
Evidence for Hypoxia-Induced Shift in ATP Production from Glycolysis to Mitochondrial Respiration in Pulmonary Artery Smooth Muscle Cells in Pulmonary Arterial Hypertension
 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation of Human Pulmonary Artery Smooth Muscle Cells
2.2. Exposure to Normoxia and Hypoxia
2.3. Quantitative RT-PCR Analysis
2.4. Western Blot Analysis
2.5. Lactate Assay
2.6. Energy Metabolism Analysis
2.7. Transmission Electron Microscopy
2.8. Animal Study Protocol
2.9. Statistical Analysis
3. Results
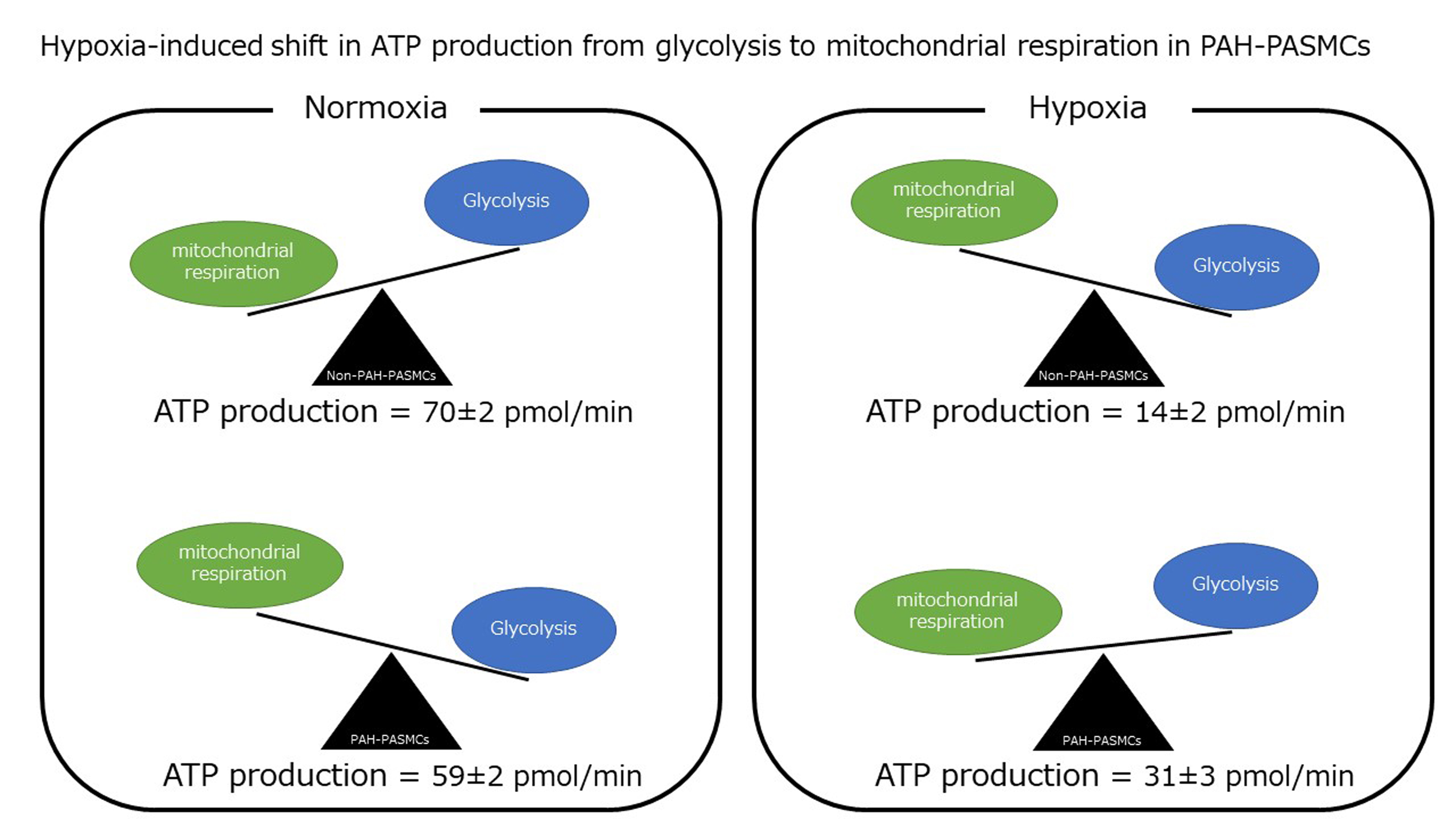
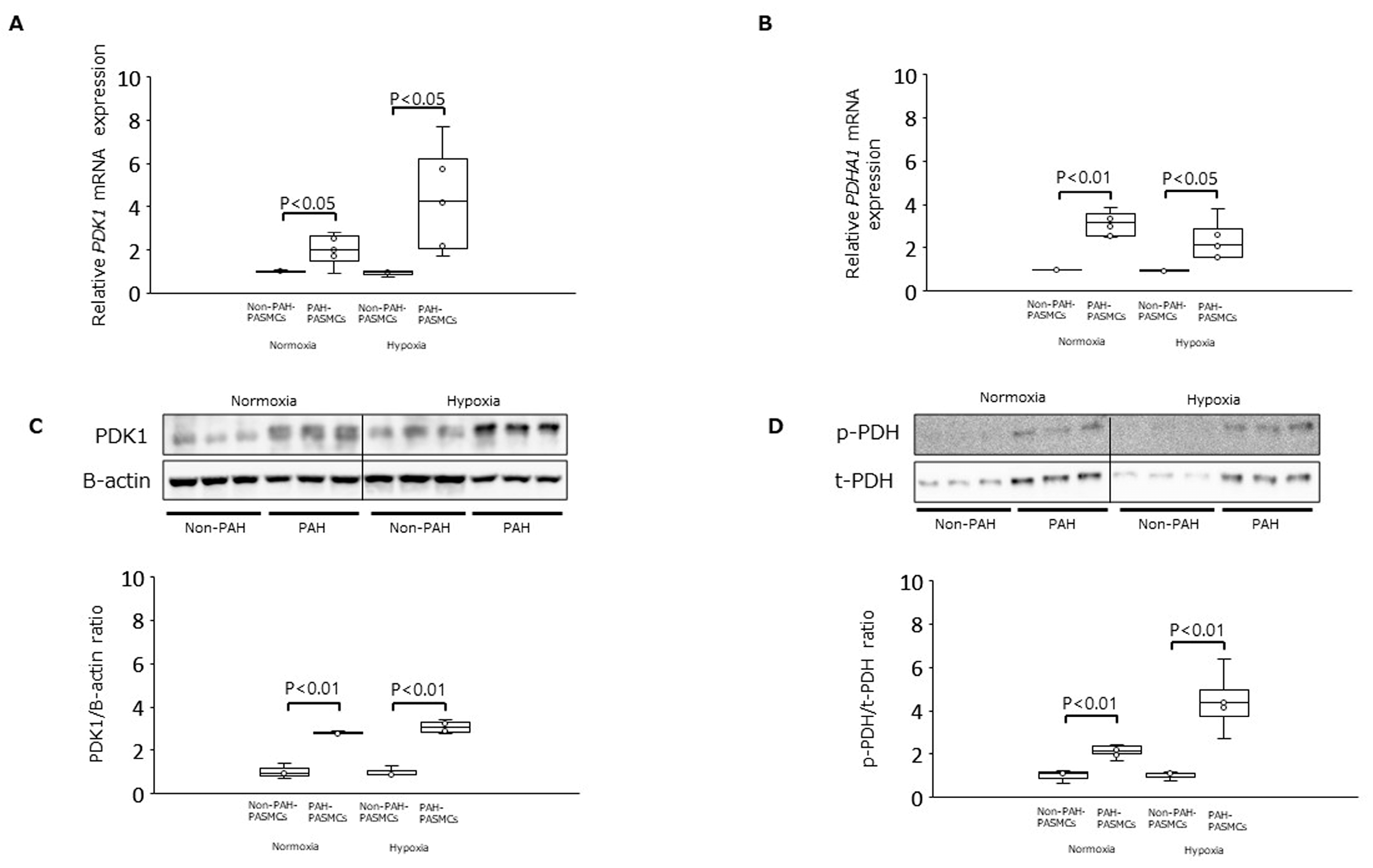
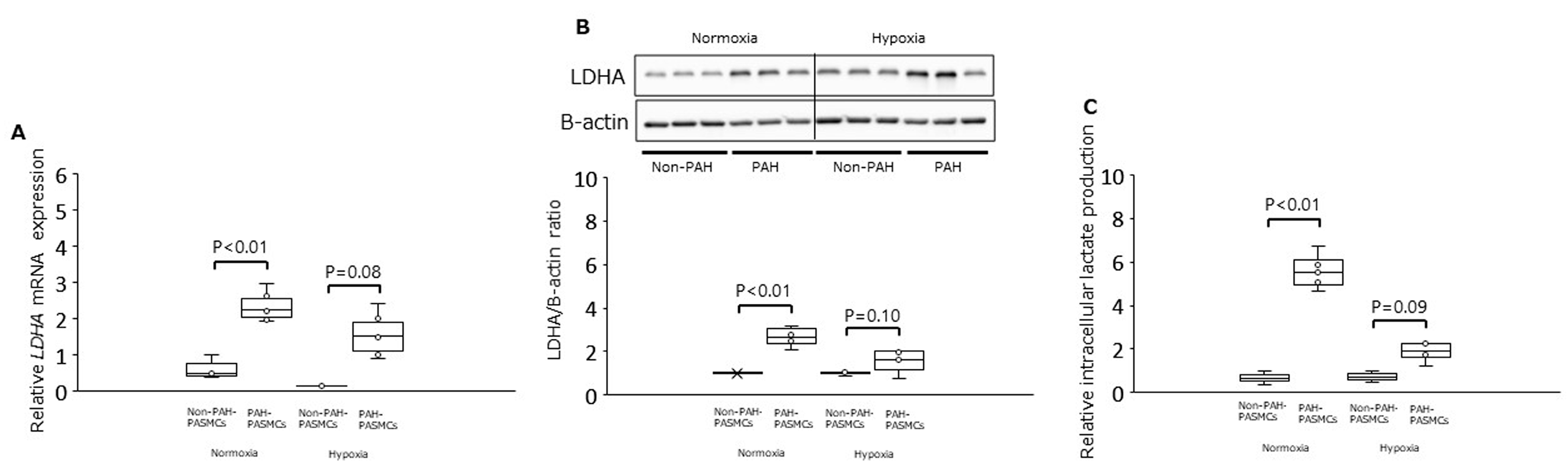
3.1. PDK1, PDHA1, and LDHA Expression and Intracellular Lactate Production under Normoxia and Hypoxia
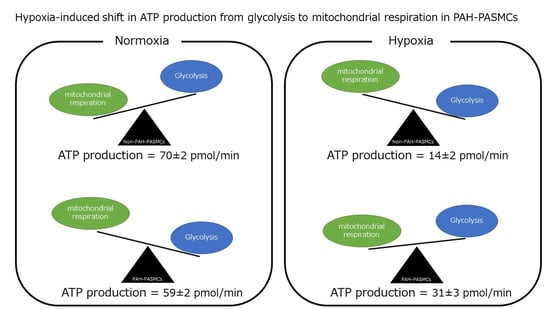
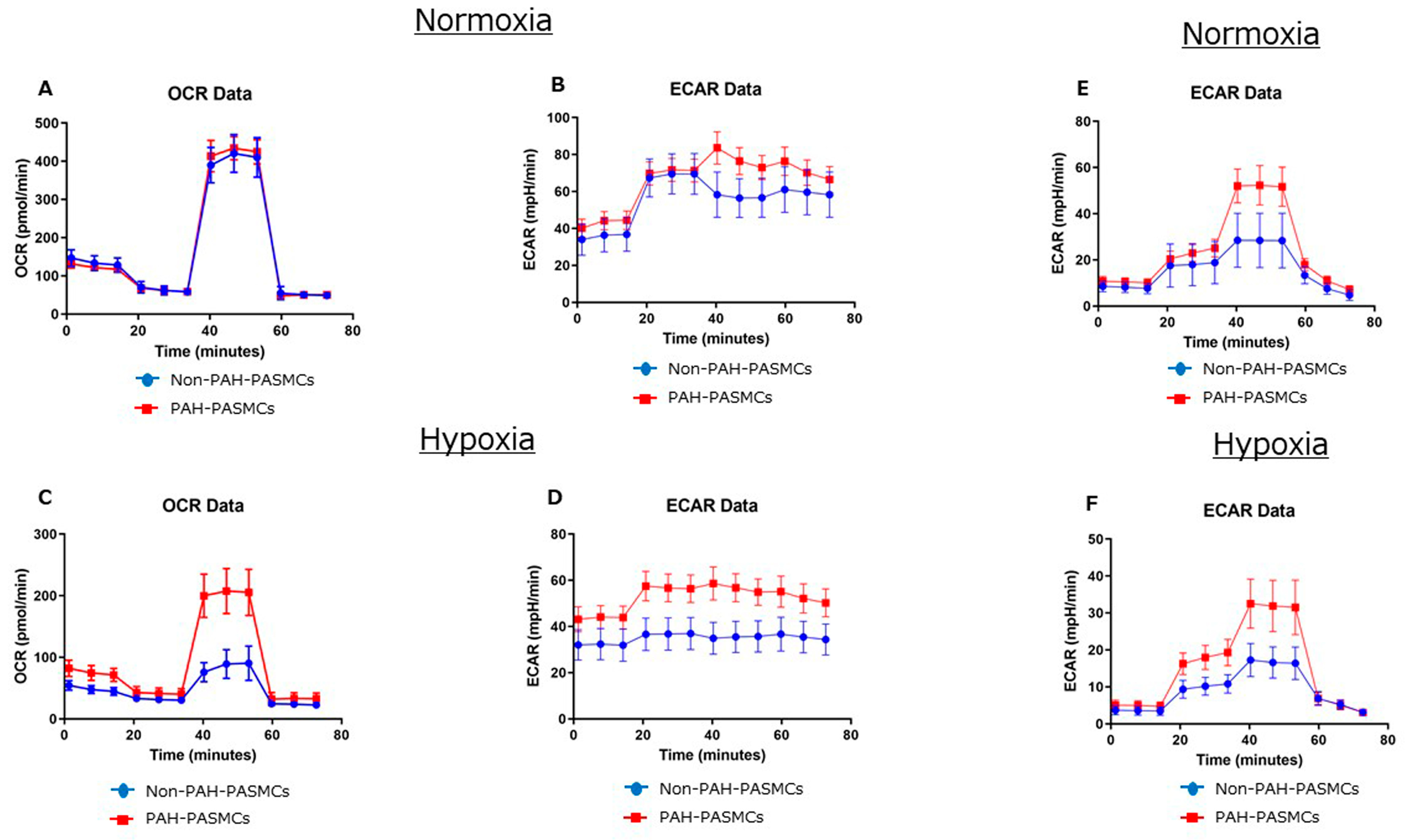
3.2. Energy Metabolism in Normoxia and Hypoxia
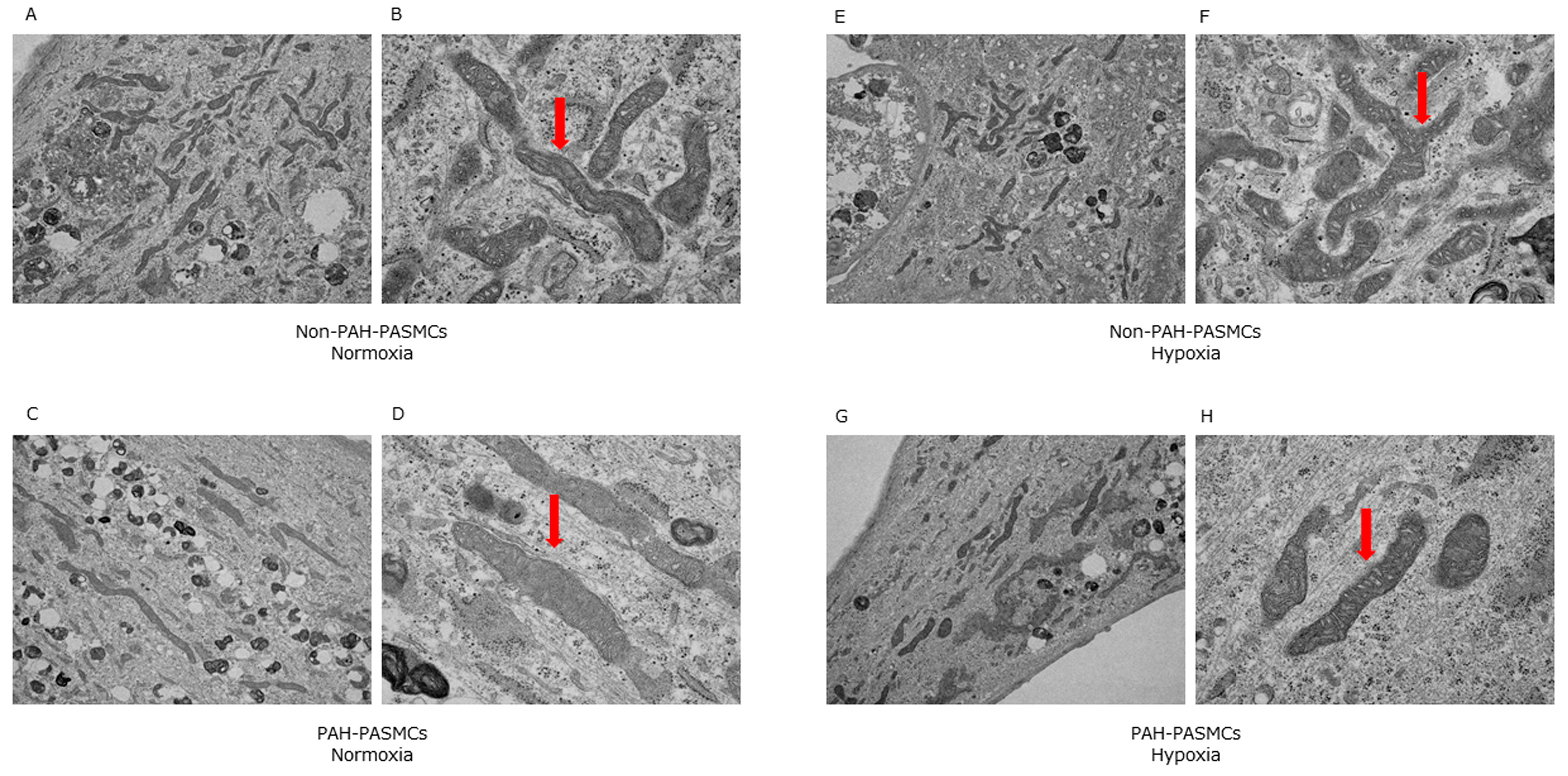
3.3. Mitochondrial Morphology in Normoxia and Hypoxia
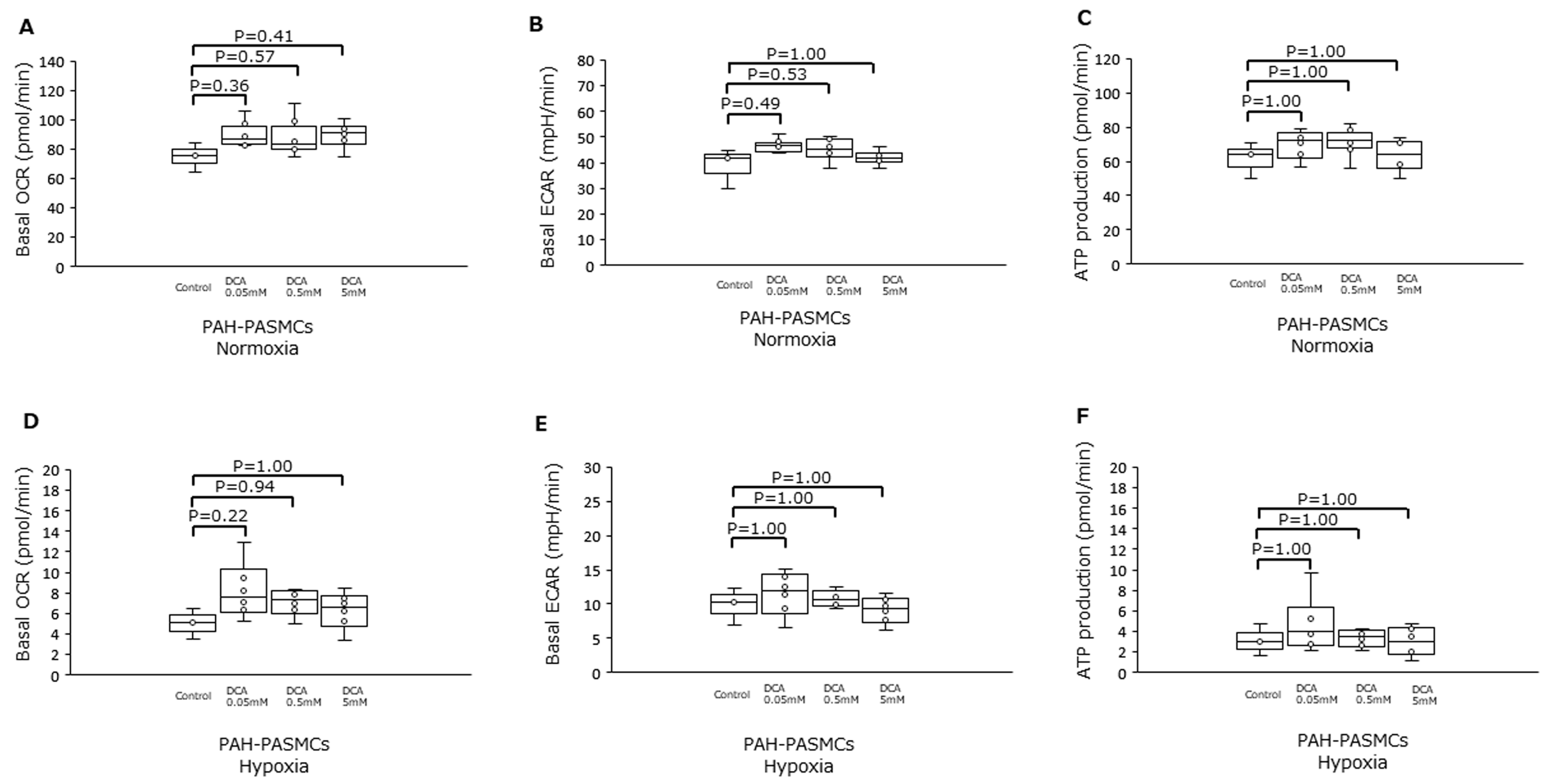
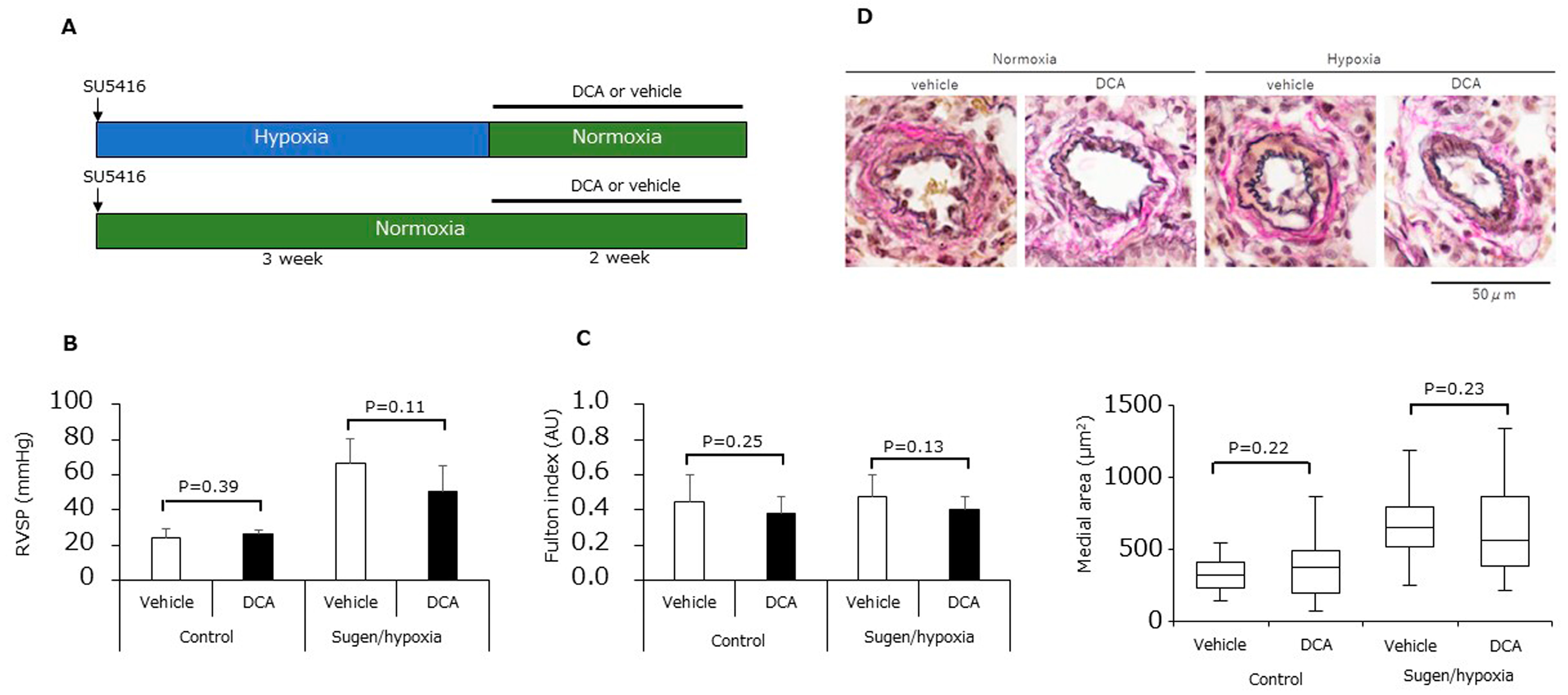
3.4. Dichloroacetic Acid Did Not Increase ATP Production and Improve Pulmonary Hypertension
4. Discussion
Limitation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Akagi, S.; Matsubara, H.; Nakamura, K.; Ito, H. Modern treatment to reduce pulmonary arterial pressure in pulmonary arterial hypertension. J. Cardiol. 2018, 72, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Sommer, N.; Ghofrani, H.A.; Pak, O.; Bonnet, S.; Provencher, S.; Sitbon, O.; Rosenkranz, S.; Hoeper, M.M.; Kiely, D.G. Current and future treatments of pulmonary arterial hypertension. Br. J. Pharmacol. 2020, 178, 6–30. [Google Scholar] [CrossRef] [PubMed]
- Pullamsetti, S.S.; Savai, R.; Seeger, W.; Goncharova, E.A. Translational Advances in the Field of Pulmonary Hypertension.From Cancer Biology to New Pulmonary Arterial Hypertension Therapeutics. Targeting Cell Growth and Proliferation Signaling Hubs. Am. J. Respir. Crit. Care Med. 2017, 195, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Pullamsetti, S.S.; Mamazhakypov, A.; Weissmann, N.; Seeger, W.; Savai, R. Hypoxia-inducible factor signaling in pulmonary hypertension. J. Clin. Investig. 2020, 130, 5638–5651. [Google Scholar] [CrossRef]
- Young, J.M.; Williams, D.R.; Thompson, A.A.R. Thin Air, Thick Vessels: Historical and Current Perspectives on Hypoxic Pulmonary Hypertension. Front. Med. 2019, 6, 93. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Han, S.; Chandel, N.S. Lessons from Cancer Metabolism for Pulmonary Arterial Hypertension and Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 65, 134–145. [Google Scholar] [CrossRef]
- Smith, K.A.; Waypa, G.B.; Dudley, V.J.; Budinger, G.R.S.; Abdala-Valencia, H.; Bartom, E.; Schumacker, P.T. Role of Hypoxia-Inducible Factors in Regulating Right Ventricular Function and Remodeling during Chronic Hypoxia–induced Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2020, 63, 652–664. [Google Scholar] [CrossRef]
- Sutendra, G.; Bonnet, S.; Rochefort, G.; Haromy, A.; Folmes, K.D.; Lopaschuk, G.D.; Dyck, J.R.B.; Michelakis, E.D. Fatty Acid Oxidation and Malonyl-CoA Decarboxylase in the Vascular Remodeling of Pulmonary Hypertension. Sci. Transl. Med. 2010, 2, 44ra58. [Google Scholar] [CrossRef]
- Akagi, S.; Nakamura, K.; Matsubara, H.; Kondo, M.; Miura, D.; Matoba, T.; Egashira, K.; Ito, H. Intratracheal Administration of Prostacyclin Analogue–incorporated Nanoparticles Ameliorates the Development of Monocrotaline and Sugen-Hypoxia-induced Pulmonary Arterial Hypertension. J. Cardiovasc. Pharmacol. 2016, 67, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, A.; Nakamura, K.; Matsubara, H.; Fujio, H.; Ikeda, T.; Kobayashi, K.; Miyazaki, I.; Asanuma, M.; Miyaji, K.; Miura, D.; et al. Prednisolone Inhibits Proliferation of Cultured Pulmonary Artery Smooth Muscle Cells of Patients With Idiopathic Pulmonary Arterial Hypertension. Circulation 2005, 112, 1806–1812. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Michelakis, E.D. The Metabolic Basis of Pulmonary Arterial Hypertension. Cell Metab. 2014, 19, 558–573. [Google Scholar] [CrossRef] [PubMed]
- Dromparis, P.; Michelakis, E.D. Mitochondria in Vascular Health and Disease. Annu. Rev. Physiol. 2013, 75, 95–126. [Google Scholar] [CrossRef]
- Xu, W.; Janocha, A.J.; Erzurum, S.C. Metabolism in Pulmonary Hypertension. Annu. Rev. Physiol. 2021, 83, 551–576. [Google Scholar] [CrossRef]
- Wujak, M.; Veith, C.; Wu, C.-Y.; Wilke, T.; Kanbagli, Z.I.; Novoyatleva, T.; Guenther, A.; Seeger, W.; Grimminger, F.; Sommer, N.; et al. Adenylate Kinase 4—A Key Regulator of Proliferation and Metabolic Shift in Human Pulmonary Arterial Smooth Muscle Cells via Akt and HIF-1α Signaling Pathways. Int. J. Mol. Sci. 2021, 22, 10371. [Google Scholar] [CrossRef]
- Dai, J.; Zhou, Q.; Chen, J.; Rexius-Hall, M.L.; Rehman, J.; Zhou, G. Alpha-enolase regulates the malignant phenotype of pulmonary artery smooth muscle cells via the AMPK-Akt pathway. Nat. Commun. 2018, 9, 3850. [Google Scholar] [CrossRef]
- Dabral, S.; Muecke, C.; Valasarajan, C.; Schmoranzer, M.; Wietelmann, A.; Semenza, G.L.; Meister, M.; Muley, T.; Seeger-Nukpezah, T.; Samakovlis, C.; et al. A RASSF1A-HIF1α loop drives Warburg effect in cancer and pulmonary hypertension. Nat. Commun. 2019, 10, 2130. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.-Y.; et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Investig. 2016, 126, 3313–3335. [Google Scholar] [CrossRef]
- Michelakis, E.D.; McMurtry, M.S.; Wu, X.-C.; Dyck, J.R.; Moudgil, R.; Hopkins, T.A.; Lopaschuk, G.D.; Puttagunta, L.; Waite, R.; Archer, S.L.; et al. Dichloroacetate, a Metabolic Modulator, Prevents and Reverses Chronic Hypoxic Pulmonary Hypertension in Rats: Role of increased expression and activity of voltage-gated potassium channels. Circulation 2002, 105, 244–250. [Google Scholar] [CrossRef] [PubMed]
- McMurtry, M.S.; Bonnet, S.; Wu, X.; Dyck, J.R.; Haromy, A.; Hashimoto, K.; Michelakis, E.D. Dichloroacetate Prevents and Reverses Pulmonary Hypertension by Inducing Pulmonary Artery Smooth Muscle Cell Apoptosis. Circ. Res. 2004, 95, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Gurtu, V.; Webster, L.; Barnes, G.; Watson, G.; Howard, L.; Cupitt, J.; Paterson, I.; Thompson, R.B.; Chow, K.; et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci. Transl. Med. 2017, 9, eaao4583. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Dromparis, P.; Bonnet, S.; Haromy, A.; McMurtry, M.S.; Bleackley, R.C.; Michelakis, E.D. Pyruvate dehydrogenase inhibition by the inflammatory cytokine TNFα contributes to the pathogenesis of pulmonary arterial hypertension. J. Mol. Med. 2011, 89, 771–783. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, X.; Wang, L.; Yang, Q.; Ma, Q.; Xu, J.; Wang, J.; Kovacs, L.; Ayon, R.J.; Liu, Z.; et al. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2019, 116, 13394–13403. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Gender | Age |
|---|---|---|
| 1 | F | 13 |
| 2 | F | 18 |
| 3 | F | 30 |
| 4 | F | 29 |
| 5 | M | 18 |
| 6 | M | 6 |
| Gene Symbol | Sequence | |
|---|---|---|
| PDK1 | F | CATGTCACGCTGGGTAATGAGG |
| R | CTCAACACGAGGTCTTGGTGCA | |
| PDHA1 | F | GGATGGTGAACAGCAATCTTGCC |
| R | TCGCTGGAGTAGATGTGGTAGC | |
| LDHA | F | GGACAAGTTGGTATGGCGTGTG |
| R | AAGCTCCCATGCTGCAGATCCA | |
| ACTB | F | CACCATTGGCAATGAGCGGTTC |
| R | AGGTCTTTGCGGATGTCCACGT |
| Normoxia | Hypoxia | |||||
|---|---|---|---|---|---|---|
| Non-PAH-PASMCs | PAH-PASMCs | p Value | Non-PAH-PASMCs | PAH-PASMCs | p Value | |
| Basal OCR (pmol/min) | 79 ± 4 | 70 ± 5 | <0.01 | 22 ± 3 | 39 ± 4 | <0.01 |
| Basal ECAR (mpH/min) | 37 ± 2 | 44 ± 3 | <0.01 | 32 ± 3 | 44 ± 3 | <0.01 |
| ATP production (pmol/min) | 70 ± 2 | 59 ± 2 | <0.01 | 14 ± 2 | 31 ± 3 | <0.01 |
| Maximal respiration | 372 ± 49 | 387 ± 31 | 0.16 | 68 ± 26 | 176 ± 34 | <0.01 |
| Spare respiratory capacity | 293 ± 46 | 317 ± 27 | <0.01 | 46 ± 25 | 136 ± 28 | <0.01 |
| Coupling efficiency | 0.89 ± 0.04 | 0.85 ± 0.07 | <0.01 | 0.65 ± 0.04 | 0.81 ± 0.10 | <0.01 |
| Normoxia | Hypoxia | |||||
|---|---|---|---|---|---|---|
| ECAR (mpH/min) | Non-PAH-PASMCs | PAH-PASMCs | p Value | Non-PAH-PASMCs | PAH-PASMCs | p Value |
| Glycolysis yield | 21 ± 10 | 42 ± 8 | <0.01 | 14 ± 4 | 28 ± 6 | <0.01 |
| Glycolytic capacity | 11 ± 7 | 15 ± 3 | <0.01 | 7 ± 1 | 15 ± 2 | <0.01 |
| Glycolytic reserve | 10 ± 4 | 27 ± 5 | <0.01 | 6 ± 3 | 13 ± 5 | <0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akagi, S.; Nakamura, K.; Kondo, M.; Hirohata, S.; Udono, H.; Nishida, M.; Saito, Y.; Yoshida, M.; Miyoshi, T.; Ito, H. Evidence for Hypoxia-Induced Shift in ATP Production from Glycolysis to Mitochondrial Respiration in Pulmonary Artery Smooth Muscle Cells in Pulmonary Arterial Hypertension. J. Clin. Med. 2023, 12, 5028. https://doi.org/10.3390/jcm12155028
Akagi S, Nakamura K, Kondo M, Hirohata S, Udono H, Nishida M, Saito Y, Yoshida M, Miyoshi T, Ito H. Evidence for Hypoxia-Induced Shift in ATP Production from Glycolysis to Mitochondrial Respiration in Pulmonary Artery Smooth Muscle Cells in Pulmonary Arterial Hypertension. Journal of Clinical Medicine. 2023; 12(15):5028. https://doi.org/10.3390/jcm12155028
Chicago/Turabian StyleAkagi, Satoshi, Kazufumi Nakamura, Megumi Kondo, Satoshi Hirohata, Heiichiro Udono, Mikako Nishida, Yukihiro Saito, Masashi Yoshida, Toru Miyoshi, and Hiroshi Ito. 2023. "Evidence for Hypoxia-Induced Shift in ATP Production from Glycolysis to Mitochondrial Respiration in Pulmonary Artery Smooth Muscle Cells in Pulmonary Arterial Hypertension" Journal of Clinical Medicine 12, no. 15: 5028. https://doi.org/10.3390/jcm12155028
APA StyleAkagi, S., Nakamura, K., Kondo, M., Hirohata, S., Udono, H., Nishida, M., Saito, Y., Yoshida, M., Miyoshi, T., & Ito, H. (2023). Evidence for Hypoxia-Induced Shift in ATP Production from Glycolysis to Mitochondrial Respiration in Pulmonary Artery Smooth Muscle Cells in Pulmonary Arterial Hypertension. Journal of Clinical Medicine, 12(15), 5028. https://doi.org/10.3390/jcm12155028