
Key Chemokine Pathways in Atherosclerosis and Their Therapeutic Potential



Abstract
1. Introduction
1.1. Chemokines
1.2. Atherosclerosis
2. Chemokines, Chemokine Receptors, and Atherosclerosis: A Complex Interplay
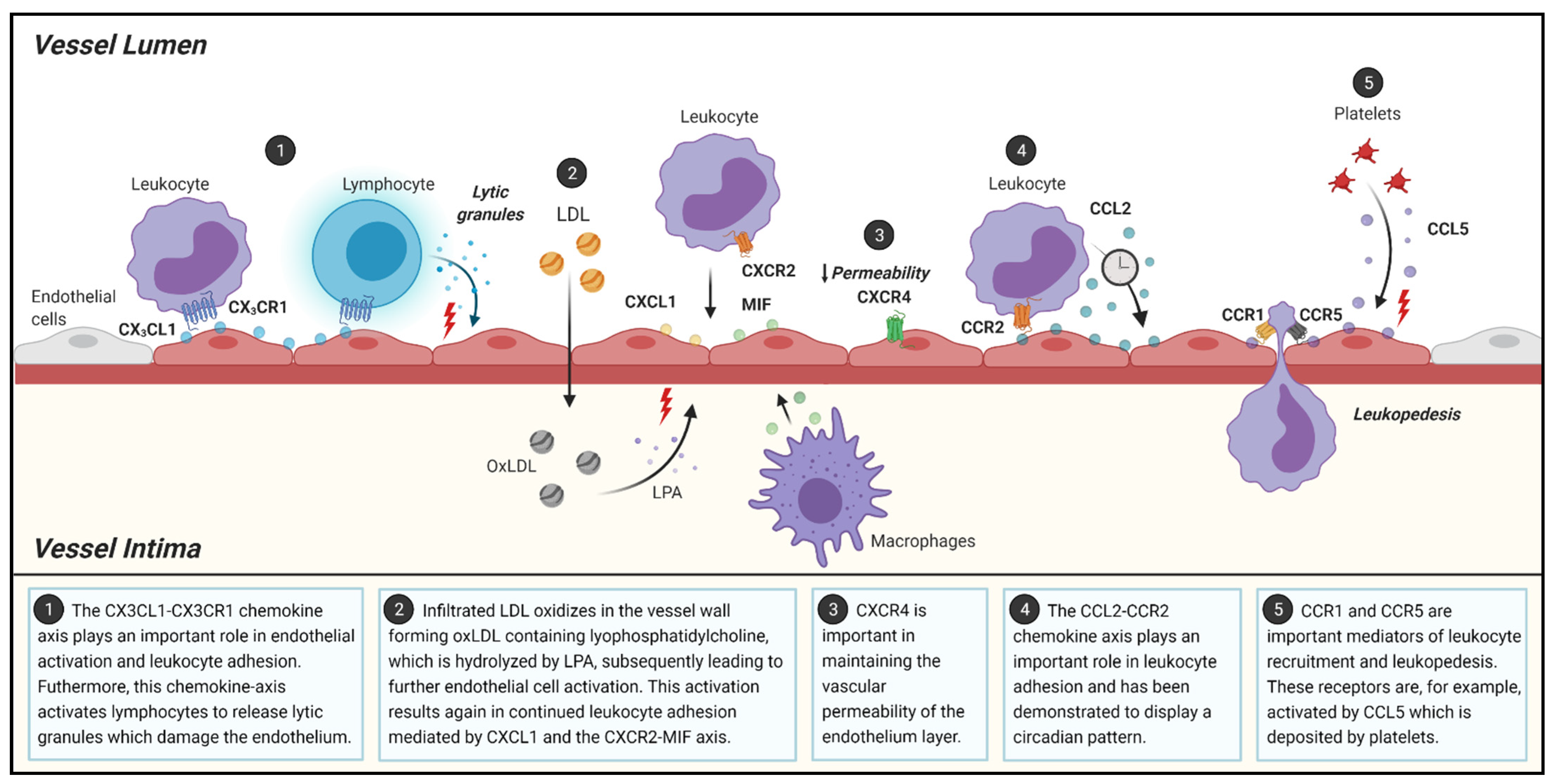
2.1. Initiation of Atherosclerosis
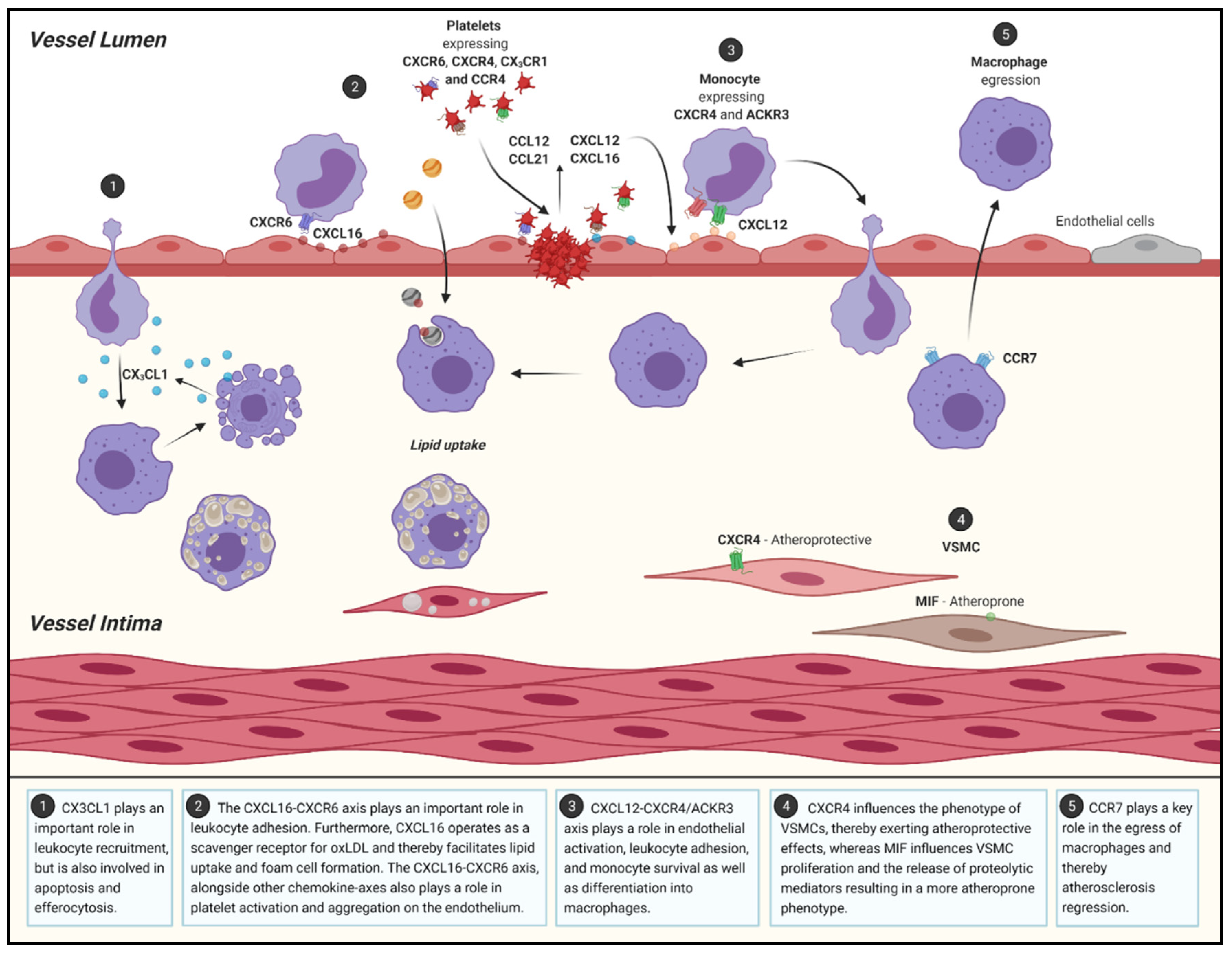
2.2. Atherosclerotic Progression
2.3. Regression in Atherosclerosis
3. Targeting Chemokines and Chemokine Receptors: Therapeutic Potential in Atherosclerosis
3.1. Preclinical Studies: Discovering the Therapeutic Potential of Chemokines and Chemokine Receptors
3.2. Clinical Trials: Targeting Chemokines and Their Receptors as Therapeutic Tools
3.3. Imaging as a Diagnostic Tool in Atherosclerosis
4. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Roth, A.G.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Frostegård, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117. [Google Scholar] [CrossRef] [PubMed]
- Noels, H.; Weber, C.; Koenen, R.R. Chemokines as therapeutic targets in cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 583–592. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting early atherosclerosis: A focus on oxidative stress and inflammation. Oxid. Med. Cell. Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-dose methotrexate for the prevention of atherosclerotic events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.K.; Hernán, M.A.; Seeger, J.D.; Robins, J.M.; Wolfe, F. Methotrexate and mortality in patients with rheumatoid arthritis: A prospective study. Lancet 2002, 359, 1173–1177. [Google Scholar] [CrossRef]
- Micha, R.; Imamura, F.; von Ballmoos, M.W.; Solomon, D.H.; Hernán, M.; Ridker, P.M.; Mozaffarian, D. Systematic review and meta-analysis of methotrexate use and risk of cardiovascular disease. Am. J. Cardiol. 2011, 108, 1362–1370. [Google Scholar] [CrossRef] [PubMed]
- Westlake, S.L.; Colebatch, A.N.; Baird, J.; Kiely, P.; Quinn, M.; Choy, E.; Ostor, A.J.K.; Edwards, C.J. The effect of methotrexate on cardiovascular disease in patients with rheumatoid arthritis: A systematic literature review. Rheumatology 2009, 49, 295–307. [Google Scholar] [CrossRef]
- Zernecke, A.; Weber, C. Chemokines in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 742–750. [Google Scholar] [CrossRef]
- Van Der Vorst, E.P.C.; Döring, Y.; Weber, C. Chemokines and their receptors in Atherosclerosis. J. Mol. Med. 2015, 93, 963–971. [Google Scholar] [CrossRef]
- Gencer, S.; Evans, B.; van der Vorst, E.; Döring, Y.; Weber, C. Inflammatory chemokines in atherosclerosis. Cells 2021, 10, 226. [Google Scholar] [CrossRef]
- Raman, D.; Sobolik-Delmaire, T.; Richmond, A. Chemokines in health and disease. Exp. Cell Res. 2011, 317, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Laing, K.J. Chemokines. Dev. Comp. Immunol. 2003, 28, 443–460. [Google Scholar] [CrossRef] [PubMed]
- Bonecchi, R.; Graham, G.J. Atypical chemokine receptors and their roles in the resolution of the inflammatory response. Front. Immunol. 2016, 7, 224. [Google Scholar] [CrossRef]
- Chen, K.; Bao, Z.; Tang, P.; Gong, W.; Yoshimura, T.; Wang, J.M. Chemokines in homeostasis and diseases. Cell. Mol. Immunol. 2018, 15, 324–334. [Google Scholar] [CrossRef]
- Blanchet, X.; Langer, M.; Weber, C.; Koenen, R.; von Hundelshausen, P. Touch of chemokines. Front. Immunol. 2012, 3, 175. [Google Scholar] [CrossRef] [PubMed]
- Van Der Vorst, E.P.C.; Peters, L.J.F.; Müller, M.; Gencer, S.; Yan, Y.; Weber, C.; Döring, Y. G-protein coupled receptor targeting on myeloid cells in atherosclerosis. Front. Pharmacol. 2019, 10, 531. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Moser, B. Chemokines: Role in inflammation and immune surveillance. Ann. Rheum. Dis. 2004, 63, ii84–ii89. [Google Scholar] [CrossRef]
- Bacon, K.; Baggiolini, M.; Broxmeyer, H.; Horuk, R.; Lindley, I.; Mantovani, A.; Matsushima, K.; Murphy, P.; Nomiyama, H.; Zoon, K. Chemokine/Chemokine receptor nomenclature. J. Leukoc. Biol. 2001, 70, 465–466. [Google Scholar] [CrossRef]
- Tang, P.; Wang, J.M. Chemokines: The past, the present and the future. Cell. Mol. Immunol. 2018, 15, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediat. Inflamm. 2013, 2013, 480739. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Ley, K. Immunity and inflammation in atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.N. Atherosclerosis—An inflammatory process. J. Insur. Med. 2005, 37, 72–75. [Google Scholar]
- Pamukcu, B.; Lip, G.Y.; Shantsila, E. The nuclear factor—Kappa B pathway in atherosclerosis: A potential therapeutic target for atherothrombotic vascular disease. Thromb. Res. 2011, 128, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sun, B. Toll-like receptor 4 in atherosclerosis. J. Cell. Mol. Med. 2007, 11, 88–95. [Google Scholar] [CrossRef]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int. J. Prevent. Med. 2014, 5, 927–946. [Google Scholar]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef]
- Insull, W. The pathology of atherosclerosis: Plaque development and plaque responses to medical treatment. Am. J. Med. 2009, 122, S3–S14. [Google Scholar] [CrossRef] [PubMed]
- Bergheanu, S.C.; Bodde, M.C.; Jukema, J.W. Pathophysiology and treatment of atherosclerosis. Neth. Heart J. 2017, 25, 231–242. [Google Scholar] [CrossRef]
- Reustle, A.; Torzewski, M. Role of p38 MAPK in atherosclerosis and aortic valve sclerosis. Int. J. Mol. Sci. 2018, 19, 3761. [Google Scholar] [CrossRef]
- De Winther, M.P.J.; Kanters, E.; Kraal, G.; Hofker, M.H. Nuclear factor κB signaling in atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 904–914. [Google Scholar] [CrossRef]
- Nording, H.M.; Seizer, P.; Langer, H.F. Platelets in inflammation and atherogenesis. Front. Immunol. 2015, 6, 98. [Google Scholar] [CrossRef] [PubMed]
- Dubland, J.; Francis, G.A. Lysosomal acid lipase: At the crossroads of normal and atherogenic cholesterol metabolism. Front. Cell Dev. Biol. 2015, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-H.; Fu, Y.-C.; Zhang, D.-W.; Yin, K.; Tang, C.-K. Foam cells in atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Macrophage-mediated cholesterol handling in atherosclerosis. J. Cell. Mol. Med. 2015, 20, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.; Grechko, A.; Orekhov, A.N. Mechanisms of foam cell formation in atherosclerosis. J. Mol. Med. 2017, 95, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Liu, H.; Jiang, D. Fractalkine/CX3CR1 and atherosclerosis. Clin. Chim. Acta 2011, 412, 1180–1186. [Google Scholar] [CrossRef]
- Apostolakis, S.; Spandidos, A.D. Chemokines and atherosclerosis: Focus on the CX3CL1/CX3CR1 pathway. Acta Pharmacol. Sin. 2013, 34, 1251–1256. [Google Scholar] [CrossRef]
- Yoneda, O.; Imai, T.; Goda, S.; Inoue, H.; Yamauchi, A.; Okazaki, T.; Imai, H.; Yoshie, O.; Bloom, E.T.; Domae, N.; et al. Fractalkine-mediated endothelial cell injury by nK cells. J. Immunol. 2000, 164, 4055–4062. [Google Scholar] [CrossRef]
- White, G.E.; Greaves, D.R. Fractalkine: A survivor’s guide. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Alon, R.; Feigelson, S. From rolling to arrest on blood vessels: Leukocyte tap dancing on endothelial integrin ligands and chemokines at sub-second contacts. Semin. Immunol. 2002, 14, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Döring, Y.; Noels, H.; Van Der Vorst, E.P.; Neideck, C.; Egea, V.; Drechsler, M.; Mandl, M.; Pawig, L.; Jansen, Y.; Schröder, K.; et al. Vascular CXCR4 limits atherosclerosis by maintaining arterial integrity. Circulation 2017, 136, 388–403. [Google Scholar] [CrossRef]
- Zhou, Z.; Subramanian, P.; Sevilmis, G.; Globke, B.; Soehnlein, O.; Karshovska, E.; Megens, R.; Heyll, K.; Chun, J.; Saulnier-Blache, J.S.; et al. Lipoprotein-derived lysophosphatidic acid promotes atherosclerosis by releasing CXCL1 from the endothelium. Cell Metab. 2011, 13, 592–600. [Google Scholar] [CrossRef]
- Sellau, J.; Groneberg, M.; Fehling, H.; Thye, T.; Hoenow, S.; Marggraff, C.; Weskamm, M.; Hansen, C.; Stanelle-Bertram, S.; Kuehl, S.; et al. Androgens predispose males to monocyte-mediated immunopathology by inducing the expression of leukocyte recruitment factor CXCL1. Nat. Commun. 2020, 11, 3459. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.; Libby, P.; Aikawa, E.; Alcaide, P.; Luscinskas, F.W.; Weissleder, R.; Pittet, M.J. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Investig. 2007, 117, 195–205. [Google Scholar] [CrossRef]
- Soehnlein, O.; Drechsler, M.; Döring, Y.; Lievens, D.; Hartwig, H.; Kemmerich, K.; Ortega-Gomez, A.; Mandl, M.; Vijayan, S.; Projahn, D.; et al. Distinct functions of chemokine receptor axes in the atherogenic mobilization and recruitment of classical monocytes. EMBO Mol. Med. 2013, 5, 471–481. [Google Scholar] [CrossRef]
- Bernhagen, J.; Krohn, R.; Lue, H.; Gregory, J.L.; Zernecke, A.; Koenen, R.; Dewor, M.; Georgiev, I.; Schober, A.; Leng, L.; et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 2007, 13, 587–596. [Google Scholar] [CrossRef]
- von Hundelshausen, P.; Weber, K.S.C.; Huo, Y.; Proudfoot, A.E.I.; Nelson, P.J.; Ley, K.; Weber, C. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation 2001, 103, 1772–1777. [Google Scholar] [CrossRef]
- Veillard, N.R.; Kwak, B.; Pelli, G.; Mulhaupt, F.; James, R.W.; Proudfoot, A.E.; Mach, F. Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ. Res. 2004, 94, 253–261. [Google Scholar] [CrossRef]
- Braunersreuther, V.; Zernecke, A.; Arnaud, C.; Liehn, E.A.; Steffens, S.; Shagdarsuren, E.; Bidzhekov, K.; Burger, F.; Pelli, G.; Luckow, B.; et al. Ccr5 but not Ccr1 deficiency reduces development of diet-induced atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 373–379. [Google Scholar] [CrossRef]
- Döring, Y.; Drechsler, M.; Soehnlein, O.; Weber, C. Neutrophils in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Georgakis, M.K.; Malik, R.; Björkbacka, H.; Pana, T.; Demissie, S.; Ayers, C.; Elhadad, M.A.; Fornage, M.; Beiser, A.S.; Benjamin, E.J.; et al. Circulating monocyte chemoattractant protein-1 and risk of stroke. Circ. Res. 2019, 125, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Okada, Y.; Clinton, S.K.; Gerard, C.; Sukhova, G.K.; Libby, P.; Rollins, B.J. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor–deficient mice. Mol. Cell 1998, 2, 275–281. [Google Scholar] [CrossRef]
- Boring, L.; Gosling, J.; Cleary, M.L.; Charo, I.F. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 1998, 394, 894–897. [Google Scholar] [CrossRef]
- Combadiere, C.; Potteaux, S.; Rodero, M.; Simon, T.; Pezard, A.; Esposito, B.; Merval, R.; Proudfoot, A.; Tedgui, A.; Mallat, Z. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C hi and Ly6C Lo monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 2008, 117, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Serbina, N.V.; Pamer, E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006, 7, 311–317. [Google Scholar] [CrossRef]
- Jung, H.; Mithal, D.S.; Park, J.E.; Miller, R.J. Localized CCR2 activation in the bone marrow niche mobilizes monocytes by desensitizing CXCR4. PLoS ONE 2015, 10, e0128387. [Google Scholar] [CrossRef]
- Winter, C.; Silvestre-Roig, C.; Ortega-Gomez, A.; Lemnitzer, P.; Poelman, H.; Schumski, A.; Winter, J.; Drechsler, M.; de Jong, R.; Immler, R.; et al. Chrono-pharmacological targeting of the CCL2-CCR2 axis ameliorates atherosclerosis. Cell Metab. 2018, 28, 175–182.e5. [Google Scholar] [CrossRef] [PubMed]
- Asam, S.; Nayar, S.; Gardner, D.; Barone, F. Stromal cells in tertiary lymphoid structures: Architects of autoimmunity. Immunol. Rev. 2021, 302, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Hill, D.G.; Jones, S.A. Understanding immune cells in tertiary lymphoid organ development: It is all starting to come together. Front. Immunol. 2016, 7, 401. [Google Scholar] [CrossRef]
- Barone, F.; Gardner, D.H.; Nayar, S.; Steinthal, N.; Buckley, C.D.; Luther, S.A. Stromal fibroblasts in tertiary lymphoid structures: A novel target in chronic inflammation. Front. Immunol. 2016, 7, 477. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Mohanta, S.; Srikakulapu, P.; Weber, C.; Habenicht, A. Artery tertiary lymphoid organs: Powerhouses of atherosclerosis immunity. Front. Immunol. 2016, 7, 387. [Google Scholar] [CrossRef]
- Lötzer, K.; Döpping, S.; Connert, S.; Gräbner, R.; Spanbroek, R.; Lemser, B.; Beer, M.; Hildner, M.; Hehlgans, T.; Van Der Wall, M.; et al. Mouse aorta smooth muscle cells differentiate into lymphoid tissue organizer-like cells on combined tumor necrosis factor receptor-1/Lymphotoxin-receptor NF-B signaling. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 395–402. [Google Scholar] [CrossRef]
- Lin, L.; Hu, X.; Zhang, H.; Hu, H. Tertiary lymphoid organs in cancer immunology: Mechanisms and the new strategy for immunotherapy. Front. Immunol. 2019, 10, 1398. [Google Scholar] [CrossRef]
- Aloisi, F.; Borrell, R.P. Lymphoid neogenesis in chronic inflammatory diseases. Nat. Rev. Immunol. 2006, 6, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Gräbner, R.; Lötzer, K.; Döpping, S.; Hildner, M.; Radke, D.; Beer, M.; Spanbroek, R.; Lippert, B.; Reardon, C.A.; Getz, G.S.; et al. Lymphotoxin β receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE−/− mice. J. Exp. Med. 2009, 206, 233–248. [Google Scholar] [CrossRef]
- Houtkamp, M.A.; de Boer, O.J.; van der Loos, C.M.; van der Wal, A.C.; Becker, A.E. Adventitial infiltrates associated with advanced atherosclerotic plaques: Structural organization suggests generation of local humoral immune responses. J. Pathol. 2001, 193, 263–269. [Google Scholar] [CrossRef]
- Moos, M.P.; John, N.; Gräbner, R.; Noßmann, S.; Günther, B.; Vollandt, R.; Funk, C.D.; Kaiser, B.; Habenicht, A.J. The lamina adventitia is the major site of immune cell accumulation in standard chow-fed apolipoprotein E–deficient mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2386–2391. [Google Scholar] [CrossRef]
- Zhao, L.; Moos, M.P.W.; Gräbner, R.; Pédrono, F.; Fan, J.; Kaiser, B.; John, N.; Schmidt, S.; Spanbroek, R.; Lötzer, K.; et al. The 5-lipoxygenase pathway promotes pathogenesis of hyperlipidemia-dependent aortic aneurysm. Nat. Med. 2004, 10, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Srikakulapu, P.; Upadhye, A.; Rosenfeld, S.M.; Marshall, M.A.; McSkimming, C.; Hickman, A.W.; Mauldin, I.; Ailawadi, G.; Lopes, M.B.S.; Taylor, A.M.; et al. Perivascular adipose tissue harbors atheroprotective IgM-producing B cells. Front. Physiol. 2017, 8, 719. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, S.M.; Perry, H.M.; Gonen, A.; Prohaska, T.; Srikakulapu, P.; Grewal, S.; Das, D.; McSkimming, C.; Taylor, A.M.; Tsimikas, S.; et al. B-1b cells secrete atheroprotective IgM and attenuate atherosclerosis. Circ. Res. 2015, 117, e28–e39. [Google Scholar] [CrossRef]
- Ait-Oufella, H.; Herbin, O.; Bouaziz, J.-D.; Binder, C.J.; Uyttenhove, C.; Laurans, L.; Taleb, S.; Van Vré, E.; Esposito, B.; Vilar, J.; et al. B cell depletion reduces the development of atherosclerosis in mice. J. Exp. Med. 2010, 207, 1579–1587. [Google Scholar] [CrossRef]
- Kyaw, T.; Tay, C.; Khan, A.; DuMouchel, V.; Cao, A.; To, K.; Kehry, M.; Dunn, R.; Agrotis, A.; Tipping, P.; et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J. Immunol. 2010, 185, 4410–4419. [Google Scholar] [CrossRef]
- Kyaw, T.; Tay, C.; Hosseini, H.; Kanellakis, P.; Gadowski, T.; Mackay, F.; Tipping, P.; Bobik, A.; Toh, B.-H. Depletion of B2 but not B1a B cells in BAFF receptor-deficient ApoE−/− mice attenuates atherosclerosis by potently ameliorating arterial inflammation. PLoS ONE 2012, 7, e29371. [Google Scholar] [CrossRef]
- Ait-Oufella, H.; Sage, A.; Mallat, Z.; Tedgui, A. Adaptive (T and B Cells) immunity and control by dendritic cells in atherosclerosis. Circ. Res. 2014, 114, 1640–1660. [Google Scholar] [CrossRef] [PubMed]
- Stein, J.V.; Nombela-Arrieta, C. Chemokine control of lymphocyte trafficking: A general overview. Immunology 2005, 116, 1–12. [Google Scholar] [CrossRef]
- Doran, A.C.; Lipinski, M.J.; Oldham, S.N.; Garmey, J.C.; Campbell, K.A.; Skaflen, M.D.; Cutchins, A.; Lee, D.J.; Glover, D.K.; Kelly, K.A.; et al. B-cell aortic homing and atheroprotection depend on ID3. Circ. Res. 2012, 110, e1–e12. [Google Scholar] [CrossRef]
- Virdis, A. Endothelial dysfunction in obesity: Role of inflammation. High Blood Press. Cardiovasc. Prev. 2016, 23, 83–85. [Google Scholar] [CrossRef]
- Omar, A.; Chatterjee, T.K.; Tang, Y.; Hui, D.Y.; Weintraub, N.L. Proinflammatory phenotype of perivascular adipocytes. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Morel, S.; Kwak, B.; Rohner-Jeanrenaud, F.; Steffens, S.; Molica, F. Adipokines at the crossroad between obesity and cardiovascular disease. Thromb. Haemost. 2015, 113, 553–566. [Google Scholar] [CrossRef]
- Ferrara, D.; Montecucco, F.; Dallegri, F.; Carbone, F. Impact of different ectopic fat depots on cardiovascular and metabolic diseases. J. Cell. Physiol. 2019, 234, 21630–21641. [Google Scholar] [CrossRef]
- Gil-Ortega, M.; Somoza, B.; Huang, Y.; Gollasch, M.; Fernández-Alfonso, M.S. Regional differences in perivascular adipose tissue impacting vascular homeostasis. Trends Endocrinol. Metab. 2015, 26, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Fitzgibbons, T.P.; Czech, M.P. Epicardial and perivascular adipose tissues and their influence on cardiovascular disease: Basic mechanisms and clinical associations. J. Am. Heart Assoc. 2014, 3, e000582. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Z.; Wang, C.; Ma, Q.; Zhao, Y. Perivascular adipose tissue-derived adiponectin inhibits collar-induced carotid atherosclerosis by promoting macrophage autophagy. PLoS ONE 2015, 10, e0124031. [Google Scholar] [CrossRef]
- Kauser, K.; Da Cunha, V.; Fitch, R.; Mallari, C.; Rubanyi, G.M. Role of endogenous nitric oxide in progression of atherosclerosis in apolipoprotein E-deficient mice. Am. J. Physiol. Circ. Physiol. 2000, 278, H1679–H1685. [Google Scholar] [CrossRef]
- Mani, S.; Li, H.; Untereiner, A.; Wu, L.; Yang, G.; Austin, R.C.; Dickhout, J.G.; Lhoták, S.; Meng, Q.H.; Wang, R. Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation 2013, 127, 2523–2534. [Google Scholar] [CrossRef]
- Manka, D.; Chatterjee, T.K.; Stoll, L.L.; Basford, J.E.; Konaniah, E.S.; Srinivasan, R.; Bogdanov, V.Y.; Tang, Y.; Blomkalns, A.L.; Hui, D.Y.; et al. Transplanted perivascular adipose tissue accelerates injury-induced neointimal hyperplasia. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1723–1730. [Google Scholar] [CrossRef]
- Konaniah, E.S.; Kuhel, D.G.; Basford, J.E.; Weintraub, N.L.; Hui, D.Y. Deficiency of LRP1 in mature adipocytes promotes diet-induced inflammation and atherosclerosis—Brief report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1046–1049. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2009, 10, 36–46. [Google Scholar] [CrossRef]
- Kojima, Y.; Weissman, I.L.; Leeper, N.J. The role of efferocytosis in atherosclerosis. Circulation 2017, 135, 476–489. [Google Scholar] [CrossRef]
- Elliott, M.R.; Koster, K.M.; Murphy, P.S. Efferocytosis signaling in the regulation of macrophage inflammatory responses. J. Immunol. 2017, 198, 1387–1394. [Google Scholar] [CrossRef]
- Landsman, L.; Bar-On, L.; Zernecke, A.; Kim, K.-W.; Krauthgamer, R.; Shagdarsuren, E.; Lira, S.A.; Weissman, I.L.; Weber, C.; Jung, S. CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood 2009, 113, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Panek, C.A.; Ramos, M.V.; Mejias, M.P.; Abrey-Recalde, M.J.; Fernandez-Brando, R.J.; Gori, M.S.; Salamone, G.V.; Palermo, M.S. Differential expression of the fractalkine chemokine receptor (CX3CR1) in human monocytes during differentiation. Cell. Mol. Immunol. 2014, 12, 669–680. [Google Scholar] [CrossRef]
- Van Vré, E.A.; Ait-Oufella, H.; Tedgui, A.; Mallat, Z. Apoptotic cell death and efferocytosis in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Lesnik, P.; Haskell, C.A.; Charo, I.F. Decreased atherosclerosis in CX3CR1–/– mice reveals a role for fractalkine in atherogenesis. J. Clin. Investig. 2003, 111, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Combadiere, C.; Potteaux, S.; Gao, J.-L.; Esposito, B.; Casanova, S.; Lee, E.J.; Debré, P.; Tedgui, A.; Murphy, P.M.; Mallat, Z. Decreased atherosclerotic lesion formation in CX3CR1/apolipoprotein E double knockout mice. Circulation 2003, 107, 1009–1016. [Google Scholar] [CrossRef]
- Scheuerer, B.; Ernst, M.; Dürrbaum-Landmann, I.; Fleischer, J.; Grage-Griebenow, E.; Brandt, E.; Flad, H.-D.; Petersen, F. The CXC-chemokine platelet factor 4 promotes monocyte survival and induces monocyte differentiation into macrophages. Blood 2000, 95, 1158–1166. [Google Scholar] [CrossRef]
- Sachais, B.S.; Turrentine, T.; Dawicki McKenna, J.M.; Rux, A.H.; Rader, D.; Kowalska, M.A. Elimination of platelet factor 4 (PF4) from platelets reduces atherosclerosis in C57Bl/6 and apoE−/− mice. Thromb. Haemost. 2007, 98, 1108–1113. [Google Scholar]
- Gutwein, P.; Abdel-Bakky, M.; Schramme, A.; Doberstein, K.; Kämpfer-Kolb, N.; Amann, K.; Hauser, I.A.; Obermüller, N.; Bartel, C.; Abdel-Aziz, A.-A.H.; et al. CXCL16 is expressed in podocytes and acts as a scavenger receptor for oxidized low-density lipoprotein. Am. J. Pathol. 2009, 174, 2061–2072. [Google Scholar] [CrossRef]
- dos Santos, S.M.; Blankenbach, K.; Scholich, K.; Dörr, A.; Monsefi, N.; Keese, M.; Linke, B.; Deckmyn, H.; Nelson, K.; Harder, S. Platelets from flowing blood attach to the inflammatory chemokine CXCL16 expressed in the endothelium of the human vessel wall. Thromb. Haemost. 2015, 114, 297–312. [Google Scholar] [CrossRef]
- Gencer, S.; Van Der Vorst, E.P.C.; Aslani, M.; Weber, C.; Döring, Y.; Duchene, J. Atypical chemokine receptors in cardiovascular disease. Thromb. Haemost. 2019, 119, 534–541. [Google Scholar] [CrossRef]
- Weber, C. Platelets and chemokines in atherosclerosis. Circ. Res. 2005, 96, 612–616. [Google Scholar] [CrossRef]
- Postea, O.; Vasina, E.M.; Cauwenberghs, S.; Projahn, D.; Liehn, E.A.; Lievens, D.; Theelen, W.; Kramp, B.K.; Butoi, E.D.; Soehnlein, O.; et al. Contribution of platelet CX3CR1 to platelet–monocyte complex formation and vascular recruitment during hyperlipidemia. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Gleissner, C.A.; von Hundelshausen, P.; Ley, K. Platelet chemokines in vascular disease. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1920–1927. [Google Scholar] [CrossRef] [PubMed]
- Gear, A.R.; Camerini, D. Platelet chemokines and chemokine receptors: Linking hemostasis, inflammation, and host defense. Microcirculation 2003, 10, 335–350. [Google Scholar] [CrossRef]
- Abi-Younes, S.; Sauty, A.; Mach, F.; Sukhova, G.K.; Libby, P.; Luster, A.D. The stromal cell–derived factor-1 chemokine is a potent platelet agonist highly expressed in atherosclerotic plaques. Circ. Res. 2000, 86, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; I Von Ungern-Sternberg, S.N.; Seizer, P.; Schlegel, F.; Büttcher, M.; Sindhu, N.A.; Muller, S.; Mack, A.F.; Gawaz, M. Platelet-derived CXCL12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of CXCR4–CXCR7. Cell Death Dis. 2015, 6, e1989. [Google Scholar] [CrossRef]
- Ma, W.; Liu, Y.; Ellison, N.; Shen, J. Induction of C-X-C chemokine receptor type 7 (Cxcr7) Switches stromal cell-derived factor-1 (Sdf-1) signaling and phagocytic activity in macrophages linked to atherosclerosis. J. Biol. Chem. 2013, 288, 15481–15494. [Google Scholar] [CrossRef]
- Wang, C.; Chen, W.; Shen, J.; Wang, C.; Chen, W.; Shen, J. CXCR7 targeting and its major disease relevance. Front. Pharmacol. 2018, 9, 641. [Google Scholar] [CrossRef]
- Quinn, K.; Mackie, D.; Caron, K. Emerging roles of atypical chemokine receptor 3 (ACKR3) in normal development and physiology. Cytokine 2018, 109, 17–23. [Google Scholar] [CrossRef]
- Gencer, S.; Döring, Y.; Jansen, Y.; Bayasgalan, S.; Schengel, O.; Müller, M.; Peters, L.; Weber, C.; van der Vorst, E. Adipocyte-specific ACKR3 regulates lipid levels in adipose tissue. Biomedicines 2021, 9, 394. [Google Scholar] [CrossRef]
- Li, X.; Zhu, M.; Penfold, M.E.; Koenen, R.R.; Thiemann, A.; Heyll, K.; Akhtar, S.; Koyadan, S.; Wu, Z.; Gremse, F.; et al. Activation of CXCR7 limits atherosclerosis and improves hyperlipidemia by increasing cholesterol uptake in adipose tissue. Circulation 2014, 129, 1244–1253. [Google Scholar] [CrossRef]
- Pan, J.-H.; Sukhova, G.K.; Yang, J.-T.; Wang, B.; Xie, T.; Fu, H.; Zhang, Y.; Satoskar, A.R.; David, J.R.; Metz, C.N.; et al. Macrophage migration inhibitory factor deficiency impairs atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2004, 109, 3149–3153. [Google Scholar] [CrossRef]
- Borne, P.V.D.; Quax, P.; Hoefer, I.E.; Pasterkamp, G. The multifaceted functions of CXCL10 in cardiovascular disease. BioMed Res. Int. 2014, 2014, 893106. [Google Scholar] [CrossRef]
- Lupieri, A.; Smirnova, N.; Solinhac, R.; Malet, N.; Benamar, M.; Saoudi, A.; Santos-Zas, I.; Zeboudj, L.; Ait-Oufella, H.; Hirsch, E.; et al. Smooth muscle cells-derived CXCL10 prevents endothelial healing through PI3Kγ-dependent T cells response. Cardiovasc. Res. 2020, 116, 438–449. [Google Scholar] [CrossRef]
- Grönberg, C.; Nilsson, J.; Wigren, M. Recent advances on CD4+ T cells in atherosclerosis and its implications for therapy. Eur. J. Pharmacol. 2017, 816, 58–66. [Google Scholar] [CrossRef]
- Zernecke, A.; Liehn, E.A.; Gao, J.-L.; Kuziel, W.A.; Murphy, P.M.; Weber, C. Deficiency in CCR5 but not CCR1 protects against neointima formation in atherosclerosis-prone mice: Involvement of IL-10. Blood 2006, 107, 4240–4243. [Google Scholar] [CrossRef]
- Galkina, E.; Harry, B.L.; Ludwig, A.; Liehn, E.A.; Sanders, J.M.; Bruce, A.; Weber, C.; Ley, K. CXCR6 promotes atherosclerosis by supporting t-cell homing, interferon-γ production, and macrophage accumulation in the aortic wall. Circulation 2007, 116, 1801–1811. [Google Scholar] [CrossRef]
- Yan, Y.; Thakur, M.; van der Vorst, E.P.; Weber, C.; Döring, Y. Targeting the chemokine network in atherosclerosis. Atherosclerosis 2021, 330, 95–106. [Google Scholar] [CrossRef]
- Ayari, H.; Bricca, G. Identification of two genes potentially associated in iron-heme homeostasis in human carotid plaque using microarray analysis. J. Biosci. 2013, 38, 311–315. [Google Scholar] [CrossRef]
- Hägg, S.; Skogsberg, J.; Lundström, J.; Noori, P.; Nilsson, R.; Zhong, H.; Maleki, S.; Shang, M.-M.; Brinne, B.; Bradshaw, M.; et al. Multi-organ expression profiling uncovers a gene module in coronary artery disease involving transendothelial migration of leukocytes and lim domain binding 2: The stockholm atherosclerosis gene expression (Stage) Study. PLoS Genet. 2009, 5, e1000754. [Google Scholar] [CrossRef]
- Döring, Y.; Manthey, H.D.; Drechsler, M.; Lievens, D.; Megens, R.; Soehnlein, O.; Busch, M.; Manca, M.; Koenen, R.; Pelisek, J.; et al. Auto-antigenic protein-dna complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation 2012, 125, 1673–1683. [Google Scholar] [CrossRef]
- Yang, R.; Yao, L.; Du, C.; Wu, Y. Identification of key pathways and core genes involved in atherosclerotic plaque progression. Ann. Transl. Med. 2021, 9, 267. [Google Scholar] [CrossRef] [PubMed]
- Rallidis, L.S.; Hamodraka, E.S.; Fountoulaki, K.; Moustogiannis, G.; Zolindaki, M.G.; Kremastinos, D.T. Simvastatin exerts its anti-inflammatory effect in hypercholesterolaemic patients by decreasing the serum levels of monocyte chemoattractant protein-1. Int. J. Cardiol. 2008, 124, 271–272. [Google Scholar] [CrossRef] [PubMed]
- Feig, J.E.; Feig, J.L. Macrophages, dendritic cells, and regression of atherosclerosis. Front. Physiol. 2012, 3, 286. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Lionakis, M.S.; Liu, Q.; Roffê, E.; Murphy, P.M. Genetic deletion of chemokine receptor Ccr7 exacerbates atherogenesis in ApoE-deficient mice. Cardiovasc. Res. 2012, 97, 580–588. [Google Scholar] [CrossRef][Green Version]
- Trogan, E.; Feig, J.E.; Dogan, S.; Rothblat, G.H.; Angeli, V.; Tacke, F.; Randolph, G.J.; Fisher, E.A. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc. Natl. Acad. Sci. USA 2006, 103, 3781–3786. [Google Scholar] [CrossRef]
- Herlea-Pana, O.; Yao, L.; Heuser-Baker, J.; Wang, Q.; Wang, Q.; Georgescu, C.; Zou, M.-H.; Barlic-Dicen, J. Chemokine receptors CXCR2 and CX3CR1 differentially regulate functional responses of bone-marrow endothelial progenitors during atherosclerotic plaque regression. Cardiovasc. Res. 2015, 106, 324–337. [Google Scholar] [CrossRef]
- Cipriani, S.; Francisci, D.; Mencarelli, A.; Renga, B.; Schiaroli, E.; D’Amore, C.; Baldelli, F.; Fiorucci, S. Efficacy of the CCR5 antagonist maraviroc in reducing early, ritonavir-induced atherogenesis and advanced plaque progression in mice. Circulation 2013, 127, 2114–2124. [Google Scholar] [CrossRef]
- Feinberg, M.W.; Moore, K.J. MicroRNA regulation of atherosclerosis. Circ. Res. 2016, 118, 703–720. [Google Scholar] [CrossRef]
- Schober, A.; Nazari-Jahantigh, M.; Weber, C. MicroRNA-mediated mechanisms of the cellular stress response in atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 361–374. [Google Scholar] [CrossRef]
- Ma, S.; Tian, X.Y.; Zhang, Y.; Mu, C.; Shen, H.; Bismuth, J.; Pownall, H.; Huang, Y.; Wong, W.T. E-selectin-targeting delivery of microRNAs by microparticles ameliorates endothelial inflammation and atherosclerosis. Sci. Rep. 2016, 6, 22910. [Google Scholar] [CrossRef]
- Kanmogne, G.; Woollard, S. Maraviroc: A review of its use in HIV infection and beyond. Drug Des. Dev. Ther. 2015, 9, 5447–5468. [Google Scholar] [CrossRef]
- Choi, W.-T.; Yang, Y.; Xu, Y.; An, J. Targeting chemokine receptor CXCR4 for treatment of HIV-1 infection, tumor progression, and metastasis. Curr. Top. Med. Chem. 2014, 14, 1574–1589. [Google Scholar] [CrossRef]
- DiPersio, J.F.; Uy, G.L.; Yasothan, U.; Kirkpatrick, P. Plerixafor. Nat. Rev. Drug Discov. 2009, 8, 105–107. [Google Scholar] [CrossRef]
- Solomon, D.H.; Goodson, N.J.; Katz, J.N.; Weinblatt, M.E.; Avorn, J.; Setoguchi, S.; Canning, C.; Schneeweiss, S. Patterns of cardiovascular risk in rheumatoid arthritis. Ann. Rheum. Dis. 2006, 65, 1608–1612. [Google Scholar] [CrossRef]
- Lopez-Candales, A.; Burgos, P.M.H.; Hernandez-Suarez, D.F.; Harris, D. Linking chronic inflammation with cardiovascular disease: From normal aging to the metabolic syndrome. J. Nat. Appl. Sci. 2017, 3, e341. [Google Scholar]
- Giza, D.E.; Iliescu, G.; Hassan, S.; Marmagkiolis, K.; Iliescu, C. Cancer as a risk factor for cardiovascular disease. Curr. Oncol. Rep. 2017, 19, 39. [Google Scholar] [CrossRef]
- Zhang, S.; Yue, J.; Ge, Z.; Xie, Y.; Zhang, M.; Jiang, L. Activation of CXCR7 alleviates cardiac insufficiency after myocardial infarction by promoting angiogenesis and reducing apoptosis. Biomed. Pharmacother. 2020, 127, 110168. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Hu, S.; Chen, H.; Bu, D.; Zhu, L.; Xu, C.; Chu, F.; Huo, X.; Tang, Y.; Sun, X.; et al. Loss of endothelial CXCR7 impairs vascular homeostasis and cardiac remodeling after myocardial infarction. Circulation 2017, 135, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Majmudar, M.D.; Keliher, E.J.; Heidt, T.; Leuschner, F.; Truelove, J.; Sena, B.F.; Gorbatov, R.; Iwamoto, Y.; Dutta, P.; Wojtkiewicz, G.; et al. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation 2013, 127, 2038–2046. [Google Scholar] [CrossRef]
- Leuschner, F.; Courties, G.; Dutta, P.; Mortensen, L.J.; Gorbatov, R.; Sena, B.; Novobrantseva, T.I.; Borodovsky, A.; Fitzgerald, K.; Koteliansky, V.; et al. Silencing of CCR2 in myocarditis. Eur. Heart J. 2014, 36, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Braunersreuther, V.; Pellieux, C.; Pelli, G.; Burger, F.; Steffens, S.; Montessuit, C.; Weber, C.; Proudfoot, A.; Mach, F.; Arnaud, C. Chemokine CCL5/RANTES inhibition reduces myocardial reperfusion injury in atherosclerotic mice. J. Mol. Cell. Cardiol. 2010, 48, 789–798. [Google Scholar] [CrossRef]
- Van Wanrooij, E.J.; Happé, H.; Hauer, A.D.; De Vos, P.; Imanishi, T.; Fujiwara, H.; Van Berkel, T.J.; Kuiper, J. HIV entry inhibitor TAK-779 attenuates atherogenesis in low-density lipoprotein receptor–deficient mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2642–2647. [Google Scholar] [CrossRef] [PubMed]
- von Hundelshausen, P.; Agten, S.M.; Eckardt, V.; Blanchet, X.; Schmitt, M.M.; Ippel, H.; Neideck, C.; Bidzhekov, K.; Leberzammer, J.; Wichapong, K.; et al. Chemokine interactome mapping enables tailored intervention in acute and chronic inflammation. Sci. Transl. Med. 2017, 9, eaah6650. [Google Scholar] [CrossRef]
- Koenen, R.; von Hundelshausen, P.; Nesmelova, I.; Zernecke, A.; Liehn, E.A.; Sarabi, A.; Kramp, B.K.; Piccinini, A.M.; Paludan, S.R.; Kowalska, M.A.; et al. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat. Med. 2009, 15, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Alard, J.-E.; Ortega-Gomez, A.; Wichapong, K.; Bongiovanni, D.; Horckmans, M.; Megens, R.T.A.; Leoni, G.; Ferraro, B.; Rossaint, J.; Paulin, N.; et al. Recruitment of classical monocytes can be inhibited by disturbing heteromers of neutrophil HNP1 and platelet CCL5. Sci. Transl. Med. 2015, 7, 317ra196. [Google Scholar] [CrossRef]
- Braunersreuther, V.; Pelli, G.; Galan, K.; Proudfoot, A.E.; Belin, A.; Vuilleumier, N.; Burger, F.; Lenglet, S.; Caffa, I.; Soncini, D.; et al. Treatment with the CC chemokine-binding protein Evasin-4 improves post-infarction myocardial injury and survival in mice. Thromb. Haemost. 2013, 110, 807–825. [Google Scholar] [CrossRef]
- Montecucco, F.; Lenglet, S.; Braunersreuther, V.; Pelli, G.; Pellieux, C.; Montessuit, C.; Lerch, R.; Deruaz, M.; Proudfoot, A.E.; Mach, F. Single administration of the CXC chemokine-binding protein evasin-3 during ischemia prevents myocardial reperfusion injury in mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1371–1377. [Google Scholar] [CrossRef]
- Ravindran, D.; Ridiandries, A.; Vanags, L.Z.; Henriquez, R.; Cartland, S.; Tan, M.-J.; Bursill, C. Chemokine binding protein ‘M3’ limits atherosclerosis in apolipoprotein E−/− mice. PLoS ONE 2017, 12, e0173224. [Google Scholar] [CrossRef]
- van Wanrooij, E.J.; de Jager, S.C.; van Es, T.; de Vos, P.; Birch, H.L.; Owen, D.A.; Watson, R.J.; Biessen, E.A.; Chapman, G.A.; van Berkel, T.J.; et al. CXCR3 antagonist NBI-74330 attenuates atherosclerotic plaque formation in LDL receptor–deficient mice. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 251–257. [Google Scholar] [CrossRef]
- Koren, L.; Barash, U.; Zohar, Y.; Karin, N.; Aronheim, A. The cardiac maladaptive ATF3-dependent cross-talk between cardiomyocytes and macrophages is mediated by the IFNγ-CXCL10-CXCR3 axis. Int. J. Cardiol. 2017, 228, 394–400. [Google Scholar] [CrossRef]
- Poupel, L.; Boissonnas, A.; Hermand, P.; Dorgham, K.; Guyon, E.; Auvynet, C.; Charles, F.S.; Lesnik, P.; Deterre, P.; Combadiere, C. Pharmacological inhibition of the chemokine receptor, CX3CR1, reduces atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2297–2305. [Google Scholar] [CrossRef]
- Low, S.; Wu, H.; Jerath, K.; Tibolla, A.; Fogal, B.; Conrad, R.; MacDougall, M.; Kerr, S.; Berger, V.; Dave, R.; et al. VHH antibody targeting the chemokine receptor CX3CR1 inhibits progression of atherosclerosis. mAbs 2020, 12, 1709322. [Google Scholar] [CrossRef] [PubMed]
- White, D.A.; Fang, L.; Chan, W.; Morand, E.F.; Kiriazis, H.; Duffy, S.J.; Taylor, A.; Dart, A.M.; Du, X.-J.; Gao, X.-M. Pro-inflammatory action of MIF in acute myocardial infarction via activation of peripheral blood mononuclear cells. PLoS ONE 2013, 8, e76206. [Google Scholar] [CrossRef]
- Gilbert, J.; Lekstrom-Himes, J.; Donaldson, D.; Lee, Y.; Hu, M.; Xu, J.; Wyant, T.; Davidson, M. Effect of CC chemokine receptor 2 CCR2 blockade on serum C-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am. J. Cardiol. 2011, 107, 906–911. [Google Scholar] [CrossRef]
- Francisci, D.; Pirro, M.; Schiaroli, E.; Mannarino, M.R.; Cipriani, S.; Bianconi, V.; Alunno, A.; Bagaglia, F.; Bistoni, O.; Falcinelli, E.; et al. Maraviroc intensification modulates atherosclerotic progression in HIV-suppressed patients at high cardiovascular risk. A randomized, crossover pilot study. Open Forum Infect. Dis. 2019, 6, ofz112. [Google Scholar] [CrossRef]
- Colombo, A.; Basavarajaiah, S.; Limbruno, U.; Picchi, A.; Lettieri, C.; Valgimigli, M.; Sciahbasi, A.; Prati, F.; Calabresi, M.; Pierucci, D.; et al. A double-blind randomised study to evaluate the efficacy and safety of bindarit in preventing coronary stent restenosis. EuroIntervention 2016, 12, e1385–e1394. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.S.; Miller, L.W.; Patel, A.N.; Anderson, R.D.; Mendelsohn, F.O.; Traverse, J.H.; Silver, K.H.; Shin, J.; Ewald, G.A.; Farr, M.J.; et al. Changes in ventricular remodelling and clinical status during the year following a single administration of stromal cell-derived factor-1 non-viral gene therapy in chronic ischaemic heart failure patients: The STOP-HF randomized Phase II trial. Eur. Heart J. 2015, 36, 2228–2238. [Google Scholar] [CrossRef]
- Shishehbor, M.H.; Rundback, J.; Bunte, M.; Hammad, T.A.; Miller, L.; Patel, P.D.; Sadanandan, S.; Fitzgerald, M.; Pastore, J.; Kashyap, V.; et al. SDF-1 plasmid treatment for patients with peripheral artery disease (STOP-PAD): Randomized, double-blind, placebo-controlled clinical trial. Vasc. Med. 2019, 24, 200–207. [Google Scholar] [CrossRef]
- Hammad, T.A.; Rundback, J.; Bunte, M.; Miller, L.; Patel, P.D.; Sadanandan, S.; Fitzgerald, M.; Pastore, J.; Kashyap, V.; Henry, T.D.; et al. Stromal cell–derived factor-1 plasmid treatment for patients with peripheral artery disease (stop-pad) trial: Six-month results. J. Endovasc. Ther. 2020, 27, 669–675. [Google Scholar] [CrossRef] [PubMed]
- van der Vorst, E.P.; Döring, Y. Tracing endothelial CXCR4 may pave the way for localized lesional treatment approaches. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 837–838. [Google Scholar] [CrossRef]
- Wei, L.; Petryk, J.; Gaudet, C.; Kamkar, M.; Gan, W.; Duan, Y.; Ruddy, T.D. Development of an inflammation imaging tracer, 111In-DOTA-DAPTA, targeting chemokine receptor CCR5 and preliminary evaluation in an ApoE−/− atherosclerosis mouse model. J. Nucl. Cardiol. 2018, 26, 1169–1178. [Google Scholar] [CrossRef]
- Li, X.; Heber, D.; Leike, T.; Beitzke, D.; Lu, X.; Zhang, X.; Wei, Y.; Mitterhauser, M.; Wadsak, W.; Kropf, S.; et al. [68Ga]Pentixafor-PET/MRI for the detection of Chemokine receptor 4 expression in atherosclerotic plaques. Eur. J. Nucl. Med. Mol. Imaging 2017, 45, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yu, W.; Wollenweber, T.; Lu, X.; Wei, Y.; Beitzke, D.; Wadsak, W.; Kropf, S.; Wester, H.J.; Haug, A.R.; et al. [68Ga]Pentixafor PET/MR imaging of chemokine receptor 4 expression in the human carotid artery. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Baba, O.; Huang, L.-H.; Elvington, A.; Szpakowska, M.; Sultan, D.; Heo, G.S.; Zhang, X.; Luehmann, H.; Detering, L.; Chevigne, A.; et al. CXCR4-binding positron emission tomography tracers link monocyte recruitment and endothelial injury in murine atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 822–836. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Woodard, P.K. Chemokine receptors: Key for molecular imaging of inflammation in atherosclerosis. J. Nucl. Cardiol. 2018, 26, 1179–1181. [Google Scholar] [CrossRef]
- Juenet, M.; Varna, M.; Aid-Launais, R.; Chauvierre, C.; Letourneur, D. Nanomedicine for the molecular diagnosis of cardiovascular pathologies. Biochem. Biophys. Res. Commun. 2015, 468, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Solari, R.; Pease, J.; Begg, M. Chemokine receptors as therapeutic targets: Why aren’t there more drugs? Eur. J. Pharmacol. 2015, 746, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Schall, T.J.; Proudfoot, A.E.I. Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat. Rev. Immunol. 2011, 11, 355–363. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Target | Treatment | Type of Treatment | Condition | Outcome | Reference |
|---|---|---|---|---|---|
| ACKR3 | CCX771 | Small-molecule receptor agonist | Atherosclerosis | ↓Lesion formation ↓Blood cholesterol | [117] |
| TC14012 | Small-molecule receptor agonist | Acute myocardial infarction | ↓Infarct size Only [144]: ↑Left ventricular internal diameter ↑Left ventricular volume ↑Vascular density | [144,145] | |
| CCR2 | RS102982 | Small-molecule receptor antagonist | Atherosclerosis | ↓Lesion size ↓Macrophage accumulation | [63] |
| Nanoparticle encased siRNA | Small silencing RNA | Myocardial infarction | ↓Ly6Chi monocytes recruitment ↓Post-MI heart failure ↓Left ventricular dilation | [146] | |
| Nanoparticle encased siRNA | Small silencing RNA | Myocarditis | ↓Ly6Chi monocytes in heart ↓Myeloid progenitor trafficking Improved ejection fraction | [147] | |
| CCL2, CCL5, CCL8, CXCL9 | miR-146a/-181b packaged in ESTA-MSV microparticles | microRNA delivery | Atherosclerosis | ↓Plaque size ↓Macrophage accumulation ↑VSMCs in plaque ↑Collagen in plaque | [137] |
| CCR5 | Maraviroc | Small–molecule receptor antagonist | Atherosclerosis | ↓Plaque size ↓Macrophage infiltration | [134] |
| [(44)AANA(47)]-RANTES | Chemokine receptor antagonist (modified chemokine) | Myocardial ischemia and reperfusion | During early myocardial reperfusion: ↓Infarct size ↓Myocardial leukocyte infiltration ↓Oxidative stress ↓Apoptosis | [148] | |
| Met-RANTES | Chemokine receptor antagonist (modified chemokine) | Atherosclerosis | ↓Lesion size ↓Leukocyte infiltration ↑Collagen content in atheroma | [54] | |
| CCR5, CXCR3 | TAK-779 | Small–molecule receptor antagonist | Atherosclerosis | ↓Lesion formation ↓T-helper 1 cell plaque infiltration | [149] |
| CCL5/CCL17 | CAN peptide | Inhibition of heteronomer formation | Atherosclerosis | ↓Lesion size in aortic root | [150] |
| CCL5/CXCL4 | MKEY peptide | Inhibition of heteronomer formation | Atherosclerosis | ↓Lesion formation ↓Macrophage accumulation | [151] |
| CCL5/HNP1 | SKY peptide | Inhibition of heteronomer formation | Myocardialinfarction | ↓Ly6Chi monocyte adhesion/recruitment ↓Macrophages ↓Inflammation | [152] |
| CCL5,CCL11 CXCL1 | Evasin-4 (CC-) Evasin-3 (CXC-) | Chemokine- binding protein (inhibits chemokin binding) | Myocardial infarction | ↑Improved survival (Evasin-4) Post infarction: ↓Leukocyte infiltration ↓ROS production ↓Neutrophil chemoattractants ↓Infarct size | [153] |
| CXCL1, CXCL2 | Evasin-3 | Chemokine-binding protein (inhibits chemokin binding) | Myocardial ischemia | ↓Infarct size In infarcted myocardium: ↓Neutrophil infiltration ↓ROS production | [154] |
| CCL2, CCL5, CX3CL1 | M3 | Chemokine-binding protein (inhibits chemokine binding) | Atherosclerosis | 12-week model: ↓Lesion area ↑Aortic smooth muscle α-actin expression 6-week model: ↓Macrophage content in plaques ↓Lipid deposition in thoracic aorta | [155] |
| CXCR3 | NBI-74330 | Small–molecule receptor antagonist | Atherosclerosis | ↓Lesion formation ↓Leukocyte migration Improved regulatory/effector T cell balance | [156] |
| AMG487 | Small–molecule receptor antagonist | Cardiac remodeling | Abrogation of ↑ in ventricle weight to body weight ratio ↓Macrophage recruitment ↓Cardiac remodeling | [157] | |
| CX3CR1 | F1 | Chemokine receptor antagonist (modified chemokine) | Atherosclerosis | ↓Monocyte adhesion ↓Macrophage accumulation in aortic sinus ↓Monocyte survival ↓Lesion size in advanced atherosclerosis | [158] |
| BI 655088 | Variable domains of camelid heavy chain-only antibody (antagonist) | Atherosclerosis | ↓Lesion formation | [159] | |
| MIF | COR100140, anti-MIF monoclonal antibody | Small–molecule receptor antagonist | Myocardial infarction | COR100140: ↓Incidence of cardiac rupture Antibody: ↓CCL2 expression ↓Leukocytes at infarct region at 24 h | [160] |
| Target | Intervention | Aim | Condition | Phase | Status and Results | Trial Identifier |
|---|---|---|---|---|---|---|
| CCR2 | MLN1202 humanized monoclonal antibody | Measuring the effects of MLN1202 on C-reactive protein levels in patients with risk factors for CV disease | Atherosclerosis | II | Completed; well tolerated in patient population and significant reduction in high-sensitivity C-reactive protein levels [161] | NCT00715169 |
| CCR5 | Maraviroc Small-molecule receptor antagonist | Augmenting rehabilitation outcomes after stroke | Stroke | II | Not yet recruiting | NCT04789616 |
| Maraviroc Small-molecule receptor antagonist | Efficacy of Maraviroc in modulating atherosclerosis in HIV patients | Atherosclerosis | IV | Significant improvement in various markers of CV risk including carotid atherosclerosis, endothelial dysfunction, and arterial stiffness. No effect on systemic inflammation apparent [162] | NCT03402815 | |
| CXCR2 | AZD5069 Small-molecule receptor antagonist | Evaluate inhibition of CXCR2 as a treatment of coronary heart disease | Coronary heart disease | II | Ongoing | EudraCT 2016-000775-24 |
| CXCR4 | POL6326 Peptidic receptor antagonist | Evaluate effects of CXCR4 inhibition in patients with large reperfused ST elevation myocardial infarction | Large reperfused ST-elevation myocardial infarction | II | Completed; results not found | NCT01905475 |
| CCL2 | Bindarit Selective inhibitor | Evaluating the efficacy and safety of different Bindarit dosages in preventing stent restenosis | Coronary restenosis | II | Primary endpoint not met. Reduction in the in-stent late loss observed. Bindarit was well tolerated [163] | NCT01269242 |
| CXCL12 (SDF-1) | JVS-100 nonviral DNA plasmid (transient CXCL12 expression) | Evaluate the safety and efficacy of a single dose of JVS-100 administered by endomyocardial injection to cohorts of adults with ischemic heart failure | Ischemic heart failure | II | Primary endpoint was not met. Safety profile supports repeat dosing with plasmid SDF-1. Potential for attenuation of left ventricular remodeling and improvement of ejection fraction [164] | NCT01643590 |
| JVS-100 nonviral DNA plasmid (transient CXCL12 expression) | Evaluate the safety and efficacy of JVS-100 administered by retrograde delivery to cohorts of adults with ischemic heart failure | Ischemic heart failure | I/II | Unknown | NCT01961726 | |
| JVS-100 nonviral DNA plasmid (transient CXCL12 expression) | Evaluate the safety and efficacy of JVS-100 administered by direct intramuscular injection to cohorts of adults with critical limb ischemia | Critical limb ischemia | II | Completed; results not found | NCT01410331 | |
| JVS-100 nonviral DNA plasmid (transient CXCL12 expression) | Evaluate the safety and efficacy of JVS-100 administered by direct intramuscular injection as adjunct to revascularization of infrapopliteal lesions in patients with advanced peripheral artery disease and tissue loss | Peripheral arterial disease | II | Primary efficacy endpoint was not met at either 3- or 6-month follow-up, intervention failed to improve wound healing [165,166] | NCT02544204 | |
| ACRX-100 nonviral DNA plasmid (transient CXCL12 expression) | Evaluate the safety of a single escalating dose of ACRX-100 administered by endomyocardial injection to cohorts of adults with ischemic heart failure | Heart failure | I | Completed; results not found | NCT01082094 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Márquez, A.B.; van der Vorst, E.P.C.; Maas, S.L. Key Chemokine Pathways in Atherosclerosis and Their Therapeutic Potential. J. Clin. Med. 2021, 10, 3825. https://doi.org/10.3390/jcm10173825
Márquez AB, van der Vorst EPC, Maas SL. Key Chemokine Pathways in Atherosclerosis and Their Therapeutic Potential. Journal of Clinical Medicine. 2021; 10(17):3825. https://doi.org/10.3390/jcm10173825
Chicago/Turabian StyleMárquez, Andrea Bonnin, Emiel P. C. van der Vorst, and Sanne L. Maas. 2021. "Key Chemokine Pathways in Atherosclerosis and Their Therapeutic Potential" Journal of Clinical Medicine 10, no. 17: 3825. https://doi.org/10.3390/jcm10173825
APA StyleMárquez, A. B., van der Vorst, E. P. C., & Maas, S. L. (2021). Key Chemokine Pathways in Atherosclerosis and Their Therapeutic Potential. Journal of Clinical Medicine, 10(17), 3825. https://doi.org/10.3390/jcm10173825