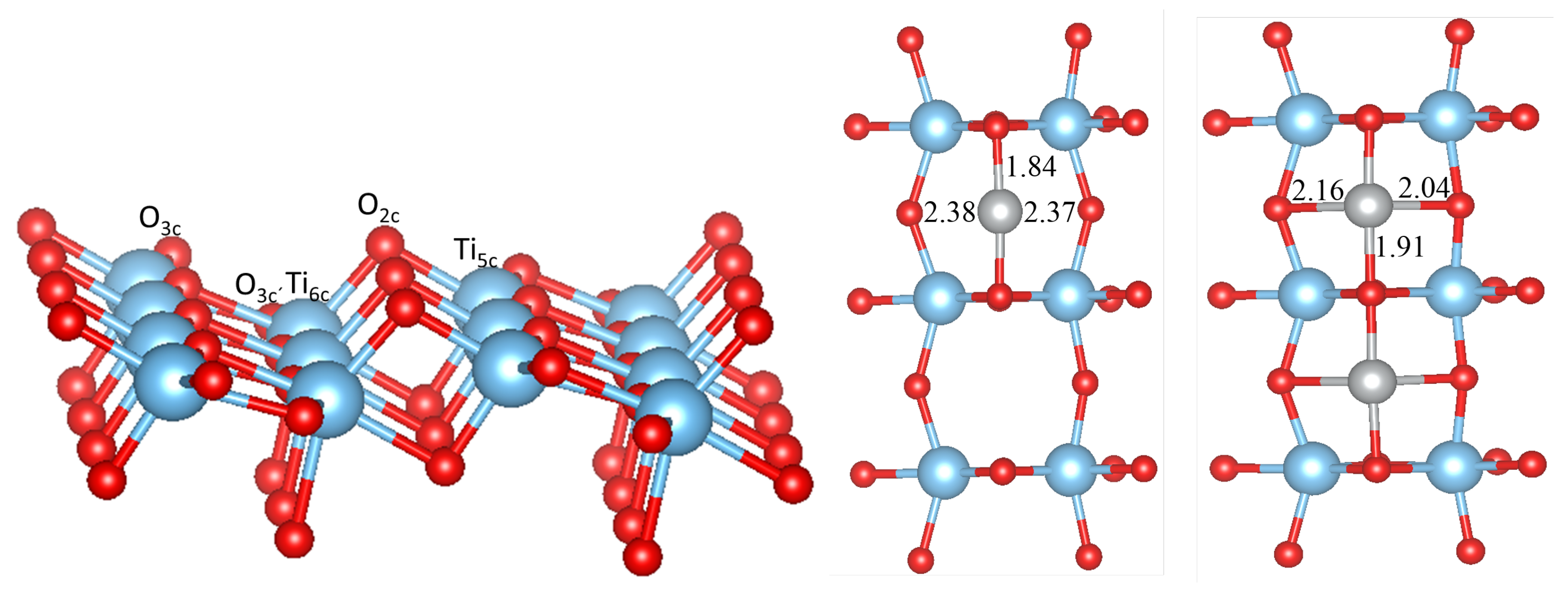
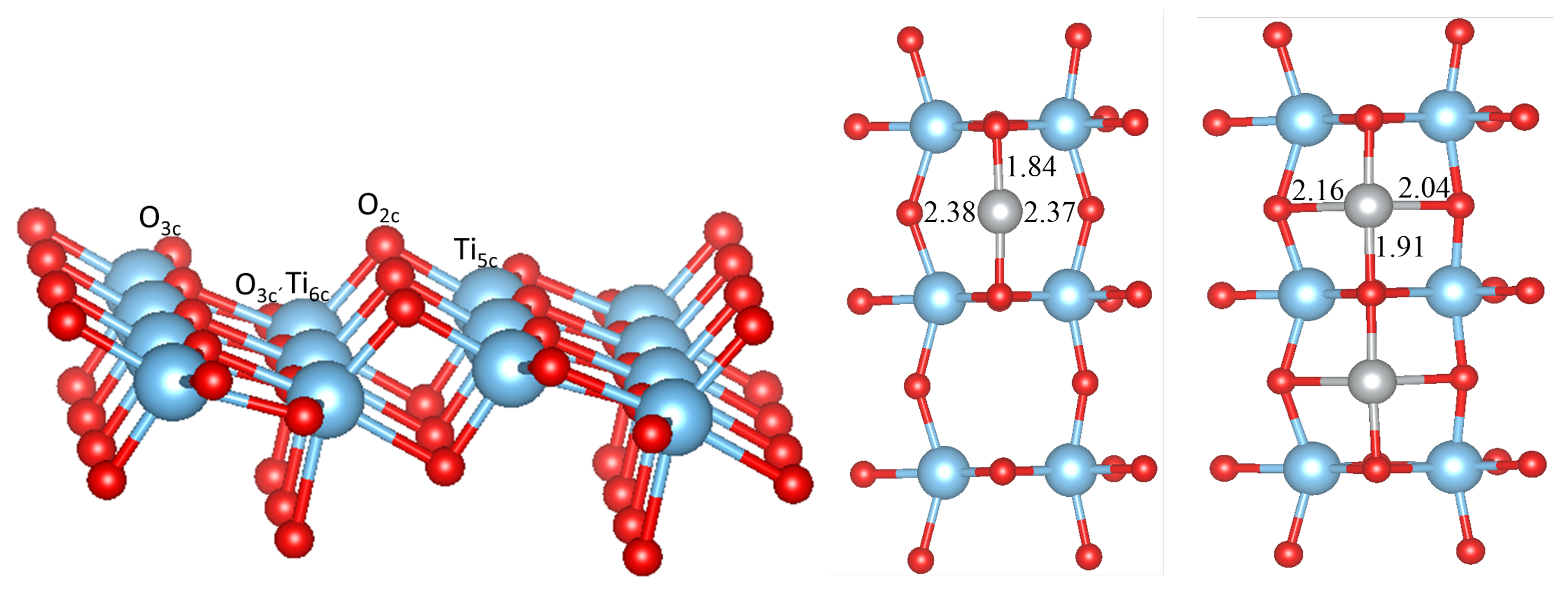
3.1. Perfect Anatase TiO2(101) Surface
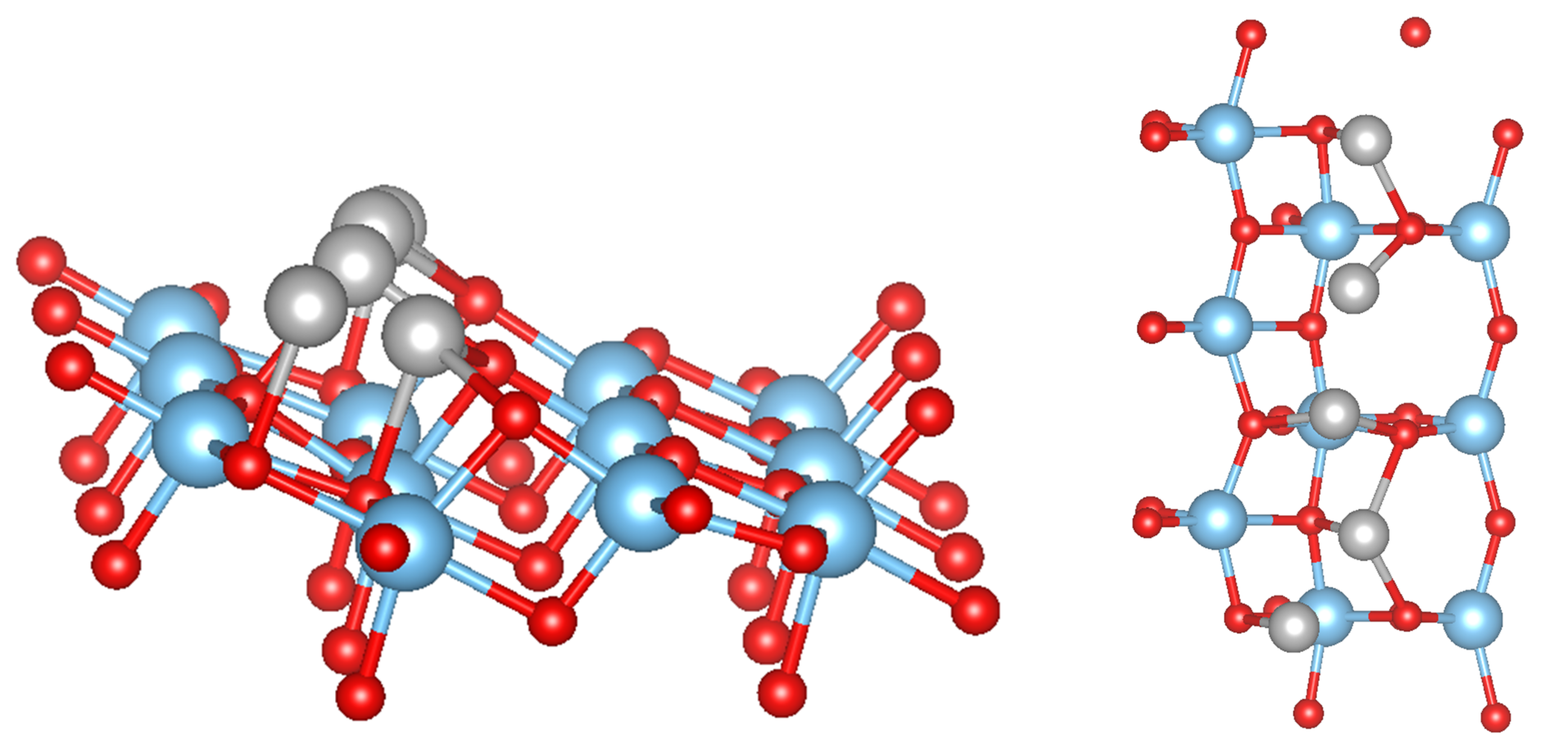
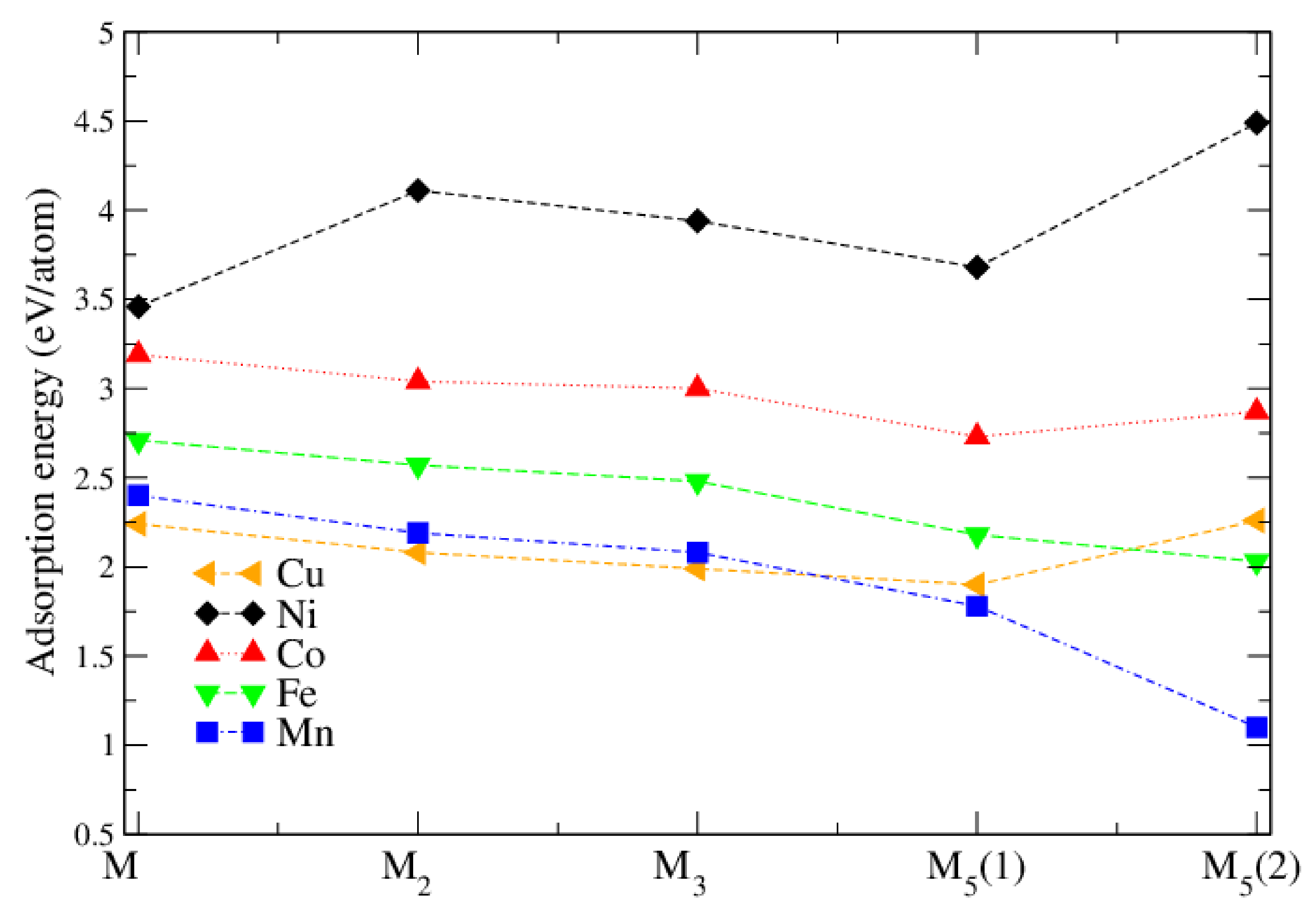
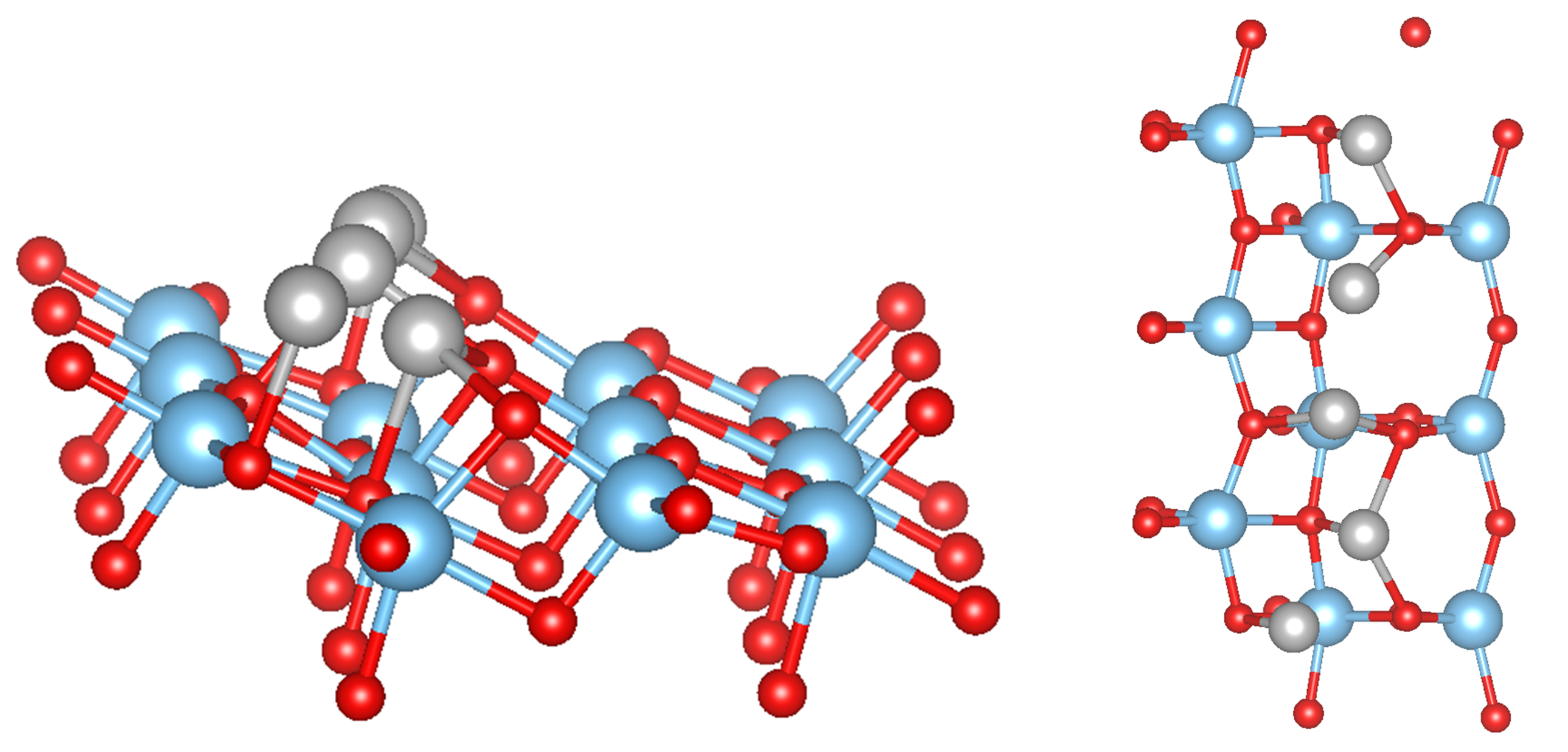
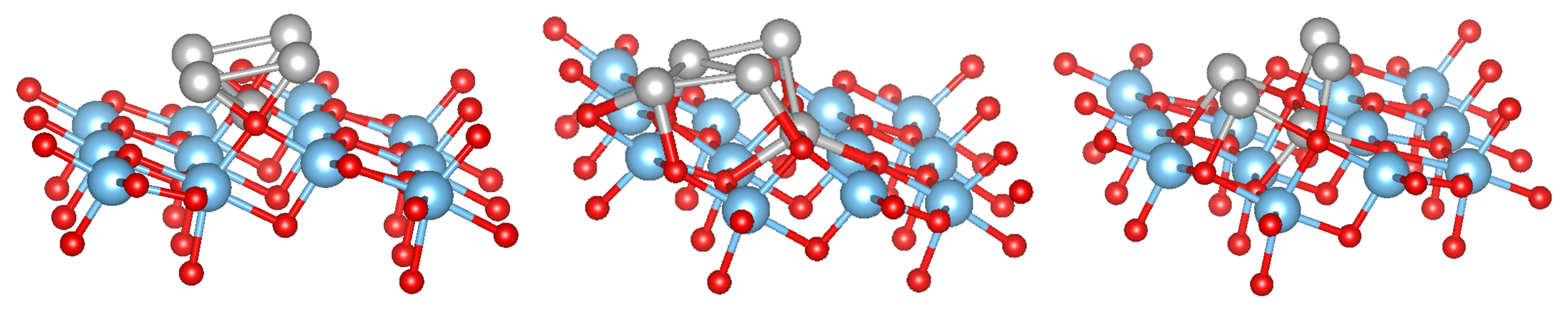
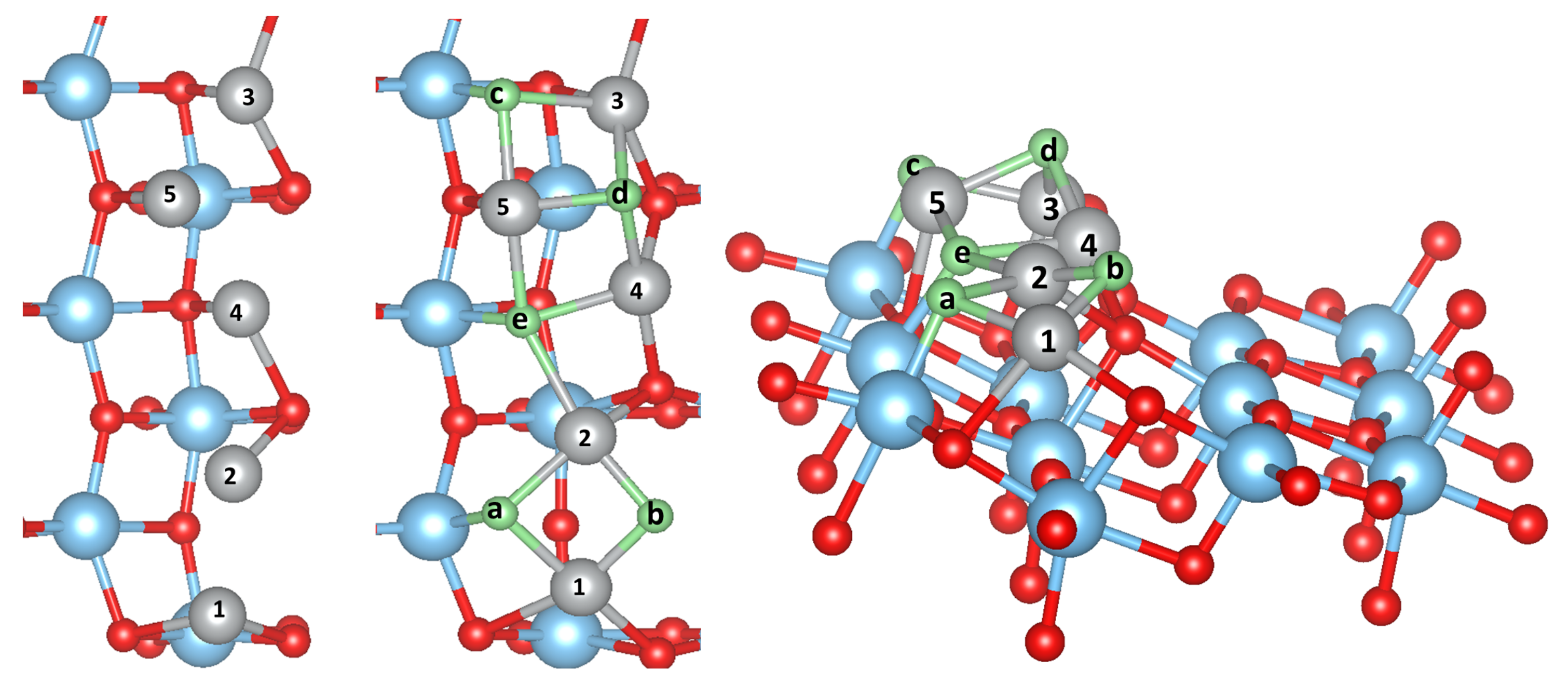
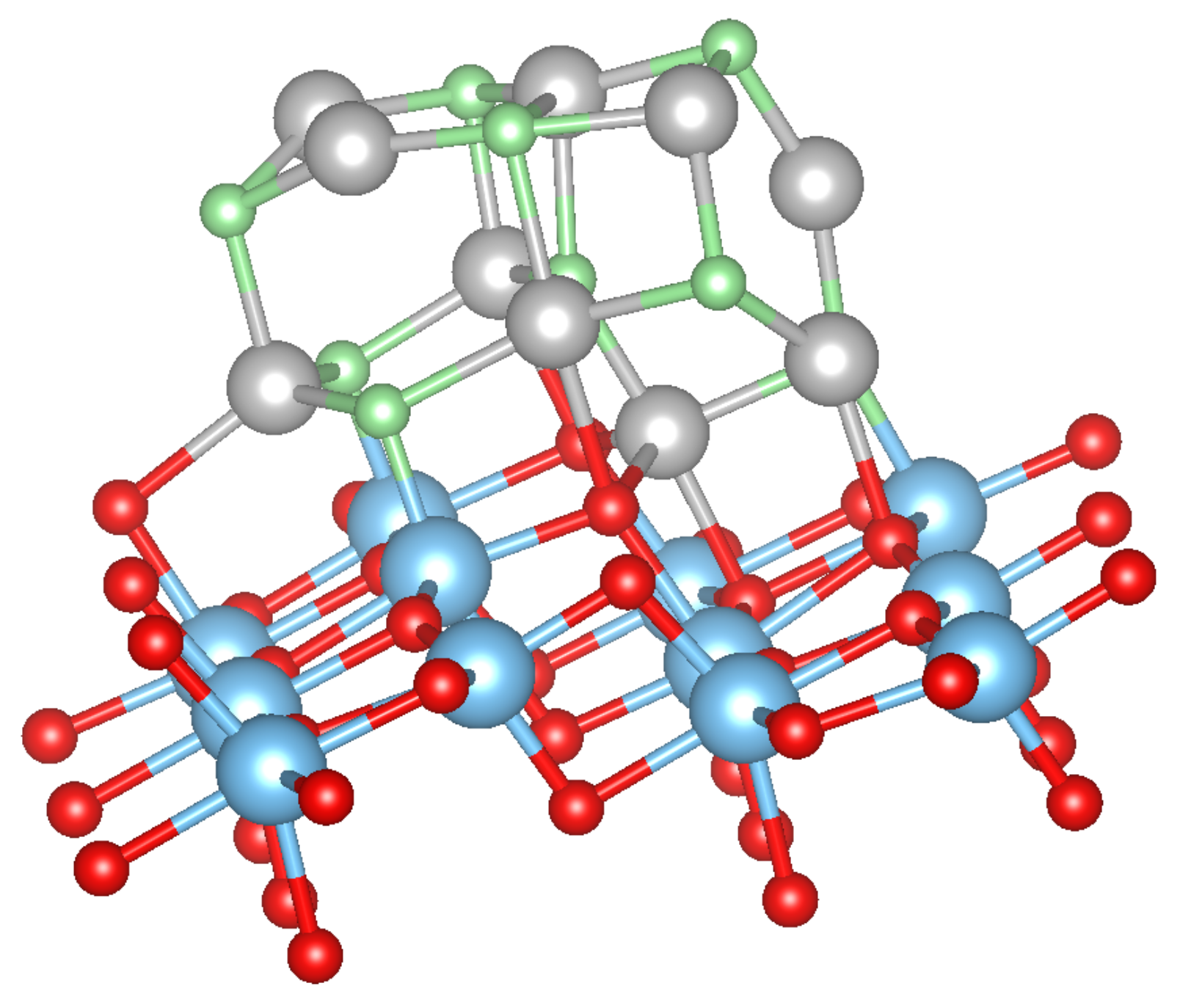
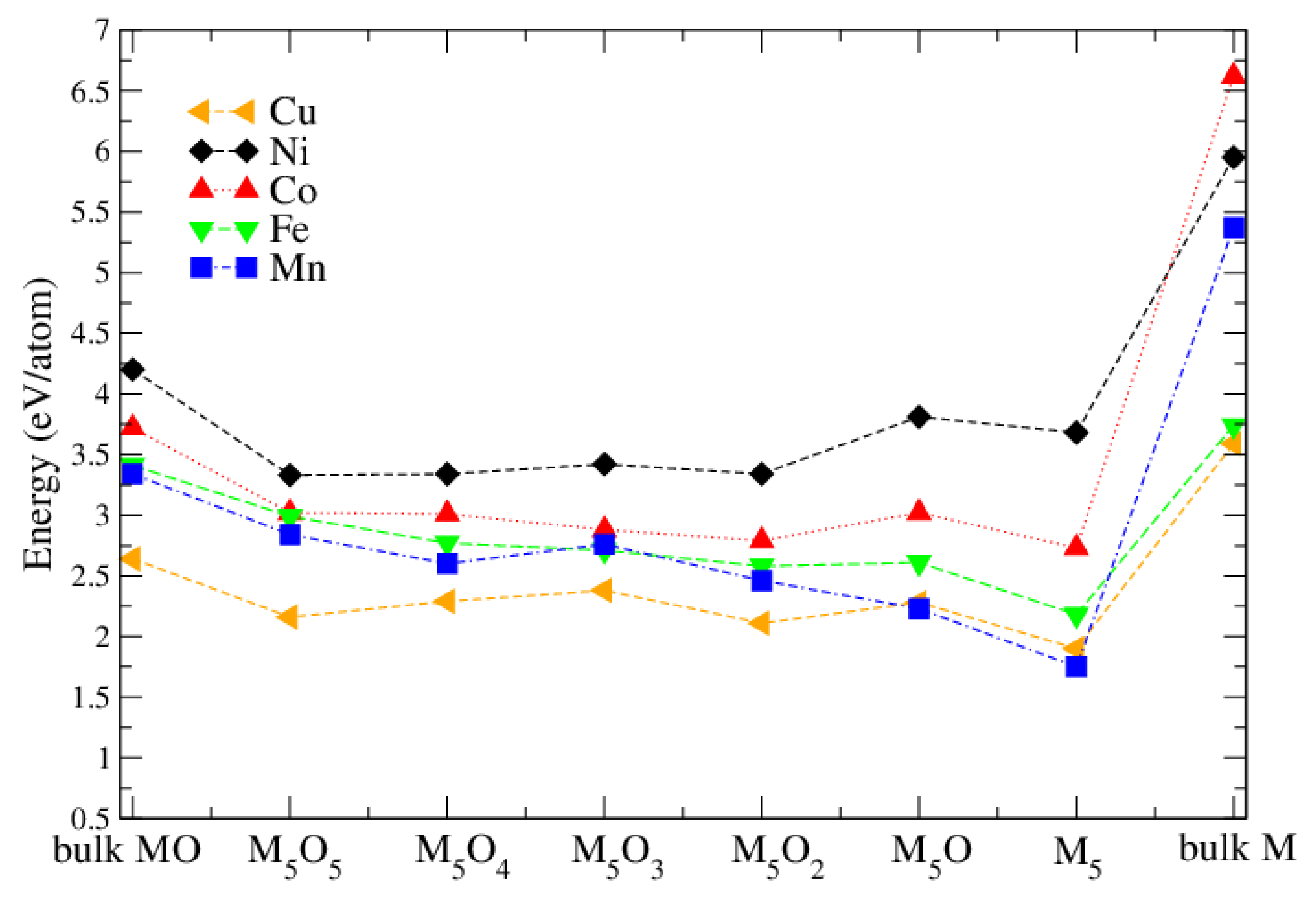
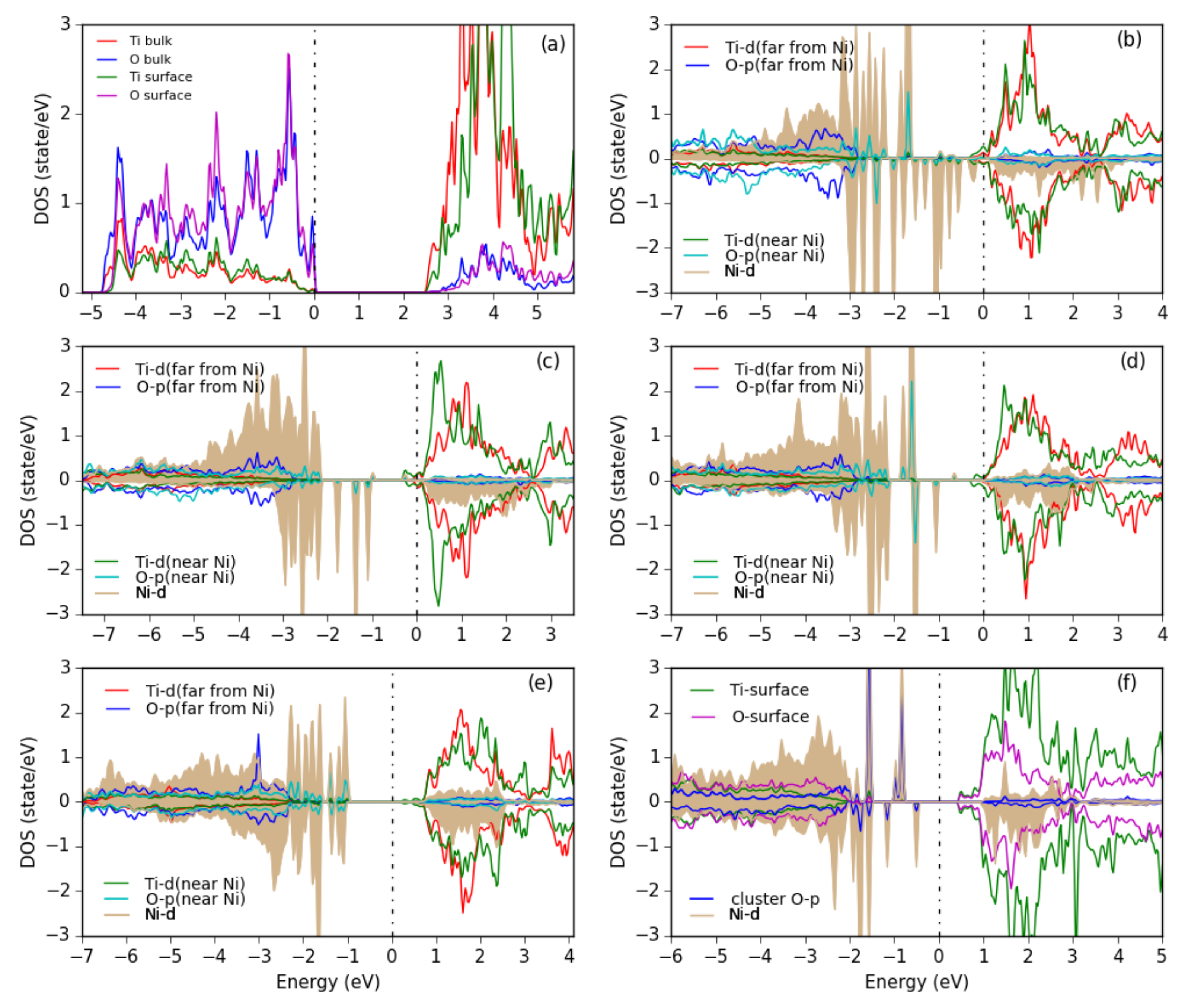
3.2. Pure Metal Clusters on the Anatase TiO2(101) Surface
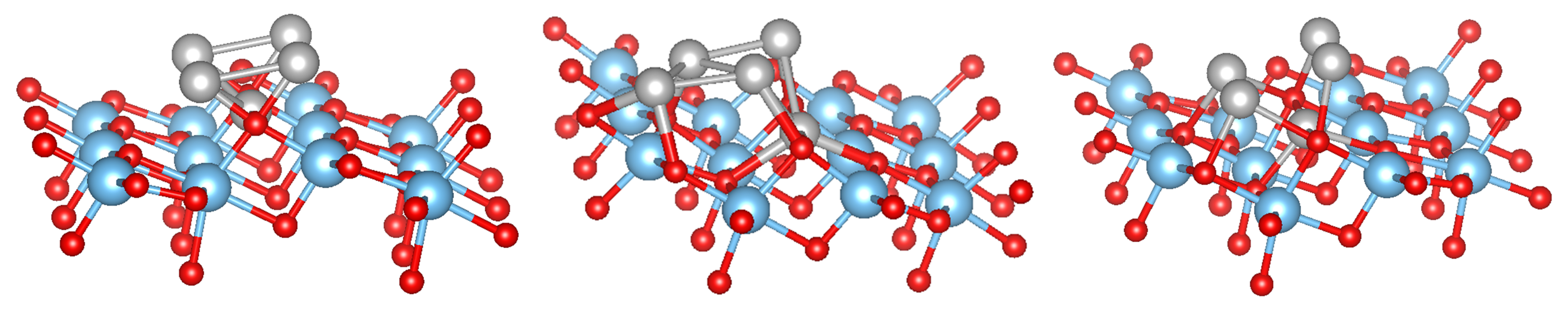
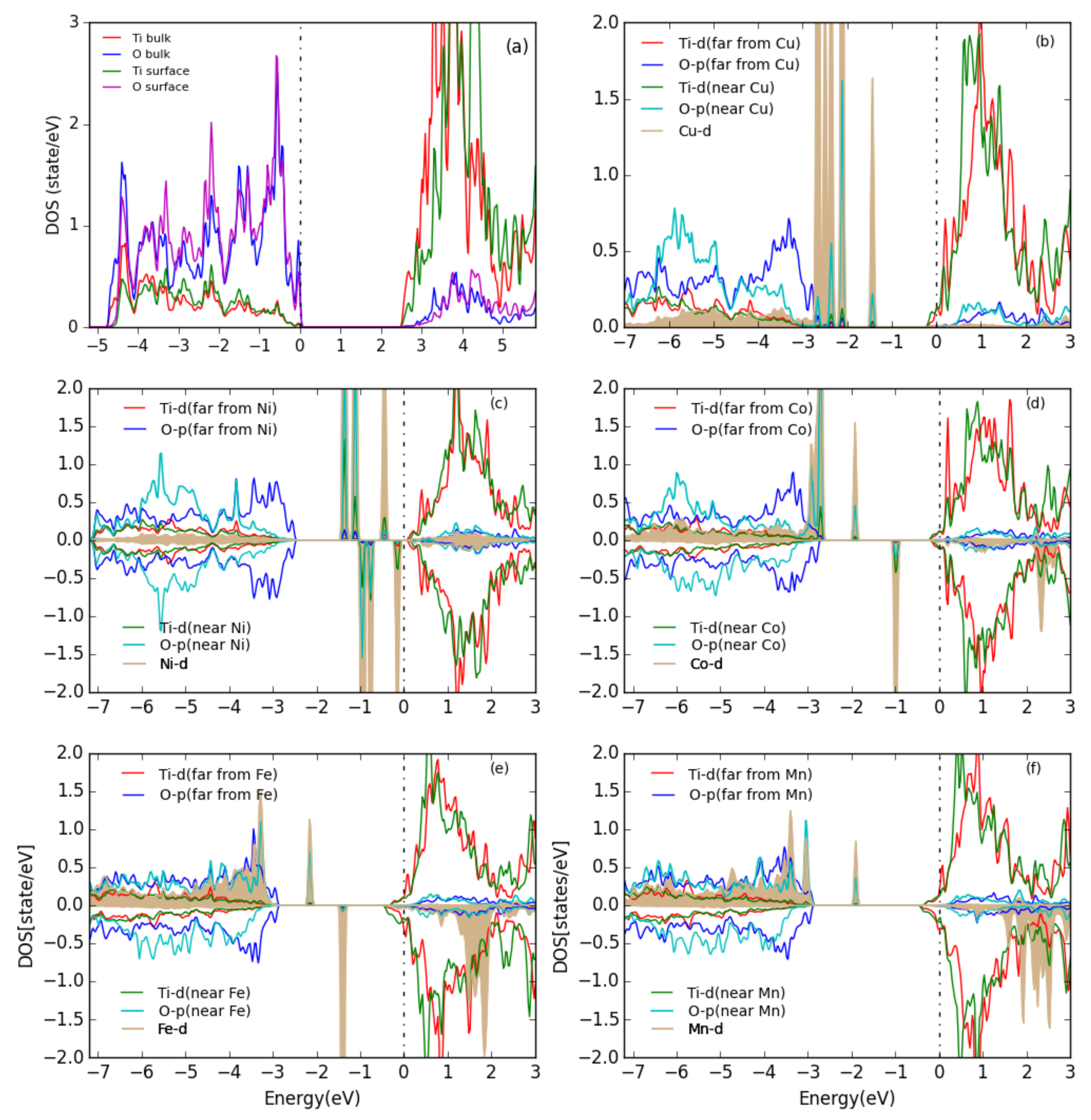
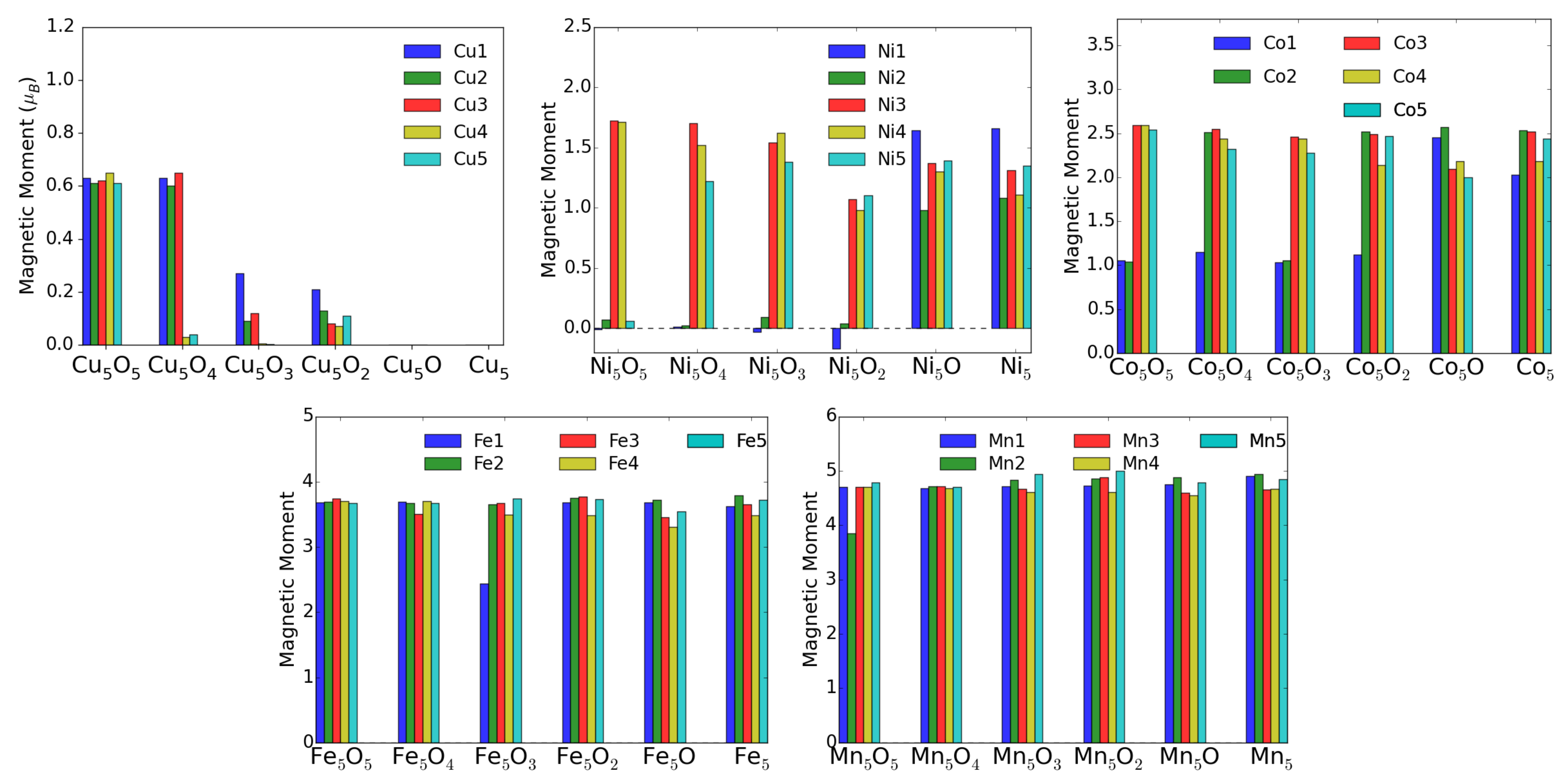
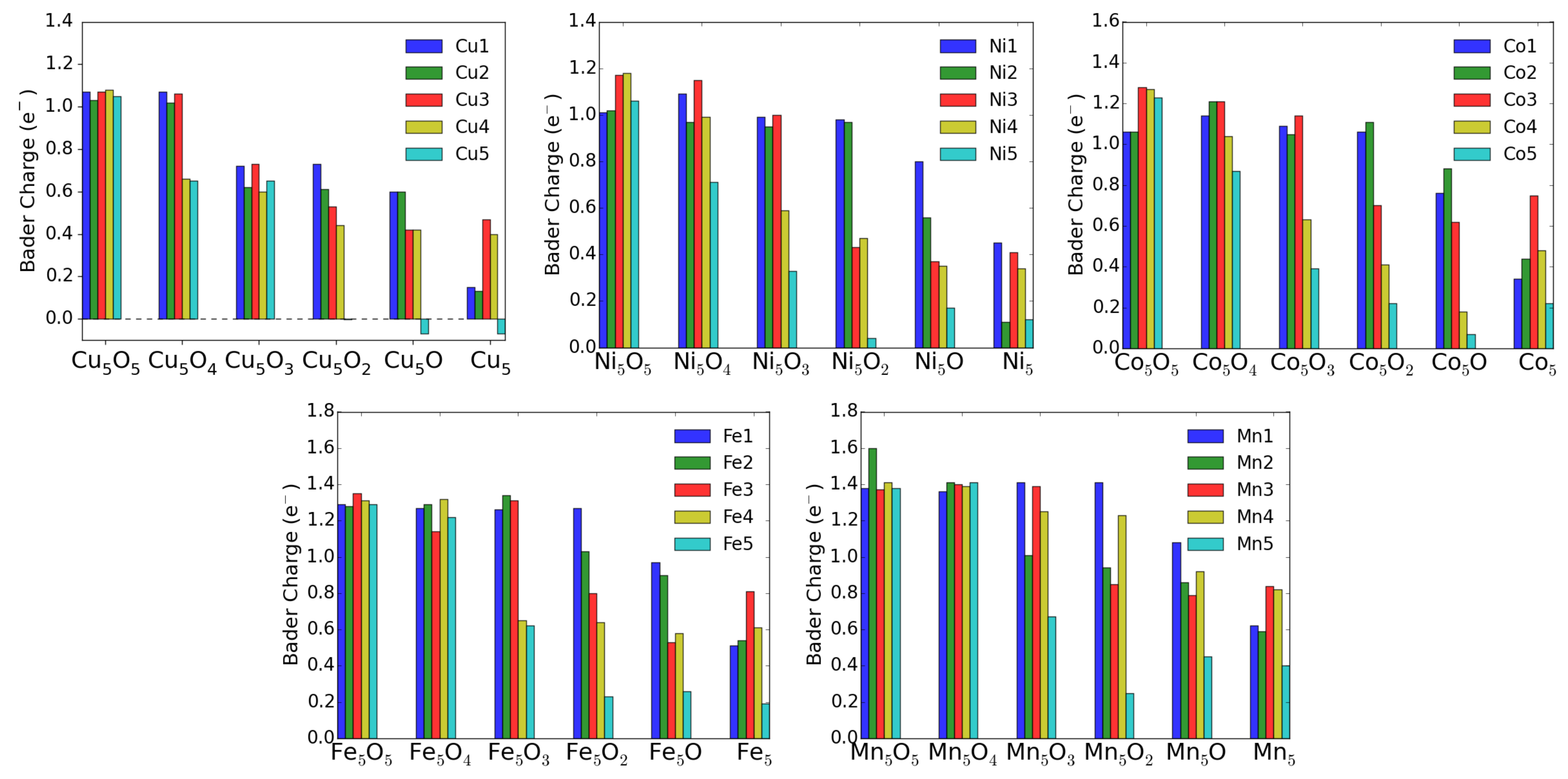
3.3. MO on the Anatase TiO2(101) Surface
4. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Diebold, U. The surface science of titanium dioxide. Surf. Sci. Rep. 2003, 48, 53–229. [Google Scholar] [CrossRef]
- Khan, M.M.; Pradhan, D.; Sohn, Y. Nanocomposites for Visible Light-Induced Photocatalysis; Springer: Cham, Switzerland, 2017; Volume 101. [Google Scholar]
- Ameta, R.; Ameta, S.C. Photocatalysis: Principles and Applications; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Ibhadon, A.O.; Fitzpatrick, P. Heterogeneous photocatalysis: Recent advances and applications. Catalysts 2013, 3, 189–218. [Google Scholar] [CrossRef] [Green Version]
- Ni, M.; Leung, M.K.; Leung, D.Y.; Sumathy, K. A review and recent developments in photocatalytic water-splitting using TiO2 for hydrogen production. Renew. Sustain. Energy Rev. 2007, 11, 401–425. [Google Scholar] [CrossRef]
- Jafari, T.; Moharreri, E.; Amin, A.S.; Miao, R.; Song, W.; Suib, S.L. Photocatalytic water splitting—The untamed dream: A review of recent advances. Molecules 2016, 21, 900. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhao, H.; Andino, J.M.; Li, Y. Photocatalytic CO2 reduction with H2O on TiO2 nanocrystals: Comparison of anatase, rutile, and brookite polymorphs and exploration of surface chemistry. ACS Catal. 2012, 2, 1817–1828. [Google Scholar] [CrossRef]
- Xu, M.; Gao, Y.; Moreno, E.M.; Kunst, M.; Muhler, M.; Wang, Y.; Idriss, H.; Wöll, C. Photocatalytic activity of bulk TiO2 anatase and rutile single crystals using infrared absorption spectroscopy. Phys. Rev. Lett. 2011, 106, 138302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, J.S.; Popovic, J.; Haselmann, G.M.; Nandan, S.P.; Wang, J.; Giesriegl, A.; Cherevan, A.S.; Eder, D. Immobilization of Co, Mn, Ni and Fe oxide co-catalysts on TiO2 for photocatalytic water splitting reactions. J. Mater. Chem. A 2019, 7, 18568–18579. [Google Scholar] [CrossRef] [Green Version]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef] [Green Version]
- Hebenstreit, W.; Ruzycki, N.; Herman, G.S.; Gao, Y.; Diebold, U. Scanning tunneling microscopy investigation of the TiO2 anatase (101) surface. Phys. Rev. B 2000, 62, R16334–R16336. [Google Scholar] [CrossRef]
- Alghannam, A.; Muhich, C.L.; Musgrave, C.B. Adatom surface diffusion of catalytic metals on the anatase TiO2(101) surface. Phys. Chem. Chem. Phys. 2017, 19, 4541–4552. [Google Scholar] [CrossRef]
- Wang, Y.; Su, Y.; Zhu, M.; Kang, L. Ni cluster nucleation and growth on the anatase TiO2(101) surface: A density functional theory study. RSC Adv. 2015, 5, 16582–16591. [Google Scholar] [CrossRef]
- Iyemperumal, S.K.; Fenton, T.G.; Gillingham, S.L.; Carl, A.D.; Grimm, R.L.; Li, G.; Deskins, N.A. The stability and oxidation of supported atomic-size Cu catalysts in reactive environments. J. Chem. Phys. 2019, 151, 054702. [Google Scholar] [CrossRef]
- Sharma, P.K.; Cortes, M.A.; Hamilton, J.; Han, Y. Surface modification of TiO2 with copper clusters for band gap narrowing. Catal. Today 2019, 321, 9–17. [Google Scholar] [CrossRef]
- Zhou, X.; Dong, H. A Theoretical perspective on charge separation and transfer in metal oxide photocatalysts for water splitting. ChemCatChem 2019, 11, 3688–3715. [Google Scholar] [CrossRef]
- Singh, D.J.; Nordström, L. Planewaves, Pseudopotentials and the LAPW Method, 2nd ed.; Springer: Berlin, Germany, 2006. [Google Scholar]
- Karsai, F.; Tran, F.; Blaha, P. On the importance of local orbitals using second energy derivatives for d and f electrons. Comput. Phys. Commun. 2017, 220, 230–238. [Google Scholar] [CrossRef]
- Blaha, P.; Schwarz, K.; Madsen, G.K.H.; Kvasnicka, D.; Luitz, J.; Laskowski, R.; Tran, F.; Marks, L.D. WIEN2k: An Augmented Plane Wave plus Local Orbitals Program for Calculating Crystal Properties; Vienna University of Technology: Vienna, Austria, 2018. [Google Scholar]
- Blaha, P.; Schwarz, K.; Tran, F.; Laskowski, R.; Madsen, G.K.H.; Marks, L.D. WIEN2k: An APW+ lo program for calculating the properties of solids. J. Chem. Phys. 2020, 152, 074101. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [Green Version]
- Schossberger, F. Über die Umwandlung des Titandioxyds. Zeitschrift Kristallographie 1942, 104, 358–374. [Google Scholar] [CrossRef]
- Anisimov, V.I.; Zaanen, J.; Andersen, O.K. Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys. Rev. B 1991, 44, 943–954. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MO Bulk | MO at the Surface | M-O at the Surface | |
|---|---|---|---|
| TiO-Cu | (CuO)1.97/(CuO)1.86 | 1.87(2) | 2.33, 2.47 |
| TiO-Cu | (1.88, 1.89); (1.88, 1.89) | (2.34, 2.52); (2.34, 2.52) | |
| TiO-Cu | 1.90(2); 1.90(2); 1.90(2) | (2.28, 2.46); (2.28, 2.46); (2.29, 2.46) | |
| TiO-Cu(1) | 1.97; 2.11; (2.09, 2.10); (2.04, 2.15); 2.83 | (2.81, 2.71); (2.31, 2.90); 2.30; 2.42; - | |
| TiO-Cu(2) | 1.86; -; -; 2.94; 1.86 | -; -; -; 2.00; 2.75 | |
| TiO-Ni | 2.08 | 1.84(2) | 2.37, 2.38 |
| TiO-Ni | (1.91, 1.92); (1.91, 1.92) | (2.04, 2.16); (2.04, 2.16) | |
| TiO-Ni | 1.89(2); 1.89(2); 1.89(2) | (2.02, 2.31); (2.02, 2.31); (2.02, 2.31) | |
| TiO-Ni(1) | 2.02; 1.99; (2.04, 2.22); (2.13, 2.46); 2.86 | 2.04; 2.75; 2.10; 2.04; 2.05 | |
| TiO-Ni(2) | (1.96, 1.94); 2.13; 1.98; -; 2.07 | (2.04, 2.16); -; -; 2.01; 2.07 | |
| TiO-Co | 2.13 | 1.89(2) | 2.03, 2.71 |
| TiO-Co | 1.97(2); (1.88, 1.89) | (2.03, 2.04); (2.03, 2.76) | |
| TiO-Co | (1.99, 2.00); (2.03, 1.91); (1.90, 2.02) | (2.05, 2.09); (2.04, 2.12); (2.04, 2.29) | |
| TiO-Co(1) | 2.08; 2.30; (2.06, 2.09); (2.03, 2.06); 2.06 | (2.15, 2.30); 2.04; 2.13; 2.15; 2.86 | |
| TiO-Co(2) | 2.02; -; 1.91; 2.90; 2.07 | 2.09; -; -; 1.96; 2.19 | |
| TiO-Fe | 2.16 | 1.89(2) | 2.08, 2.23 |
| TiO-Fe | (1.88, 1.91); (1.88, 1.91) | (2.10, 2.11); (2.10, 2.11) | |
| TiO-Fe | 1.88(2); 1.88(2); 1.88(2) | (2.11, 2.14); (2.11, 2.14); (2.11, 2.14) | |
| TiO-Fe(1) | 2.03; 2.09; (2.00, 2.09); (2.05, 2.15); 2.98 | (2.21, 2.25); 2.09; 2.14; 2.06; 2.02 | |
| TiO-Fe(2) | (2.06, 2.05); 2.04; 2.04; 2.22; 2.25 | (2.14, 2.04); -; -; 2.11; 2.08 | |
| TiO-Mn | 2.22 | 1.93(2) | 2.14, 2.30 |
| TiO-Mn | 1.94(2); 1.94(2) | (2.11, 2.28); (2.11, 2.28) | |
| TiO-Mn | (1.91, 1.92); (1.92, 1.93); (1.92, 1.93) | (2.16, 2.30); (2.16, 2.27); (2.16, 2.27) | |
| TiO-Mn(1) | 2.17; 2.19; (2.03, 2.08); (2.10, 2.03); 3.27 | 2.12; 2.13; 2.16; 2.16; 2.07 | |
| TiO-Mn(2) | (2.02, 2.02); 2.08; 2.08; 2.17; 2.18 | (2.03, 2.04); -; -; 2.09 | |
| TiO-Mn(2a) | (2.14, 2.15); 2.09; 2.09; 2.07; 2.07 | 2.15; -; -; 2.15; 2.16 |
| Solid | Magnetic Moment | |
|---|---|---|
| TiO-Cu | 2.24 | 0 |
| TiO-Cu | 2.08 | 0 |
| TiO-Cu | 1.99 | 0 |
| TiO-Cu(1) | 1.90 | 0 |
| TiO-Cu(2) | 2.26 | 0 |
| TiO-Ni | 3.46 | 0.25 |
| TiO-Ni | 4.11 | 0.97(2) |
| TiO-Ni | 3.94 | 0.98(3) |
| TiO-Ni(1) | 3.68 | 1.66; 1.08; 1.31; 1.11; 1.35 |
| TiO-Ni(2) | 4.49 | −0.97; 1.22; 0.74; −1.10; 0.87 |
| TiO-Co | 3.19 | 2.12 |
| TiO-Co | 3.04 | 2.12; 2.11 |
| TiO-Co | 3.00 | 2.15; 2.28; 2.28 |
| TiO-Co(1) | 2.73 | 2.03; 2.53; 2.52; 2.18; 2.44 |
| TiO-Co(2) | 2.87 | 2.34; 2.00; 2.66; 1.96; 2.04 |
| TiO-Fe | 2.71 | 3.61 |
| TiO-Fe | 2.57 | 3.51; 3.50 |
| TiO-Fe | 2.48 | 3.52(3) |
| TiO-Fe(1) | 2.18 | 3.62; 3.79; 3.64; 3.48; 3.71 |
| TiO-Fe(2) | 2.03 | 3.00; 3.75; 3.72; 3.58; 3.57 |
| TiO-Mn | 2.40 | 4.66 |
| TiO-Mn | 2.19 | 4.66(2) |
| TiO-Mn | 2.08 | 4.61; 4.61; 4.63 |
| TiO-Mn(1) | 1.78 | 4.90; 4.94; 4.66; 4.66; 4.83 |
| TiO-Mn(2) | 1.10 | 0.45; 5.00; 4.99; 4.57; 4.57 |
| TiO-Mn(2a) | 1.44 | 4.90; −5.04; 5.03; −4.73; −4.73 |
| Cluster | |
|---|---|
| NiO(1) | 3.55 |
| NiO(2) | 3.14 |
| NiO(3) | 3.19 |
| NiO(4) | 3.14 |
| NiO(1) | 3.07 |
| NiO(2) | 2.59 |
| NiO(3) | 3.58 |
| NiO(1) | 3.32 |
| NiO(2) | 3.17 |
| NiO(3) | 2.75 |
| NiO(4) | 3.51 |
| NiO(1) | 3.42 |
| NiO(2) | 2.87 |
| NiO | 3.35 |
| NiO | 3.63 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalantari, L.; Tran, F.; Blaha, P. Density Functional Theory Study of Metal and Metal-Oxide Nucleation and Growth on the Anatase TiO2(101) Surface. Computation 2021, 9, 125. https://doi.org/10.3390/computation9110125
Kalantari L, Tran F, Blaha P. Density Functional Theory Study of Metal and Metal-Oxide Nucleation and Growth on the Anatase TiO2(101) Surface. Computation. 2021; 9(11):125. https://doi.org/10.3390/computation9110125
Chicago/Turabian StyleKalantari, Leila, Fabien Tran, and Peter Blaha. 2021. "Density Functional Theory Study of Metal and Metal-Oxide Nucleation and Growth on the Anatase TiO2(101) Surface" Computation 9, no. 11: 125. https://doi.org/10.3390/computation9110125
APA StyleKalantari, L., Tran, F., & Blaha, P. (2021). Density Functional Theory Study of Metal and Metal-Oxide Nucleation and Growth on the Anatase TiO2(101) Surface. Computation, 9(11), 125. https://doi.org/10.3390/computation9110125