
Immunoglobulin Disorders and the Oral Cavity: A Narrative Review

 , , , ,
, , , ,  ,
,  ,
,  , and
, and
Abstract
:1. Introduction
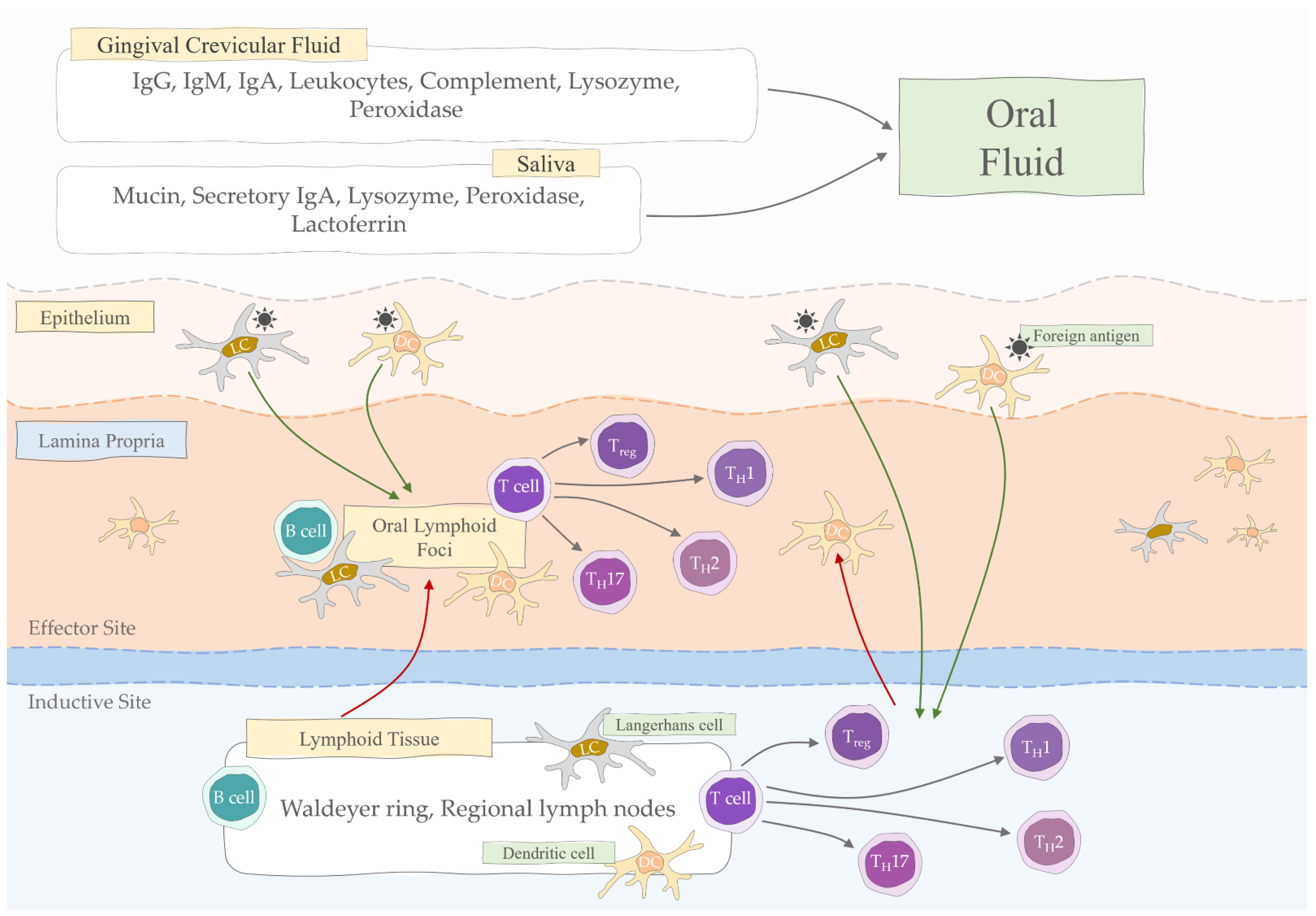
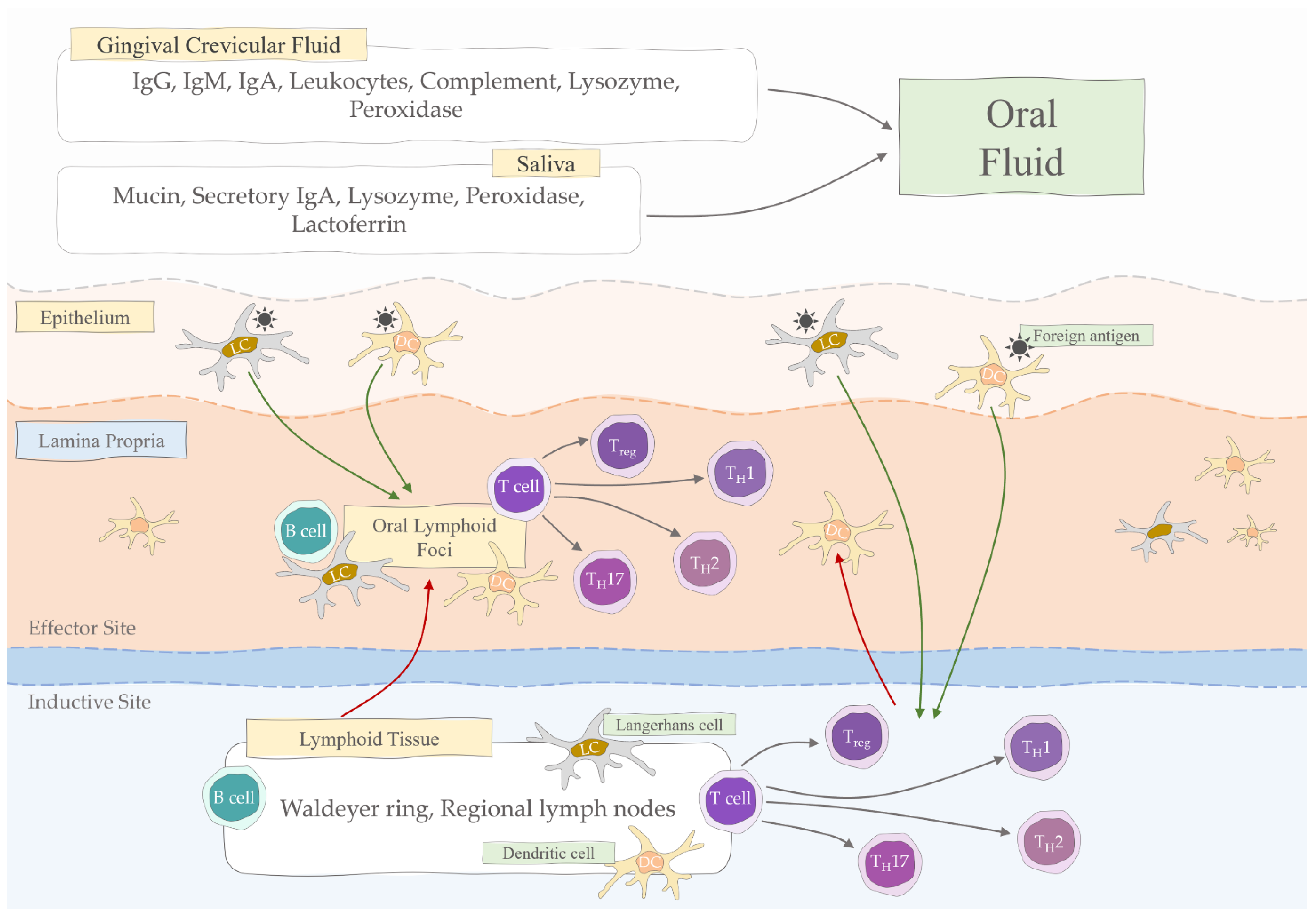
2. Salivary Immunoglobulins
3. Oral Mucosa Immunity
4. Mucosal Humoral Immune Response
5. Oral Manifestations in Selected Diseases
5.1. Mucous Membrane Pemphigoid
5.2. Pemphigus Vulgaris (PV)
5.3. Linear IgA Bullous Dermatosis (LABD)
5.4. Epidermolysis Bullosa Aquisita (EBA)
5.5. Hyper-IgE Syndrome
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Groeger, S.; Meyle, J. Oral Mucosal Epithelial Cells. Front. Immunol. 2019, 10, 208. [Google Scholar] [CrossRef] [PubMed]
- Mainas, G.; Ide, M.; Rizzo, M.; Magan-Fernandez, A.; Mesa, F.; Nibali, L. Managing the Systemic Impact of Periodontitis. Medicina 2022, 58, 621. [Google Scholar] [CrossRef] [PubMed]
- Roa, I.; Del Sol, M. Obesity, salivary glands and oral pathology. Colomb. Med. 2018, 49, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Lynge Pedersen, A.M.; Belstrøm, D. The role of natural salivary defences in maintaining a healthy oral microbiota. J. Dent. 2019, 80, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Almubarak, A.; Tanagala, K.K.K.; Papapanou, P.N.; Lalla, E.; Momen-Heravi, F. Disruption of Monocyte and Macrophage Homeostasis in Periodontitis. Front. Immunol. 2020, 11, 330. [Google Scholar] [CrossRef]
- Marshall, A.; Celentano, A.; Cirillo, N.; McCullough, M.; Porter, S. Oral keratinocytes synthesize CTACK: A new insight into the pathophysiology of the oral mucosa. Exp. Dermatol. 2018, 27, 207–210. [Google Scholar] [CrossRef]
- Fatima, T.; Khurshid, Z.; Rehman, A.; Imran, E.; Srivastava, K.C.; Shrivastava, D. Gingival Crevicular Fluid (GCF): A Diagnostic Tool for the Detection of Periodontal Health and Diseases. Molecules 2021, 26, 1208. [Google Scholar] [CrossRef]
- Heboyan, A.; Manrikyan, M.; Zafar, M.S.; Rokaya, D.; Nushikyan, R.; Vardanyan, I.; Vardanyan, A.; Khurshid, Z. Bacteriological Evaluation of Gingival Crevicular Fluid in Teeth Restored Using Fixed Dental Prostheses: An In Vivo Study. Int. J. Mol. Sci. 2021, 22, 5463. [Google Scholar] [CrossRef]
- Linden, G.J.; Herzberg, M.C. Working group 4 of joint EFP/AAP workshop. Periodontitis and systemic diseases: A record of discussions of working group 4 of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J. Clin. Periodontol. 2013, 40, 520–523. [Google Scholar] [CrossRef]
- Preshaw, P.M.; Alba, A.L.; Herrera, D.; Jepsen, S.; Konstantinidis, A.; Makrilakis, K.; Taylor, R. Periodontitis and diabetes: A two-way relationship. Diabetologia 2012, 55, 21–31. [Google Scholar] [CrossRef]
- Lauritano, D.; Boccalari, E.; Di Stasio, D.; Della Vella, F.; Carinci, F.; Lucchese, A.; Petruzzi, M. Prevalence of Oral Lesions and Correlation with Intestinal Symptoms of Inflammatory Bowel Disease: A Systematic Review. Diagnostics 2019, 9, 77. [Google Scholar] [CrossRef] [PubMed]
- Nijakowski, K.; Gruszczyński, D.; Surdacka, A. Oral Health Status in Patients with Inflammatory Bowel Diseases: A Systematic Review. Int. J. Environ. Res. Public Health 2021, 18, 11521. [Google Scholar] [CrossRef] [PubMed]
- Sojod, B.; Pidorodeski Nagano, C.; Garcia Lopez, G.M.; Zalcberg, A.; Dridi, S.M.; Anagnostou, F. Systemic Lupus Erythematosus and Periodontal Disease: A Complex Clinical and Biological Interplay. J. Clin. Med. 2021, 10, 1957. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Tseng, C.F.; Liu, J.M.; Chuang, H.C.; Lei, W.T.; Liu, L.Y.; Yu, Y.C.; Hsu, R.J. Association between Periodontal Disease and Subsequent Sjögren’s Syndrome: A Nationwide Population-Based Cohort Study. Int. J. Environ. Res. Public Health 2019, 16, 771. [Google Scholar] [CrossRef]
- Nakib, S.; Han, J.; Li, T.; Joshipura, K.; Qureshi, A.A. Periodontal disease and risk of psoriasis among nurses in the United States. Acta Odontol. Scand. 2013, 71, 1423–1429. [Google Scholar] [CrossRef]
- Thilagar, S.; Theyagarajan, R.; Sudhakar, U.; Suresh, S.; Saketharaman, P.; Ahamed, N. Comparison of serum tumor necrosis factor-α levels in rheumatoid arthritis individuals with and without chronic periodontitis: A biochemical study. J. Indian Soc. Periodontol. 2018, 22, 116–121. [Google Scholar] [CrossRef]
- Liccardo, D.; Cannavo, A.; Spagnuolo, G.; Ferrara, N.; Cittadini, A.; Rengo, C.; Rengo, G. Periodontal Disease: A Risk Factor for Diabetes and Cardiovascular Disease. Int. J. Mol. Sci. 2019, 20, 1414. [Google Scholar] [CrossRef]
- Kapellas, K.; Singh, A.; Bertotti, M.; Nascimento, G.G.; Jamieson, L.M. Perio-CKD collaboration. Periodontal and chronic kidney disease association: A systematic review and meta-analysis. Nephrology 2019, 24, 202–212. [Google Scholar] [CrossRef]
- Carpenter, G.H. Salivary Factors That Maintain the Normal Oral Commensal Microflora. J. Dent. Res. 2020, 99, 644–649. [Google Scholar] [CrossRef]
- Gupta, G. Gingival Crevicular Fluid as a Periodontal Diagnostic Indicator- II: Inflammatory Mediators, Host-Response Modifiers and Chair Side Diagnostic Aids. J. Med. Life 2013, 6, 7–13. [Google Scholar]
- Brandtzaeg, P. Secretory Immunity with Special Reference to the Oral Cavity. J. Oral. Microbiol. 2013, 5, 20401. [Google Scholar] [CrossRef] [PubMed]
- Delacroix, D.L.; Vaerman, J.P. Function of the Human Liver in IgA Homeostasis in Plasma. Ann. N. Y. Acad. Sci. 1983, 409, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Nijakowski, K.; Rutkowski, R.; Eder, P.; Simon, M.; Korybalska, K.; Witowski, J.; Surdacka, A. Potential Salivary Markers for Differential Diagnosis of Crohn’s Disease and Ulcerative Colitis. Life 2021, 11, 943. [Google Scholar] [CrossRef] [PubMed]
- Nijakowski, K.; Rutkowski, R.; Eder, P.; Korybalska, K.; Witowski, J.; Surdacka, A. Changes in Salivary Parameters of Oral Immunity after Biologic Therapy for Inflammatory Bowel Disease. Life 2021, 11, 1409. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef]
- Palmeira, P.; Quinello, C.; Silveira-Lessa, A.L.; Zago, C.A.; Carneiro-Sampaio, M. IgG Placental Transfer in Healthy and Pathological Pregnancies. Clin. Dev. Immunol. 2011, 2012, e985646. [Google Scholar] [CrossRef]
- Srinivasan, P.C. Immunoglobulin Levels and Periodontal Diseases-A Clinical Immunological Study. J. Clin. Cell Immunol. 2012, 4, 1–4. [Google Scholar] [CrossRef]
- Molnar, C.; Gair, J. Antibodies. In Concepts of Biology—1st Canadian Edition; BCcampus: Victoria, BC, Canada, 2015. [Google Scholar]
- Jotwani, R.; Palucka, A.K.; Al-Quotub, M.; Nouri-Shirazi, M.; Kim, J.; Bell, D.; Banchereau, J.; Cutler, C.W. Mature Dendritic Cells Infiltrate the T Cell-Rich Region of Oral Mucosa in Chronic Periodontitis: In Situ, In Vivo, and In Vitro Studies. J. Immunol. 2001, 167, 4693–4700. [Google Scholar] [CrossRef]
- Sahingur, S.E.; Yeudall, W.A. Chemokine Function in Periodontal Disease and Oral Cavity Cancer. Front. Immunol. 2015, 6, 214. [Google Scholar] [CrossRef]
- Feller, L.; Altini, M.; Khammissa, R.A.G.; Chandran, R.; Bouckaert, M.; Lemmer, J. Oral Mucosal Immunity. Oral Surg Oral Med. Oral Pathol. Oral Radiol. 2013, 116, 576–583. [Google Scholar] [CrossRef]
- Moutsopoulos, N.M.; Konkel, J.E. Tissue-Specific Immunity at the Oral Mucosal Barrier. Trends Immunol. 2018, 39, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.M. Oral Mucosal Immunology: An Overview. Ann. Acad. Med. Singap. 2004, 33, 27–30. [Google Scholar] [PubMed]
- Billings, M.; Dye, B.; Iafolla, T.; Grisius, M.; Alevizos, I. Elucidating the Role of Hyposalivation and Autoimmunity in Oral Candidiasis. Oral Dis. 2017, 23, 387–394. [Google Scholar] [CrossRef]
- Gooty, J.R.; Kannam, D.; Guntakala, V.R.; Palaparthi, R. Distribution of Dendritic Cells and Langerhans Cells in Peri-Implant Mucosa. Contemp. Clin. Dent. 2018, 9, 548–553. [Google Scholar] [CrossRef]
- Hans, M.; Hans, V.M. Toll-like Receptors and Their Dual Role in Periodontitis: A Review. J. Oral Sci. 2011, 53, 263–271. [Google Scholar] [CrossRef]
- Azzolino, D.; Passarelli, P.C.; De Angelis, P.; Piccirillo, G.B.; D’Addona, A.; Cesari, M. Poor Oral Health as a Determinant of Malnutrition and Sarcopenia. Nutrients 2019, 11, 2898. [Google Scholar] [CrossRef] [PubMed]
- Schröder, N.W.J.; Meister, D.; Wolff, V.; Christan, C.; Kaner, D.; Haban, V.; Purucker, P.; Hermann, C.; Moter, A.; Göbel, U.B.; et al. Chronic Periodontal Disease Is Associated with Single-Nucleotide Polymorphisms of the Human TLR-4 Gene. Genes Immun. 2005, 6, 448–451. [Google Scholar] [CrossRef]
- Hou, L.; Sasaki, H.; Stashenko, P. Toll-like Receptor 4-Deficient Mice Have Reduced Bone Destruction Following Mixed Anaerobic Infection. Infect. Immun. 2000, 68, 4681–4687. [Google Scholar] [CrossRef]
- Belstrøm, D.; Holmstrup, P.; Fiehn, N.E.; Rosing, K.; Bardow, A.; Paster, B.J.; Lynge Pedersen, A.M. Bacterial composition in whole saliva from patients with severe hyposalivation--a case-control study. Oral Dis. 2016, 22, 330–337. [Google Scholar] [CrossRef]
- Kawai, T.; Eisen-Lev, R.; Seki, M.; Eastcott, J.W.; Wilson, M.E.; Taubman, M.A. Requirement of B7 Costimulation for Th1-Mediated Inflammatory Bone Resorption in Experimental Periodontal Disease. J. Immunol. 2000, 164, 2102–2109. [Google Scholar] [CrossRef]
- Challacombe, S.J.; Shirlaw, P.J.; Thornhill, M.H. Chapter 102—Immunology of Diseases of the Oral Cavity. In Mucosal Immunology, 4th ed.; Mestecky, J., Strober, W., Russell, M.W., Kelsall, B.L., Cheroutre, H., Lambrecht, B.N., Eds.; Academic Press: Boston, MA, USA, 2015; pp. 1943–1983. ISBN 978-0-12-415847-4. [Google Scholar]
- Ebersole, J.L.; Al-Sabbagh, M.; Gonzalez, O.A.; Dawson, D.R. Ageing Effects on Humoral Immune Responses in Chronic Periodontitis. J. Clin. Periodontol. 2018, 45, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, N.; Bhateja, S.; Arora, G.; Yasmin, T. Candidiasis- The Most Common Fungal Infection of Oral Cavity. Biomed. J. Sci. Tech. Res. 2018, 8, 001–003. [Google Scholar] [CrossRef]
- Zdziarski, P.; Paściak, M.; Rogala, K.; Korzeniowska-Kowal, A.; Gamian, A. Elizabethkingia miricola as an Opportunistic Oral Pathogen Associated with Superinfectious Complications in Humoral Immunodeficiency: A Case Report. BMC Infect. Dis. 2017, 17, 763. [Google Scholar] [CrossRef] [PubMed]
- Kamaguchi, M.; Iwata, H. The Diagnosis and Blistering Mechanisms of Mucous Membrane Pemphigoid. Front. Immunol. 2019, 10, 34. [Google Scholar] [CrossRef]
- Gaudin, O.; Seta, V.; Alexandre, M.; Bohelay, G.; Aucouturier, F.; Mignot-Grootenboer, S.; Ingen-Housz-Oro, S.; Bernardeschi, C.; Schneider, P.; Mellottee, B.; et al. Gliptin Accountability in Mucous Membrane Pemphigoid Induction in 24 Out of 313 Patients. Front. Immunol. 2018, 9, 1030. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-Y.; Kuo, Y.-W.; Chen, C.-W.; Huang, Y.-F.; Hsu, C.-H.; Lin, J.-H.; Liu, C.-R.; Chen, J.-F.; Hsia, K.-C.; Ho, H.-H. Viable and Heat-Killed Probiotic Strains Improve Oral Immunity by Elevating the IgA Concentration in the Oral Mucosa. Curr. Microbiol. 2021, 78, 3541–3549. [Google Scholar] [CrossRef]
- Santi, C.G.; Gripp, A.C.; Roselino, A.M.; Mello, D.S.; Gordilho, J.O.; de Marsillac, P.F.; Porro, A.M. Consensus on the Treatment of Autoimmune Bullous Dermatoses: Bullous Pemphigoid, Mucous Membrane Pemphigoid and Epidermolysis Bullosa Acquisita—Brazilian Society of Dermatology. An. Bras. Dermatol. 2019, 94, 33–47. [Google Scholar] [CrossRef]
- Ali, S.; Kelly, C.; Challacombe, S.J.; Donaldson, A.N.A.; Dart, J.K.G.; Gleeson, M.; MMP Study Group 2009-14; Setterfield, J.F. Salivary IgA and IgG Antibodies to Bullous Pemphigoid 180 Noncollagenous Domain 16a as Diagnostic Biomarkers in Mucous Membrane Pemphigoid. Br. J. Dermatol. 2016, 174, 1022–1029. [Google Scholar] [CrossRef]
- Alkan, A.; Günhan, Ö.; Alkan, A.; Otan, F. A Clinical Study of Oral Mucous Membrane Pemphigoid. J. Int. Med. Res. 2003, 31, 340–344. [Google Scholar] [CrossRef]
- Carey, B.; Setterfield, J. Mucous Membrane Pemphigoid and Oral Blistering Diseases. Clin. Exp. Dermatol. 2019, 44, 732–739. [Google Scholar] [CrossRef]
- Xu, H.-H.; Werth, V.P.; Parisi, E.; Sollecito, T.P. Mucous Membrane Pemphigoid. Dent. Clin. N. Am. 2013, 57, 611–630. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.; Lamberts, A.; Diercks, G.F.H.; Pas, H.H.; Meijer, J.M.; Bolling, M.C.; Horváth, B. Oral Lesions in Autoimmune Bullous Diseases: An Overview of Clinical Characteristics and Diagnostic Algorithm. Am. J. Clin. Dermatol. 2019, 20, 847–861. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.; Rashid, H.; Marzano, A.V.; Lamberts, A.; Di Zenzo, G.; Diercks, G.F.H.; Alberti-Violetti, S.; Barry, R.J.; Borradori, L.; Caproni, M.; et al. European Guidelines (S3) on Diagnosis and Management of Mucous Membrane Pemphigoid, Initiated by the European Academy of Dermatology and Venereology—Part II. J. Eur Acad. Dermatol. Venereol. 2021, 35, 1926–1948. [Google Scholar] [CrossRef] [PubMed]
- Saccucci, M.; Di Carlo, G.; Bossù, M.; Giovarruscio, F.; Salucci, A.; Polimeni, A. Autoimmune Diseases and Their Manifestations on Oral Cavity: Diagnosis and Clinical Management. J. Immunol. Res. 2018, 2018, e6061825. [Google Scholar] [CrossRef] [PubMed]
- Gomes, R.R. Isolated Oral Pemphigus Vulgaris. Int. J. Clin. Stud. Med. Case 2021, 8, 1–4. [Google Scholar] [CrossRef]
- Mortazavi, H.; Saficac, Y.; Baharvand, M.; Rahmani, S. Diagnostic Features of Common Oral Ulcerative Lesions: An Updated Decision Tree. Int. J. Dent. 2016, 2016, e7278925. [Google Scholar] [CrossRef] [PubMed]
- Porro, A.M.; Seque, C.A.; Ferreira, M.C.C.; Enokihara, M.M.S.E.S. Pemphigus Vulgaris. An. Bras. Dermatol. 2019, 94, 264–278. [Google Scholar] [CrossRef]
- Kridin, K.; Bergman, R. Assessment of the Prevalence of Mucosal Involvement in Bullous Pemphigoid. JAMA Dermatol. 2019, 155, 166. [Google Scholar] [CrossRef]
- Waschke, J.; Bruggeman, P.; Baumgartner, W.; Zillikens, D.; Drenckhahn, D. Pemphigus Foliaceus IgG Causes Dissociation of Desmoglein 1–Containing Junctions without Blocking Desmoglein 1 Transinteraction. J. Clin. Investig. 2005, 115, 3157–3165. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Ndoye, A.; Shultz, L.D.; Pittelkow, M.R.; Grando, S.A. Antibodies against Keratinocyte Antigens Other than Desmogleins 1 and 3 Can Induce Pemphigus Vulgaris–like Lesions. J. Clin. Investig. 2000, 106, 1467–1479. [Google Scholar] [CrossRef]
- Harman, K.E.; Seed, P.T.; Gratian, M.J.; Bhogal, B.S.; Challacombe, S.J.; Black, M.M. The Severity of Cutaneous and Oral Pemphigus Is Related to Desmoglein 1 and 3 Antibody Levels. Br. J. Dermatol 2001, 144, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, M.G.; Wang, Z.; Rothenberger, K.; Koch, P.J.; Amagai, M.; Stanley, J.R. Explanations for the Clinical and Microscopic Localization of Lesions in Pemphigus Foliaceus and Vulgaris. J. Clin. Investig. 1999, 103, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Melchionda, V.; Harman, K.E. Pemphigus Vulgaris and Pemphigus Foliaceus: An Overview of the Clinical Presentation, Investigations and Management. Clin. Exp. Dermatol. 2019, 44, 740–746. [Google Scholar] [CrossRef]
- Genovese, G.; Venegoni, L.; Fanoni, D.; Muratori, S.; Berti, E.; Marzano, A.V. Linear IgA Bullous Dermatosis in Adults and Children: A Clinical and Immunopathological Study of 38 Patients. Orphanet J. Rare Dis. 2019, 14, 115. [Google Scholar] [CrossRef] [PubMed]
- Beyzaee, A.M.; Rahmatpour Rokni, G.; Patil, A.; Goldust, M. Rituximab as the Treatment of Pemphigus Vulgaris in the COVID-19 Pandemic Era: A Narrative Review. Dermatol. Ther. 2021, 34, e14405. [Google Scholar] [CrossRef]
- Bernett, C.N.; Fong, M.; Yadlapati, S.; Rosario-Collazo, J.A. Linear IGA Dermatosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Lammer, J.; Hein, R.; Roenneberg, S.; Biedermann, T.; Volz, T. Drug-Induced Linear IgA Bullous Dermatosis: A Case Report and Review of the Literature. Acta Derm. Venereol. 2019, 99, 508–515. [Google Scholar] [CrossRef]
- Shin, L.; Gardner, J.T.; Dao, H. Updates in the Diagnosis and Management of Linear IgA Disease: A Systematic Review. Medicina 2021, 57, 818. [Google Scholar] [CrossRef]
- do Vale, E.C.S.; Dimatos, O.C.; Porro, A.M.; Santi, C.G. Consensus on the Treatment of Autoimmune Bullous Dermatoses: Dermatitis Herpetiformis and Linear IgA Bullous Dermatosis—Brazilian Society of Dermatology. An. Bras. Dermatol. 2019, 94, 48–55. [Google Scholar] [CrossRef]
- Kridin, K.; Kneiber, D.; Kowalski, E.H.; Valdebran, M.; Amber, K.T. Epidermolysis Bullosa Acquisita: A Comprehensive Review. Autoimmun. Rev. 2019, 18, 786–795. [Google Scholar] [CrossRef]
- Chen, M.; Kim, G.H.; Prakash, L.; Woodley, D.T. Epidermolysis Bullosa Acquisita: Autoimmunity to Anchoring Fibril Collagen. Autoimmunity 2012, 45, 91–101. [Google Scholar] [CrossRef]
- Gupta, R.; Woodley, D.T.; Chen, M. Epidermolysis Bullosa Acquitsita. Clin. Dermatol. 2012, 30, 60–69. [Google Scholar] [CrossRef]
- Pratasava, V.; Sahni, V.N.; Suresh, A.; Huang, S.; Are, A.; Hsu, S.; Motaparthi, K. Bullous Pemphigoid and Other Pemphigoid Dermatoses. Medicina 2021, 57, 1061. [Google Scholar] [CrossRef]
- Xiao, A.; Tsuchiya, A. Epidermolysis Bullosa Acquisita. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Kulkarni, R.; Payne, A.S.; Werth, V.P.; Sollecito, T.P.; Stoopler, E.T. Custom dental trays with topical corticosteroids for management of gingival lesions of mucous membrane pemphigoid. Int. J. Dermatol. 2020, 59, e211–e213. [Google Scholar] [CrossRef]
- Vorobyev, A.; Ludwig, R.J.; Schmidt, E. Clinical Features and Diagnosis of Epidermolysis Bullosa Acquisita. Expert Rev. Clin. Immunol. 2017, 13, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Mysorekar, V.V.; Sumathy, T.K.; Shyam Prasad, A.L. Role of Direct Immunofluorescence in Dermatological Disorders. Indian Dermatol. Online J. 2015, 6, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.F.; Holland, S.M. The Hyper IgE Syndromes. Immunol. Allergy Clin. N. Am. 2008, 28, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Koga, H.; Prost-Squarcioni, C.; Iwata, H.; Jonkman, M.F.; Ludwig, R.J.; Bieber, K. Epidermolysis Bullosa Acquisita: The 2019 Update. Front. Med. 2019, 5, 362. [Google Scholar] [CrossRef]
- Minegishi, Y. Hyper-IgE Syndrome, 2021 Update. Allergol. Int. 2021, 70, 407–414. [Google Scholar] [CrossRef]
- Gerreth, K.; Szczawinska-Poplonyk, A.; Kycler, Z.; Adamski, Z.; Borysewicz-Lewicka, M.; Breborowicz, A. Factors Causing Oral and Skin Pathological Features in the Hyperimmunoglobulin E Syndrome Patient Including the Environmental Component: A Review of the Literature and Own Experience. Postepy Dermatol. Alergol. 2020, 37, 326–332. [Google Scholar] [CrossRef]
- Al-Shaikhly, T.; Ochs, H.D. Hyper IgE Syndromes: Clinical and Molecular Characteristics. Immunol. Cell Biol. 2019, 97, 368–379. [Google Scholar] [CrossRef]
- Vila, T.; Sultan, A.S.; Montelongo-Jauregui, D.; Jabra-Rizk, M.A. Oral Candidiasis: A Disease of Opportunity. J. Fungi 2020, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.F.; Domingo, D.L.; Holland, S.M. Hyper IgE (Job’s) Syndrome: A Primary Immune Deficiency with Oral Manifestations. Oral Dis. 2009, 15, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Domingo, D.L.; Freeman, A.F.; Davis, J.; Puck, J.M.; Tianxia, W.; Holland, S.M.; Hart, T.C. Novel Intraoral Phenotypes in Hyperimmunoglobulin-E Syndrome. Oral Dis. 2008, 14, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Park, B.; Liu, G.Y. Staphylococcus Aureus and Hyper-IgE Syndrome. Int. J. Mol. Sci. 2020, 21, 9152. [Google Scholar] [CrossRef]
- O’Connell, A.C.; Puck, J.M.; Grimbacher, B.; Facchetti, F.; Majorana, A.; Gallin, J.I.; Malech, H.L.; Holland, S.M. Delayed Eruption of Permanent Teeth in Hyperimmunoglobulinemia E Recurrent Infection Syndrome. Oral Surg Oral Med. Oral Pathol. Oral Radiol. Endod. 2000, 89, 177–185. [Google Scholar] [CrossRef]
- Gharehzadehshirazi, A.; Amini, A.; Rezaei, N. Hyper IgE Syndromes: A Clinical Approach. Clin. Immunol. 2022, 237, 108988. [Google Scholar] [CrossRef]
- Marcotte, H.; Lavoie, M.C. Oral Microbial Ecology and the Role of Salivary Immunoglobulin A. Microbiol. Mol. Biol. Rev. 1998, 62, 71–109. [Google Scholar] [CrossRef]
- Ruggeri, F.M.; Johansen, K.; Basile, G.; Kraehenbuhl, J.-P.; Svensson, L. Antirotavirus Immunoglobulin A Neutralizes Virus In Vitro after Transcytosis through Epithelial Cells and Protects Infant Mice from Diarrhea. J. Virol. 1998, 72, 2708–2714. [Google Scholar] [CrossRef]
- Millet, N.; Solis, N.V.; Swidergall, M. Mucosal IgA Prevents Commensal Candida Albicans Dysbiosis in the Oral Cavity. Front. Immunol. 2020, 11, 555363. [Google Scholar] [CrossRef]
- Chang, E.; Kobayashi, R.; Fujihashi, K.; Komiya, M.; Kurita-Ochiai, T. Impaired Salivary SIgA Antibodies Elicit Oral Dysbiosis and Subsequent Induction of Alveolar Bone Loss. Inflamm. Res. 2021, 70, 151–158. [Google Scholar] [CrossRef]
- Arisawa, E.A.L.; Almeida, J.D.; Carvalho, Y.R.; Cabral, L.A.G. Clinicopathological Analysis of Oral Mucous Autoimmune Disease: A 27-Year Study. Med. Oral Patol. Oral Cir. Bucal. 2008, 13, E94–E97. [Google Scholar] [PubMed]
- Femiano, F.; Gombos, F.; Nunziata, M.; Esposito, V.; Scully, C. Pemphigus Mimicking Aphthous Stomatitis. J. Oral Pathol. Med. 2005, 34, 508–510. [Google Scholar] [CrossRef] [PubMed]
- Scully, C.; Paes De Almeida, O.; Porter, S.R.; Gilkes, J.J. Pemphigus Vulgaris: The Manifestations and Long-Term Management of 55 Patients with Oral Lesions. Br. J. Dermatol. 1999, 140, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.C.; Lozada-Nur, F.; Frieden, I. Oral Pemphigus Vulgaris: A Review of the Literature and a Report on the Management of 12 Cases. Oral Surg Oral Med. Oral Pathol. Oral Radiol. Endod. 1997, 84, 349–355. [Google Scholar] [CrossRef]
- Sultan, A.S.; Villa, A.; Saavedra, A.P.; Treister, N.S.; Woo, S.-B. Oral Mucous Membrane Pemphigoid and Pemphigus Vulgaris-a Retrospective Two-Center Cohort Study. Oral Dis. 2017, 23, 498–504. [Google Scholar] [CrossRef]
- Palmer, R.A.; Ogg, G.; Allen, J.; Banerjee, A.; Ryatt, K.S.; Ratnavel, R.; Wojnarowska, F. Vancomycin-Induced Linear IgA Disease with Autoantibodies to BP180 and LAD285. Br. J. Dermatol. 2001, 145, 816–820. [Google Scholar] [CrossRef]
- Buckley, R.H.; Wray, B.B.; Belmaker, E.Z. Extreme Hyperimmunoglobulinemia E and Undue Susceptibility to Infection. Pediatrics 1972, 49, 59–70. [Google Scholar] [CrossRef]
- Grimbacher, B.; Holland, S.M.; Gallin, J.I.; Greenberg, F.; Hill, S.C.; Malech, H.L.; Miller, J.A.; O’Connell, A.C.; Puck, J.M. Hyper-IgE Syndrome with Recurrent Infections--an Autosomal Dominant Multisystem Disorder. N. Engl. J. Med. 1999, 340, 692–702. [Google Scholar] [CrossRef]
- Allen, J.; Wojnarowska, F. Linear IgA Disease: The IgA and IgG Response to Dermal Antigens Demonstrates a Chiefly IgA Response to LAD285 and a Dermal 180-KDa Protein. Br. J. Dermatol. 2003, 149, 1055–1058. [Google Scholar] [CrossRef]
- Zhang, Q.; Boisson, B.; Béziat, V.; Puel, A.; Casanova, J.-L. Human Hyper-IgE Syndrome: Singular or Plural? Mamm. Genome 2018, 29, 603–617. [Google Scholar] [CrossRef]
- Epstein, J.B.; Güneri, P.; Boyacioglu, H.; Abt, E. The Limitations of the Clinical Oral Examination in Detecting Dysplastic Oral Lesions and Oral Squamous Cell Carcinoma. J. Am. Dent. Assoc. 2012, 143, 1332–1342. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Disease | Clinical Manifestations | Diagnostic Tests | Treatment |
|---|---|---|---|
| mucous membrane pemphigoid (MMP) | desquamative gingivitis, erythematous plaques and erosions covered by pseudomembranes located on the gingiva, palate, lips, tongue and cheek mucosa | clinical, histological and immunopathological findings, direct immunofluorescence (DIF) for immunoglobulin G, A and M (IgG, IgA, IgM) or complement component 3 (C3) | systemic corticosteroids (prednisone), immunosuppressive drugs (cyclophosphamide, azathioprine) |
| Pemphigus vulgaris (PV) | painful mucosal ulceration | immunoblotting with immunoprecipitation | systemic corticosteroids |
| Linear IgA bullous dermatosis (LABD) | blisters and erosions of oral mucosa, desquamative gingivitis | biopsies for histopathological examination, indirect immunofluorescence (IIF) | dapsone, an immunomodulatory sulphone |
| Epidermolysis Bullosa Aquisita (EBA) | vesicle and bullae formation on the skin and erosions within the mucous membranes | detection of autoantibodies bound to the basement membrane zone and against collagen VII in the serum, biopsy (direct immunofluorescence) | non-specific immunosuppression |
| Hyper-IgE syndrome (HIES) | skin and pulmonary abscesses, eczematoid dermatitis, markedly elevated levels of serum IgE and eosinophilia, reduced neutrophil chemotaxis | the National Institutes of Health (NIH) scoring system | antibiotic prophylaxis with trimethoprim-sulfamethoxazole |
| Disease | Type | Immune Factors | Risk Factors | Symptoms |
|---|---|---|---|---|
| MMP | Autoimmune dermatoses disease | Linear deposition of IgG, IgA, C3, rarely IgG Target antigens in the basement membrane region of the epithelium: BP230, BP180, laminin 5, laminin 6, type VII collagen and β4 | Age > 50 years, female sex | Subepidermal bullous lesions |
| PV | Autoimmune disease | Autoantibodies react to Dsg 1 and 3 | - | Ulceration of the oral and esophageal mucosa, followed by skin involvement |
| LABD | Subepidermal vesiculobullous disease | Deposition of IgA rather than IgG in the basement membrane | Genetic factors, medication (vancomycin) and antibiotic use (cephalosporins, penicillins) | Oral lesions clinically identical to MMPs |
| EBA | Autoimmune blistering disease | IgG autoantibodies directed against type VII collagen | - | Blisters and blisters on the skin and erosions on the mucous membranes |
| HIES | Multisystem disorder | - | STAT3 mutations | Facial asymmetry, fleshy nose tip, deep-set eyes, prominent forehead, triad of eczema, recurrent skin and lung infections, high serum IgE levels, preserved primary dentition, oral mucosal and gingival lesions, oral candidiasis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ptasiewicz, M.; Bębnowska, D.; Małkowska, P.; Sierawska, O.; Poniewierska-Baran, A.; Hrynkiewicz, R.; Niedźwiedzka-Rystwej, P.; Grywalska, E.; Chałas, R. Immunoglobulin Disorders and the Oral Cavity: A Narrative Review. J. Clin. Med. 2022, 11, 4873. https://doi.org/10.3390/jcm11164873
Ptasiewicz M, Bębnowska D, Małkowska P, Sierawska O, Poniewierska-Baran A, Hrynkiewicz R, Niedźwiedzka-Rystwej P, Grywalska E, Chałas R. Immunoglobulin Disorders and the Oral Cavity: A Narrative Review. Journal of Clinical Medicine. 2022; 11(16):4873. https://doi.org/10.3390/jcm11164873
Chicago/Turabian StylePtasiewicz, Maja, Dominika Bębnowska, Paulina Małkowska, Olga Sierawska, Agata Poniewierska-Baran, Rafał Hrynkiewicz, Paulina Niedźwiedzka-Rystwej, Ewelina Grywalska, and Renata Chałas. 2022. "Immunoglobulin Disorders and the Oral Cavity: A Narrative Review" Journal of Clinical Medicine 11, no. 16: 4873. https://doi.org/10.3390/jcm11164873
APA StylePtasiewicz, M., Bębnowska, D., Małkowska, P., Sierawska, O., Poniewierska-Baran, A., Hrynkiewicz, R., Niedźwiedzka-Rystwej, P., Grywalska, E., & Chałas, R. (2022). Immunoglobulin Disorders and the Oral Cavity: A Narrative Review. Journal of Clinical Medicine, 11(16), 4873. https://doi.org/10.3390/jcm11164873