Integrative Analysis and Machine Learning Based Characterization of Single Circulating Tumor Cells

 , , ,
, , ,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Description of Datasets
2.2. Data Pre-Processing
2.3. Construction of Epithelial and Mesenchymal Signatures and E:M Score
2.4. Simulation of E-M Continuum
2.5. Classification of Cancer and Blood Transcriptomes
2.6. Sample Collection
2.7. CTC Enrichment
2.8. Immunofluorescence Suspension Staining
2.9. Integrated Fluidic Circuit (IFC) Operation
2.10. mRNA-Seq Library Preparation and Sequencing
2.11. Reference Component Analysis of CTCs and PBMCs
2.12. Data and Code Availability
3. Results
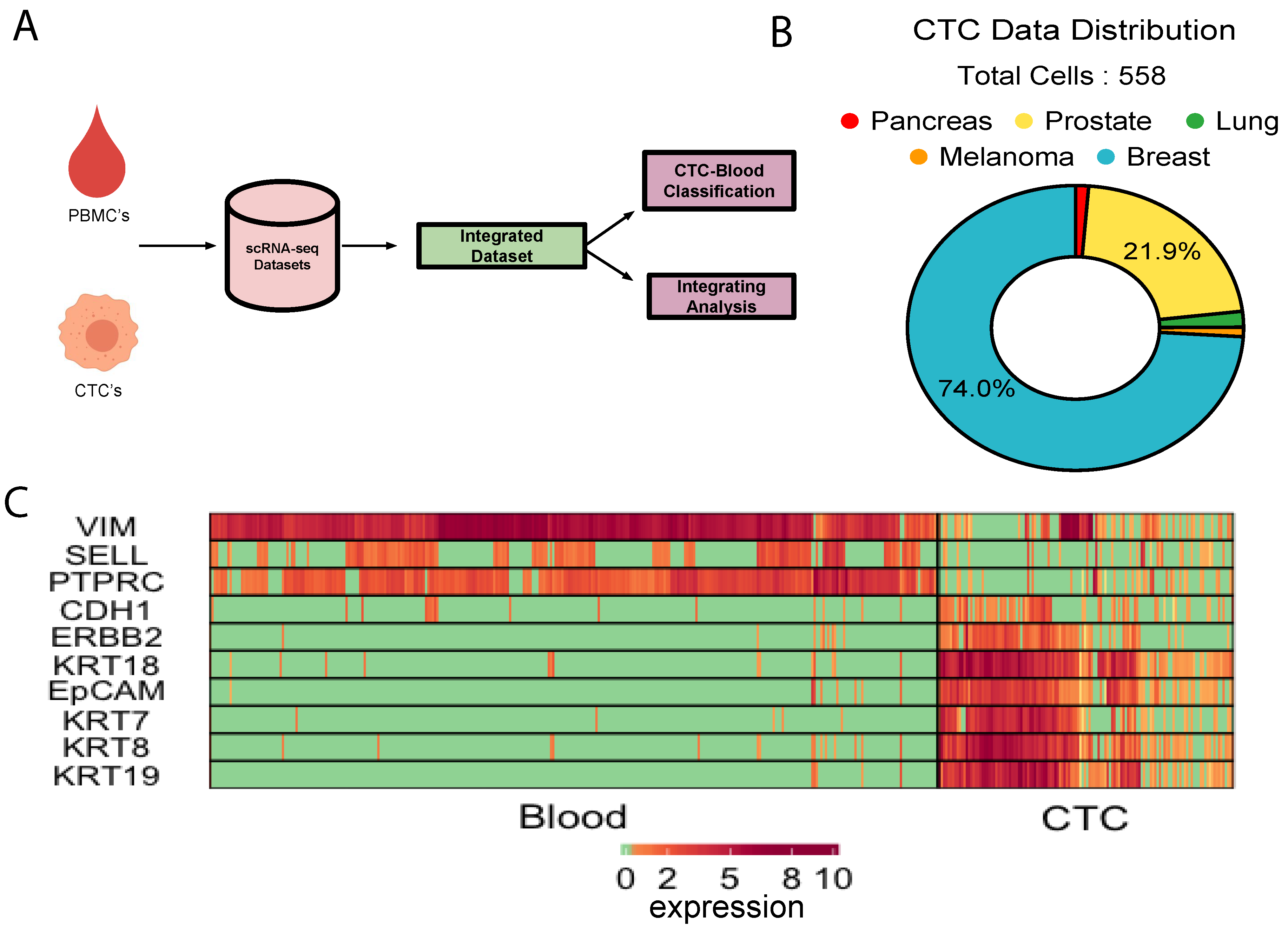
3.1. Integration of Single Cell Expression Datasets of Circulating Tumor Cells
3.2. Ubiquity of Epithelial-Mesenchymal Transition in Cancer Metastasis
3.3. Clear Patterns Observed in Expression Gradient of Immune Check-Point Inhibitor and Stemness Marker
3.4. CTC-PBMC Classification System
3.5. Identification of CTCs Captured Using Novel Label-Free Microfluidic Workflow
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Tian, T.; Shi, Y.; Liu, W.; Zou, Y.; Khajvand, T.; Wang, S.; Zhu, Z.; Yang, C. Enrichment and single-cell analysis of circulating tumor cells. Chem. Sci. 2017, 8, 1736–1751. [Google Scholar] [CrossRef]
- Dive, C.; Brady, G. SnapShot: Circulating tumor cells. Cell 2017, 168, 742. [Google Scholar] [CrossRef] [PubMed]
- Andreopoulou, E.; Yang, L.Y.; Rangel, K.; Reuben, J.; Hsu, L.; Krishnamurthy, S.; Valero, V.; Fritsche, H.; Cristofanilli, M. Comparison of assay methods for detection of circulating tumor cells in metastatic breast cancer: AdnaGen AdnaTest BreastCancer Select/Detect™ versus Veridex CellSearch™ system. Int. J. Cancer 2012, 130, 1590–1597. [Google Scholar] [CrossRef] [PubMed]
- Mikolajczyk, S.D.; Millar, L.S.; Tsinberg, P.; Coutts, S.M.; Zomorrodi, M.; Pham, T.; Bischoff, F.Z.; Pircher, T.J. Detection of EpCAM-negative and cytokeratin-negative circulating tumor cells in peripheral blood. J. Oncol. 2011, 2011, 252361. [Google Scholar] [CrossRef]
- Miller, M.C.; Doyle, G.V.; Terstappen, L.W. Significance of circulating tumor cells detected by the CellSearch system in patients with metastatic breast colorectal and prostate cancer. J. Oncol. 2010, 2010, 617421. [Google Scholar] [CrossRef]
- Farace, F.; Massard, C.; Vimond, N.; Drusch, F.; Jacques, N.; Billiot, F.; Laplanche, A.; Chauchereau, A.; Lacroix, L.; Planchard, D.; et al. A direct comparison of CellSearch and ISET for circulating tumour-cell detection in patients with metastatic carcinomas. Br. J. Cancer 2011, 105, 847–853. [Google Scholar] [CrossRef]
- Wang, L.; Balasubramanian, P.; Chen, A.P.; Kummar, S.; Evrard, Y.A.; Kinders, R.J. Promise and limits of the CellSearch platform for evaluating pharmacodynamics in circulating tumor cells. Semin. Oncol. 2016, 43, 464–475. [Google Scholar] [CrossRef]
- Gabriel, M.T.; Calleja, L.R.; Chalopin, A.; Ory, B.; Heymann, D. Circulating tumor cells: A review of non–EpCAM-based approaches for cell enrichment and isolation. Clin. Chem. 2016, 62, 571–581. [Google Scholar] [CrossRef]
- Ferreira, M.M.; Ramani, V.C.; Jeffrey, S.S. Circulating tumor cell technologies. Mol. Oncol. 2016, 10, 374–394. [Google Scholar] [CrossRef]
- Cheng, Y.H.; Chen, Y.C.; Lin, E.; Brien, R.; Jung, S.; Chen, Y.T.; Lee, W.; Hao, Z.; Sahoo, S.; Kang, H.M.; et al. Hydro-Seq enables contamination-free high-throughput single-cell RNA-sequencing for circulating tumor cells. Nat. Commun. 2019, 10, 2163. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.X.; Bai, F. Single-cell analyses of circulating tumor cells. Cancer Biol. Med. 2015, 12, 184. [Google Scholar]
- Sarioglu, A.F.; Aceto, N.; Kojic, N.; Donaldson, M.C.; Zeinali, M.; Hamza, B.; Engstrom, A.; Zhu, H.; Sundaresan, T.K.; Miyamoto, D.T.; et al. A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat. Methods 2015, 12, 685. [Google Scholar] [CrossRef] [PubMed]
- Warkiani, M.E.; Guan, G.; Luan, K.B.; Lee, W.C.; Bhagat, A.A.S.; Chaudhuri, P.K.; Tan, D.S.W.; Lim, W.T.; Lee, S.C.; Chen, P.C.; et al. Slanted spiral microfluidics for the ultra-fast, label-free isolation of circulating tumor cells. Lab A Chip 2014, 14, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Karabacak, N.M.; Spuhler, P.S.; Fachin, F.; Lim, E.J.; Pai, V.; Ozkumur, E.; Martel, J.M.; Kojic, N.; Smith, K.; Chen, P.i.; et al. Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nat. Protoc. 2014, 9, 694. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Mao, X.; Imrali, A.; Syed, F.; Mutsvangwa, K.; Berney, D.; Cathcart, P.; Hines, J.; Shamash, J.; Lu, Y.J. Optimization and evaluation of a novel size based circulating tumor cell isolation system. PLoS ONE 2015, 10, e0138032. [Google Scholar] [CrossRef] [PubMed]
- Warkiani, M.E.; Khoo, B.L.; Wu, L.; Tay, A.K.P.; Bhagat, A.A.S.; Han, J.; Lim, C.T. Ultra-fast, label-free isolation of circulating tumor cells from blood using spiral microfluidics. Nat. Protoc. 2016, 11, 134. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Ramalingam, N.; Fowler, B.; Szpankowski, L.; Leyrat, A.A.; Hukari, K.; Maung, M.T.; Yorza, W.; Norris, M.; Cesar, C.; Shuga, J.; et al. Fluidic logic used in a systems approach to enable integrated single-cell functional analysis. Front. Bioeng. Biotechnol. 2017, 4, 70. [Google Scholar] [CrossRef]
- Lin, E.; Cao, T.; Nagrath, S.; King, M.R. Circulating tumor cells: Diagnostic and therapeutic applications. Annu. Rev. Biomed. Eng. 2018, 20, 329–352. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Wittner, B.S.; Donaldson, M.C.; O’Keefe, R.; Engstrom, A.; Bersani, F.; Zheng, Y.; Comaills, V.; Niederhoffer, K.; et al. AR expression in breast cancer CTCs associates with bone metastases. Mol. Cancer Res. 2018, 16, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Miyamoto, D.T.; Wittner, B.S.; Sullivan, J.P.; Aceto, N.; Jordan, N.V.; Yu, M.; Karabacak, N.M.; Comaills, V.; Morris, R.; et al. Expression of β-globin by cancer cells promotes cell survival during blood-borne dissemination. Nat. Commun. 2017, 8, 14344. [Google Scholar] [CrossRef] [PubMed]
- Ting, D.T.; Wittner, B.S.; Ligorio, M.; Jordan, N.V.; Shah, A.M.; Miyamoto, D.T.; Aceto, N.; Bersani, F.; Brannigan, B.W.; Xega, K.; et al. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep. 2014, 8, 1905–1918. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, D.T.; Zheng, Y.; Wittner, B.S.; Lee, R.J.; Zhu, H.; Broderick, K.T.; Desai, R.; Fox, D.B.; Brannigan, B.W.; Trautwein, J.; et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015, 349, 1351–1356. [Google Scholar] [CrossRef]
- Van der Wijst, M.G.; Brugge, H.; de Vries, D.H.; Deelen, P.; Swertz, M.A.; Franke, L. Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs. Nat. Genet. 2018, 50, 493–497. [Google Scholar] [CrossRef]
- Jordan, N.V.; Bardia, A.; Wittner, B.S.; Benes, C.; Ligorio, M.; Zheng, Y.; Yu, M.; Sundaresan, T.K.; Licausi, J.A.; Desai, R.; et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature 2016, 537, 102–106. [Google Scholar] [CrossRef]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112. [Google Scholar] [CrossRef]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Jindal, A.; Gupta, P.; Sengupta, D. Discovery of rare cells from voluminous single cell expression data. Nat. Commun. 2018, 9, 1–9. [Google Scholar]
- Srivastava, D.; Iyer, A.; Kumar, V.; Sengupta, D. CellAtlasSearch: A scalable search engine for single cells. Nucleic Acids Res. 2018, 46, W141–W147. [Google Scholar]
- Sinha, D.; Sinha, P.; Saha, R.; Bandyopadhyay, S.; Sengupta, D. Improved dropClust R package with integrative analysis support for scRNA-seq data. Bioinformatics 2020, 36, 1946–1947. [Google Scholar]
- Zhang, X.; Lan, Y.; Xu, J.; Quan, F.; Zhao, E.; Deng, C.; Luo, T.; Xu, L.; Liao, G.; Yan, M.; et al. CellMarker: A manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2018, 47, D721–D728. [Google Scholar] [CrossRef]
- Huang, B.; Jia, D.; Feng, J.; Levine, H.; Onuchic, J.N.; Lu, M. RACIPE: A computational tool for modeling gene regulatory circuits using randomization. BMC Syst. Biol. 2018, 12, 74. [Google Scholar] [CrossRef]
- Pearson, K.L., III. On lines and planes of closest fit to systems of points in space. London Edinburgh Dublin Philos. Mag. J. Sci. 1901, 2, 559–572. [Google Scholar] [CrossRef]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nature Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Rish, I. An empirical study of the naive Bayes classifier. In Proceedings of the IJCAI 2001 workshop on empirical methods in artificial intelligence, Seattle, DC, USA, 4 August 2001; Volume 3, pp. 41–46. [Google Scholar]
- Friedman, J.H. Greedy function approximation: A gradient boosting machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Ho, T.K. Random decision forests. In Proceedings of the 3rd international conference on document analysis and recognition, Montreal, QC, Canada, 14–16 August 1995; Volume 1, pp. 278–282. [Google Scholar]
- Lee, Y.; Guan, G.; Bhagat, A.A. ClearCell® FX, a label-free microfluidics technology for enrichment of viable circulating tumor cells. Cytom. Part A 2018, 93, 1251–1254. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Bailey, C.M.; Morrison, J.A.; Kulesa, P.M. Melanoma revives an embryonic migration program to promote plasticity and invasion. Pigment Cell Melanoma Res. 2012, 25, 573–583. [Google Scholar] [CrossRef]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, T.; Mori, S.; Huang, R.Y.J.; Thiery, J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef]
- Garrido, F.; Ruiz-Cabello, F.; Aptsiauri, N. Rejection versus escape: The tumor MHC dilemma. Cancer Immunol. Immunother. 2017, 66, 259–271. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iyer, A.; Gupta, K.; Sharma, S.; Hari, K.; Lee, Y.F.; Ramalingam, N.; Yap, Y.S.; West, J.; Bhagat, A.A.; Subramani, B.V.; et al. Integrative Analysis and Machine Learning Based Characterization of Single Circulating Tumor Cells. J. Clin. Med. 2020, 9, 1206. https://doi.org/10.3390/jcm9041206
Iyer A, Gupta K, Sharma S, Hari K, Lee YF, Ramalingam N, Yap YS, West J, Bhagat AA, Subramani BV, et al. Integrative Analysis and Machine Learning Based Characterization of Single Circulating Tumor Cells. Journal of Clinical Medicine. 2020; 9(4):1206. https://doi.org/10.3390/jcm9041206
Chicago/Turabian StyleIyer, Arvind, Krishan Gupta, Shreya Sharma, Kishore Hari, Yi Fang Lee, Neevan Ramalingam, Yoon Sim Yap, Jay West, Ali Asgar Bhagat, Balaram Vishnu Subramani, and et al. 2020. "Integrative Analysis and Machine Learning Based Characterization of Single Circulating Tumor Cells" Journal of Clinical Medicine 9, no. 4: 1206. https://doi.org/10.3390/jcm9041206
APA StyleIyer, A., Gupta, K., Sharma, S., Hari, K., Lee, Y. F., Ramalingam, N., Yap, Y. S., West, J., Bhagat, A. A., Subramani, B. V., Sabuwala, B., Tan, T. Z., Thiery, J. P., Jolly, M. K., Ramalingam, N., & Sengupta, D. (2020). Integrative Analysis and Machine Learning Based Characterization of Single Circulating Tumor Cells. Journal of Clinical Medicine, 9(4), 1206. https://doi.org/10.3390/jcm9041206