
The Syndromes of Thrombotic Microangiopathy: A Critical Appraisal on Complement Dysregulation


Abstract
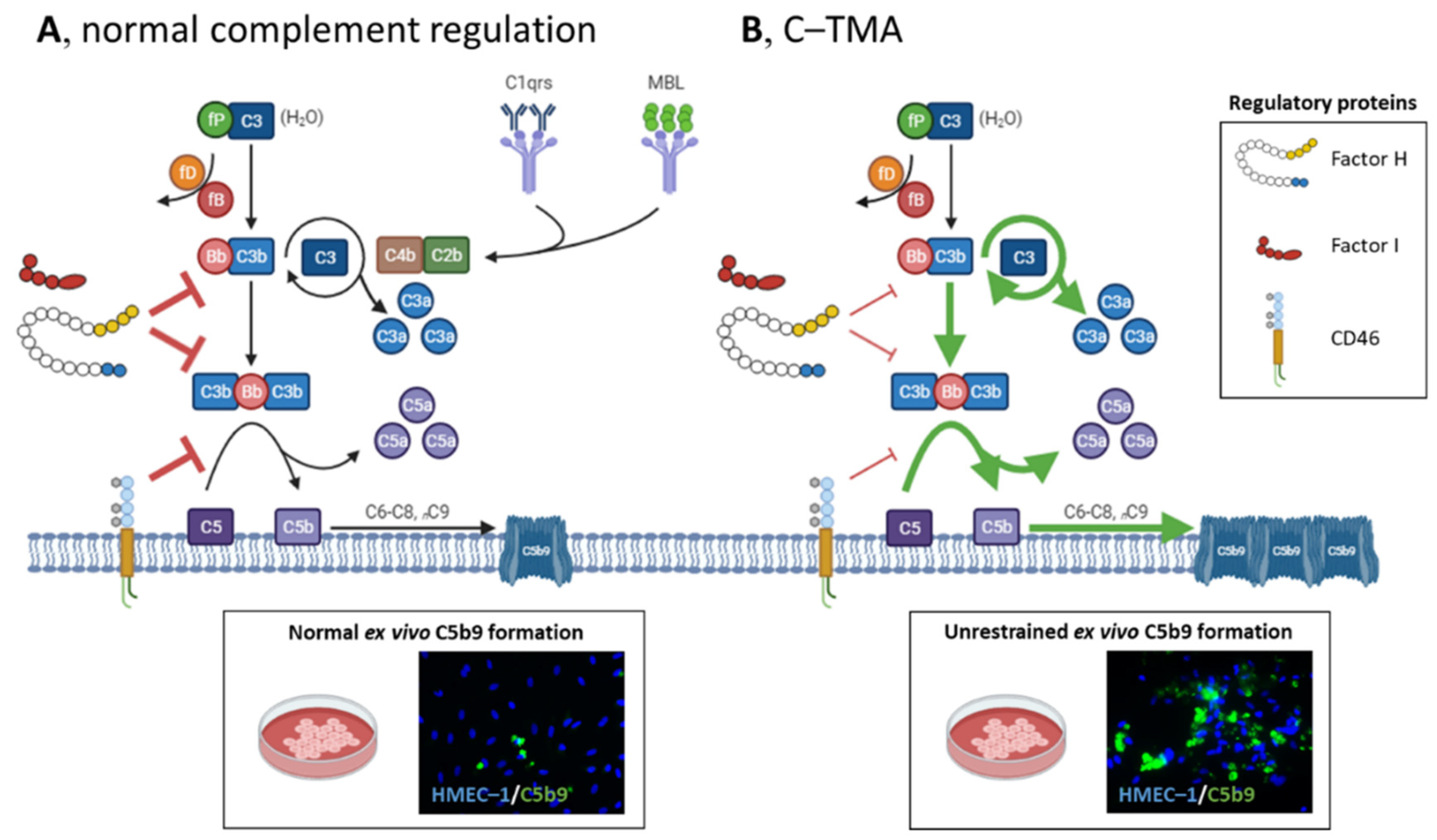
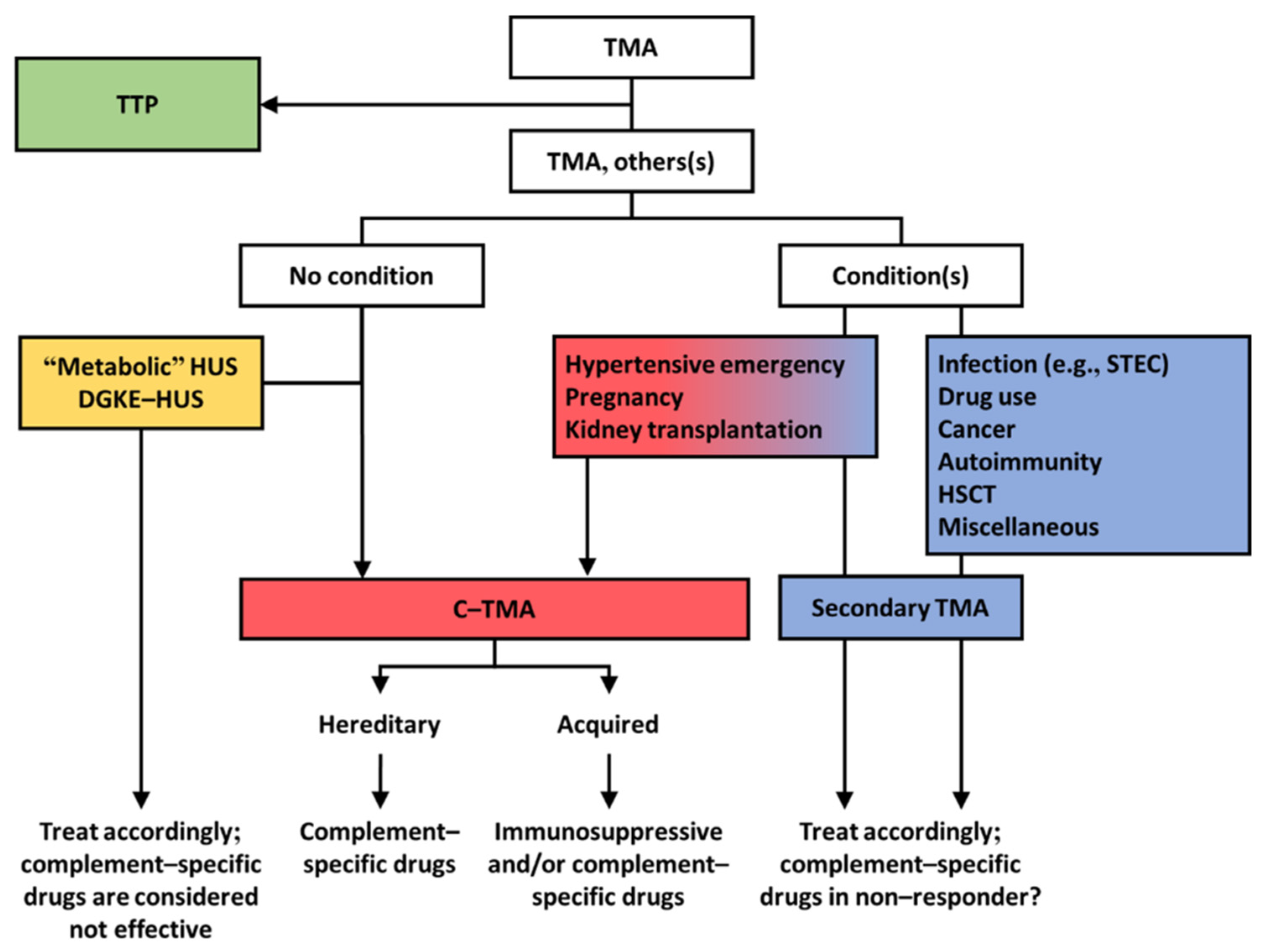
1. Integrated Discussion
2. Primary Atypical HUS, a Prototypic C-TMA
3. Patients with TMA and Coexisting Conditions May Present with Complement Dysregulation
3.1. Hypertensive Emergency
3.2. Pregnancy

| Presentation | Genotyping | Outcome at 12 Months | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Eculizumab, n/N | Creatinine, mg/dL | Dialysis (%) | Rare Variants (%) | Pathogenic (%) | Renal Response (%) | ESKD (%) | Death (%) | ESKD in Untreated Patients | |
| Hypertensive emergency | |||||||||
| Combined data | 29/122 | Unknown | Unknown | 14 (48) | Unknown | 21 (72) | 7 (24) | 1 (3) | |
| Cavero et al. [42] | 9/19 | 8 (IQR, 7–9) | 8 (89) | 5 (56) | 3 (33) | 7 (78) | 2 (22) | 0 | 60% at 1 year (N = 10) |
| El Karoui et al. [41] | 13/76 | Unknown | Unknown | 7 (54) | Unknown | 9 (69) | 4 (31) | 0 | 64% at 1 year (N = 61) |
| Timmermans et al. [8] | 7/26 | 7 (IQR, 4–9) | 4 (57) | 2 (29) | 2 (29) | 5 (71) | 1 (14) | 1 (14) | 75% at 1 year (N = 16) a |
| Pregnancy-associated atypical HUS | |||||||||
| Combined data | 17/116 | Unknown | Unknown | 7 (41) | Unknown | 15 (88) | 2 (25) | 0 | |
| Bruel et al. [9] | 4/87 | Unknown | Unknown | 2 (50) | Unknown | 3 (75) | 1 (25) | 0 | 49% at last follow-up (N = 71) |
| Huerta et al. [48] | 10/22 | 4 (IQR, 3–5) | 3 (30) | 4 (40) | 4 (40) | 10 (100) | 0 | 0 | 55% at last follow-up (N = 11) |
| Timmermans et al. [49] | 3/7 | 5 (IQR, 4–6) | 3 (100) | 1 (33) | 0 | 2 (67) | 1 (33) | 0 | 50% at last follow-up (N = 4) |
3.3. Kidney Transplantation
4. TMAs Unrelated to Complement Dysregulation
4.1. Shiga Toxin-Producing E. coli (STEC) and Other Bacterial Infections
4.2. Drug-Induced TMA
4.3. Cancer
4.4. Autoimmunity
4.5. HSCT-TMA
4.6. Miscellaneous Conditions
5. Proposal for a Pragmatic Approach to Diagnosis of TMA
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Loirat, C.; Fakhouri, F.; Ariceta, G.; Besbas, N.; Bitzan, M.; Bjerre, A.; Coppo, R.; Emma, F.; Johnson, S.; Karpman, D.; et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr. Nephrol. 2016, 31, 15–39. [Google Scholar] [CrossRef] [PubMed]
- Goodship, T.H.; Cook, H.T.; Fakhouri, F.; Fervenza, F.C.; Fremeaux-Bacchi, V.; Kavanagh, D.; Nester, C.M.; Noris, M.; Pickering, M.C.; Rodriguez de Cordoba, S.; et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017, 91, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Furlan, M.; Robles, R.; Galbusera, M.; Remuzzi, G.; Kyrle, P.A.; Brenner, B.; Krause, M.; Scharrer, I.; Aumann, V.; Mittler, U.; et al. Von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N. Engl. J. Med. 1998, 339, 1578–1584. [Google Scholar] [CrossRef]
- Noris, M.; Caprioli, J.; Bresin, E.; Mossali, C.; Pianetti, G.; Gamba, S.; Daina, E.; Fenili, C.; Castelletti, F.; Sorosina, A.; et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin. J. Am. Soc. Nephrol. 2010, 5, 1844–1859. [Google Scholar] [CrossRef]
- Fremeaux-Bacchi, V.; Fakhouri, F.; Garnier, A.; Bienaime, F.; Dragon-Durey, M.A.; Ngo, S.; Moulin, B.; Servais, A.; Provot, F.; Rostaing, L.; et al. Genetics and outcome of atypical hemolytic uremic syndrome: A nationwide French series comparing children and adults. Clin. J. Am. Soc. Nephrol. 2013, 8, 554–562. [Google Scholar] [CrossRef]
- Sheerin, N.S.; Kavanagh, D.; Goodship, T.H.; Johnson, S. A national specialized service in England for atypical haemolytic uraemic syndrome-the first year’s experience. QJM 2016, 109, 27–33. [Google Scholar] [CrossRef]
- Bayer, G.; von Tokarski, F.; Thoreau, B.; Bauvois, A.; Barbet, C.; Cloarec, S.; Merieau, E.; Lachot, S.; Garot, D.; Bernard, L.; et al. Etiology and Outcomes of Thrombotic Microangiopathies. Clin. J. Am. Soc. Nephrol. 2019, 14, 557–566. [Google Scholar] [CrossRef]
- Timmermans, S.; Werion, A.; Damoiseaux, J.; Morelle, J.; Reutelingsperger, C.P.; van Paassen, P. Diagnostic and Risk Factors for Complement Defects in Hypertensive Emergency and Thrombotic Microangiopathy. Hypertension 2020, 75, 422–430. [Google Scholar] [CrossRef]
- Bruel, A.; Kavanagh, D.; Noris, M.; Delmas, Y.; Wong, E.K.S.; Bresin, E.; Provot, F.; Brocklebank, V.; Mele, C.; Remuzzi, G.; et al. Hemolytic Uremic Syndrome in Pregnancy and Postpartum. Clin. J. Am. Soc. Nephrol. 2017, 12, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Le Quintrec, M.; Lionet, A.; Kamar, N.; Karras, A.; Barbier, S.; Buchler, M.; Fakhouri, F.; Provost, F.; Fridman, W.H.; Thervet, E.; et al. Complement mutation-associated de novo thrombotic microangiopathy following kidney transplantation. Am. J. Transplant. 2008, 8, 1694–1701. [Google Scholar] [CrossRef]
- Timmermans, S.; Werion, A.; Morelle, J.; van Paassen, P. Defects in complement and “secondary” hemolytic uremic syndrome. Kidney Int. 2019, 96, 517. [Google Scholar] [CrossRef] [PubMed]
- Legendre, C.M.; Licht, C.; Muus, P.; Greenbaum, L.A.; Babu, S.; Bedrosian, C.; Bingham, C.; Cohen, D.J.; Delmas, Y.; Douglas, K.; et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2013, 368, 2169–2181. [Google Scholar] [CrossRef] [PubMed]
- Licht, C.; Greenbaum, L.A.; Muus, P.; Babu, S.; Bedrosian, C.L.; Cohen, D.J.; Delmas, Y.; Douglas, K.; Furman, R.R.; Gaber, O.A.; et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int. 2015, 87, 1061–1073. [Google Scholar] [CrossRef]
- Fakhouri, F.; Hourmant, M.; Campistol, J.M.; Cataland, S.R.; Espinosa, M.; Gaber, A.O.; Menne, J.; Minetti, E.E.; Provot, F.; Rondeau, E.; et al. Terminal Complement Inhibitor Eculizumab in Adult Patients with Atypical Hemolytic Uremic Syndrome: A Single-Arm, Open-Label Trial. Am. J. Kidney Dis. 2016, 68, 84–93. [Google Scholar] [CrossRef]
- Thompson, R.A.; Winterborn, M.H. Hypocomplementaemia due to a genetic deficiency of beta 1H globulin. Clin. Exp. Immunol. 1981, 46, 110–119. [Google Scholar]
- Warwicker, P.; Goodship, T.H.; Donne, R.L.; Pirson, Y.; Nicholls, A.; Ward, R.M.; Turnpenny, P.; Goodship, J.A. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 1998, 53, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, V.P.; Herbert, A.P.; Cortes, C.; McKee, K.A.; Blaum, B.S.; Esswein, S.T.; Uhrin, D.; Barlow, P.N.; Pangburn, M.K.; Kavanagh, D. The binding of factor H to a complex of physiological polyanions and C3b on cells is impaired in atypical hemolytic uremic syndrome. J. Immunol. 2009, 182, 7009–7018. [Google Scholar] [CrossRef]
- Osborne, A.J.; Breno, M.; Borsa, N.G.; Bu, F.; Fremeaux-Bacchi, V.; Gale, D.P.; van den Heuvel, L.P.; Kavanagh, D.; Noris, M.; Pinto, S.; et al. Statistical Validation of Rare Complement Variants Provides Insights into the Molecular Basis of Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy. J. Immunol. 2018, 200, 2464–2478. [Google Scholar] [CrossRef]
- Bu, F.; Zhang, Y.; Wang, K.; Borsa, N.G.; Jones, M.B.; Taylor, A.O.; Takanami, E.; Meyer, N.C.; Frees, K.; Thomas, C.P.; et al. Genetic Analysis of 400 Patients Refines Understanding and Implicates a New Gene in Atypical Hemolytic Uremic Syndrome. J. Am. Soc. Nephrol. 2018, 29, 2809–2819. [Google Scholar] [CrossRef] [PubMed]
- Jozsi, M.; Licht, C.; Strobel, S.; Zipfel, S.L.; Richter, H.; Heinen, S.; Zipfel, P.F.; Skerka, C. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 2008, 111, 1512–1514. [Google Scholar] [CrossRef] [PubMed]
- Jozsi, M.; Strobel, S.; Dahse, H.M.; Liu, W.S.; Hoyer, P.F.; Oppermann, M.; Skerka, C.; Zipfel, P.F. Anti factor H autoantibodies block C-terminal recognition function of factor H in hemolytic uremic syndrome. Blood 2007, 110, 1516–1518. [Google Scholar] [CrossRef]
- Sullivan, M.; Erlic, Z.; Hoffmann, M.M.; Arbeiter, K.; Patzer, L.; Budde, K.; Hoppe, B.; Zeier, M.; Lhotta, K.; Rybicki, L.A.; et al. Epidemiological approach to identifying genetic predispositions for atypical hemolytic uremic syndrome. Ann. Hum. Genet. 2010, 74, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Bresin, E.; Rurali, E.; Caprioli, J.; Sanchez-Corral, P.; Fremeaux-Bacchi, V.; Rodriguez de Cordoba, S.; Pinto, S.; Goodship, T.H.; Alberti, M.; Ribes, D.; et al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J. Am. Soc. Nephrol. 2013, 24, 475–486. [Google Scholar] [CrossRef]
- de Jorge, E.G.; Macor, P.; Paixao-Cavalcante, D.; Rose, K.L.; Tedesco, F.; Cook, H.T.; Botto, M.; Pickering, M.C. The development of atypical hemolytic uremic syndrome depends on complement C5. J. Am. Soc. Nephrol. 2011, 22, 137–145. [Google Scholar] [CrossRef]
- Smith-Jackson, K.; Yang, Y.; Denton, H.; Pappworth, I.Y.; Cooke, K.; Barlow, P.N.; Atkinson, J.P.; Liszewski, M.K.; Pickering, M.C.; Kavanagh, D.; et al. Hyperfunctional complement C3 promotes C5-dependent atypical hemolytic uremic syndrome in mice. J. Clin. Investig. 2019, 129, 1061–1075. [Google Scholar] [CrossRef]
- Tedesco, F.; Pausa, M.; Nardon, E.; Introna, M.; Mantovani, A.; Dobrina, A. The cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. J. Exp. Med. 1997, 185, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Nagasawa, K.; Horiuchi, T.; Tsuru, T.; Nishizaka, H.; Niho, Y. C5a induces tissue factor activity on endothelial cells. Thromb. Haemost. 1997, 77, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Huber-Lang, M.; Sarma, J.V.; Zetoune, F.S.; Rittirsch, D.; Neff, T.A.; McGuire, S.R.; Lambris, J.D.; Warner, R.L.; Flierl, M.A.; Hoesel, L.M.; et al. Generation of C5a in the absence of C3: A new complement activation pathway. Nat. Med. 2006, 12, 682–687. [Google Scholar] [CrossRef]
- Foley, J.H.; Walton, B.L.; Aleman, M.M.; O’Byrne, A.M.; Lei, V.; Harrasser, M.; Foley, K.A.; Wolberg, A.S.; Conway, E.M. Complement Activation in Arterial and Venous Thrombosis is Mediated by Plasmin. EBioMedicine 2016, 5, 175–182. [Google Scholar] [CrossRef]
- Foreman, K.E.; Vaporciyan, A.A.; Bonish, B.K.; Jones, M.L.; Johnson, K.J.; Glovsky, M.M.; Eddy, S.M.; Ward, P.A. C5a-induced expression of P-selectin in endothelial cells. J. Clin. Investig. 1994, 94, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Ritis, K.; Doumas, M.; Mastellos, D.; Micheli, A.; Giaglis, S.; Magotti, P.; Rafail, S.; Kartalis, G.; Sideras, P.; Lambris, J.D. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J. Immunol. 2006, 177, 4794–4802. [Google Scholar] [CrossRef]
- Cataland, S.R.; Holers, V.M.; Geyer, S.; Yang, S.; Wu, H.M. Biomarkers of terminal complement activation confirm the diagnosis of aHUS and differentiate aHUS from TTP. Blood 2014, 123, 3733–3738. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, S.; Damoiseaux, J.; Werion, A.; Reutelingsperger, C.P.; Morelle, J.; van Paassen, P. Functional and Genetic Landscape of Complement Dysregulation Along the Spectrum of Thrombotic Microangiopathy and its Potential Implications on Clinical Outcomes. Kidney Int. Rep. 2021, 6, 1099–1109. [Google Scholar] [CrossRef]
- Van den Born, B.J.; Koopmans, R.P.; van Montfrans, G.A. The renin-angiotensin system in malignant hypertension revisited: Plasma renin activity, microangiopathic hemolysis, and renal failure in malignant hypertension. Am. J. Hypertens. 2007, 20, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Bekassy, Z.D.; Kristoffersson, A.C.; Rebetz, J.; Tati, R.; Olin, A.I.; Karpman, D. Aliskiren inhibits renin-mediated complement activation. Kidney Int. 2018, 94, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Raij, L.; Dalmasso, A.P.; Staley, N.A.; Fish, A.J. Renal injury in DOCA-salt hypertensive C5-sufficient and C5-deficient mice. Kidney Int. 1989, 36, 582–592. [Google Scholar] [CrossRef]
- Weiss, S.; Rosendahl, A.; Czesla, D.; Meyer-Schwesinger, C.; Stahl, R.A.; Ehmke, H.; Kurts, C.; Zipfel, P.F.; Kohl, J.; Wenzel, U.O. The complement receptor C5aR1 contributes to renal damage but protects the heart in angiotensin II-induced hypertension. Am. J. Physiol. Renal Physiol. 2016, 310, F1356–F1365. [Google Scholar] [CrossRef]
- Van den Born, B.J.; Honnebier, U.P.; Koopmans, R.P.; van Montfrans, G.A. Microangiopathic hemolysis and renal failure in malignant hypertension. Hypertension 2005, 45, 246–251. [Google Scholar] [CrossRef]
- Gonzalez, R.; Morales, E.; Segura, J.; Ruilope, L.M.; Praga, M. Long-term renal survival in malignant hypertension. Nephrol. Dial. Transplant. 2010, 25, 3266–3272. [Google Scholar] [CrossRef]
- Timmermans, S.; Abdul-Hamid, M.A.; Vanderlocht, J.; Damoiseaux, J.; Reutelingsperger, C.P.; van Paassen, P.; Limburg Renal, R. Patients with hypertension-associated thrombotic microangiopathy may present with complement abnormalities. Kidney Int. 2017, 91, 1420–1425. [Google Scholar] [CrossRef]
- El Karoui, K.; Boudhabhay, I.; Petitprez, F.; Vieira-Martins, P.; Fakhouri, F.; Zuber, J.; Aulagnon, F.; Matignon, M.; Rondeau, E.; Mesnard, L.; et al. Impact of hypertensive emergency and complement rare variants on presentation and outcome of atypical hemolytic uremic syndrome. Haematologica 2019, 104, 2501–2511. [Google Scholar] [CrossRef]
- Cavero, T.; Arjona, E.; Soto, K.; Caravaca-Fontan, F.; Rabasco, C.; Bravo, L.; de la Cerda, F.; Martin, N.; Blasco, M.; Avila, A.; et al. Severe and malignant hypertension are common in primary atypical hemolytic uremic syndrome. Kidney Int. 2019, 96, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, S.; Abdul-Hamid, M.A.; Potjewijd, J.; Theunissen, R.; Damoiseaux, J.; Reutelingsperger, C.P.; van Paassen, P.; Limburg Renal, R. C5b9 Formation on Endothelial Cells Reflects Complement Defects among Patients with Renal Thrombotic Microangiopathy and Severe Hypertension. J. Am. Soc. Nephrol. 2018, 29, 2234–2243. [Google Scholar] [CrossRef]
- Timmermans, S.; van Paassen, P.; Limburg Renal, R. The Authors Reply. Kidney Int. 2017, 92, 267–268. [Google Scholar] [CrossRef]
- Dashe, J.S.; Ramin, S.M.; Cunningham, F.G. The long-term consequences of thrombotic microangiopathy (thrombotic thrombocytopenic purpura and hemolytic uremic syndrome) in pregnancy. Obstet. Gynecol. 1998, 91, 662–668. [Google Scholar] [CrossRef]
- Moatti-Cohen, M.; Garrec, C.; Wolf, M.; Boisseau, P.; Galicier, L.; Azoulay, E.; Stepanian, A.; Delmas, Y.; Rondeau, E.; Bezieau, S.; et al. Unexpected frequency of Upshaw-Schulman syndrome in pregnancy-onset thrombotic thrombocytopenic purpura. Blood 2012, 119, 5888–5897. [Google Scholar] [CrossRef]
- Fakhouri, F.; Roumenina, L.; Provot, F.; Sallee, M.; Caillard, S.; Couzi, L.; Essig, M.; Ribes, D.; Dragon-Durey, M.A.; Bridoux, F.; et al. Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J. Am. Soc. Nephrol. 2010, 21, 859–867. [Google Scholar] [CrossRef]
- Huerta, A.; Arjona, E.; Portoles, J.; Lopez-Sanchez, P.; Rabasco, C.; Espinosa, M.; Cavero, T.; Blasco, M.; Cao, M.; Manrique, J.; et al. A retrospective study of pregnancy-associated atypical hemolytic uremic syndrome. Kidney Int. 2018, 93, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, S.; Werion, A.; Spaanderman, M.E.A.; Reutelingsperger, C.P.; Damoiseaux, J.; Morelle, J.; van Paassen, P. The natural course of pregnancies in women with primary atypical haemolytic uraemic syndrome and asymptomatic relatives. Br. J. Haematol. 2020, 190, 442–449. [Google Scholar] [CrossRef]
- Gaggl, M.; Aigner, C.; Csuka, D.; Szilagyi, A.; Prohaszka, Z.; Kain, R.; Haninger, N.; Knechtelsdorfer, M.; Sunder-Plassmann, R.; Sunder-Plassmann, G.; et al. Maternal and Fetal Outcomes of Pregnancies in Women with Atypical Hemolytic Uremic Syndrome. J. Am. Soc. Nephrol. 2018, 29, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Salmon, J.E.; Heuser, C.; Triebwasser, M.; Liszewski, M.K.; Kavanagh, D.; Roumenina, L.; Branch, D.W.; Goodship, T.; Fremeaux-Bacchi, V.; Atkinson, J.P. Mutations in complement regulatory proteins predispose to preeclampsia: A genetic analysis of the PROMISSE cohort. PLoS Med. 2011, 8, e1001013. [Google Scholar] [CrossRef]
- Fakhouri, F.; Jablonski, M.; Lepercq, J.; Blouin, J.; Benachi, A.; Hourmant, M.; Pirson, Y.; Durrbach, A.; Grunfeld, J.P.; Knebelmann, B.; et al. Factor H, membrane cofactor protein, and factor I mutations in patients with hemolysis, elevated liver enzymes, and low platelet count syndrome. Blood 2008, 112, 4542–4545. [Google Scholar] [CrossRef] [PubMed]
- Vaught, A.J.; Braunstein, E.M.; Jasem, J.; Yuan, X.; Makhlin, I.; Eloundou, S.; Baines, A.C.; Merrill, S.A.; Chaturvedi, S.; Blakemore, K.; et al. Germline mutations in the alternative pathway of complement predispose to HELLP syndrome. JCI Insight 2018, 3, e99128. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Servais, A.; Devillard, N.; Fremeaux-Bacchi, V.; Hummel, A.; Salomon, L.; Contin-Bordes, C.; Gomer, H.; Legendre, C.; Delmas, Y. Atypical haemolytic uraemic syndrome and pregnancy: Outcome with ongoing eculizumab. Nephrol. Dial. Transplant. 2016, 31, 2122–2130. [Google Scholar] [CrossRef] [PubMed]
- Jager, N.M.; Poppelaars, F.; Daha, M.R.; Seelen, M.A. Complement in renal transplantation: The road to translation. Mol. Immunol. 2017, 89, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Le Quintrec, M.; Zuber, J.; Moulin, B.; Kamar, N.; Jablonski, M.; Lionet, A.; Chatelet, V.; Mousson, C.; Mourad, G.; Bridoux, F.; et al. Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome. Am. J. Transplant. 2013, 13, 663–675. [Google Scholar] [CrossRef]
- Reynolds, J.C.; Agodoa, L.Y.; Yuan, C.M.; Abbott, K.C. Thrombotic microangiopathy after renal transplantation in the United States. Am. J. Kidney Dis. 2003, 42, 1058–1068. [Google Scholar] [CrossRef]
- Timmermans, S.; van Paassen, P.; Limburg Renal, R. Mother and Child Reunion in “Hypertensive” End-Stage Renal Disease: Will They Complement Each Other? Nephron 2019, 142, 253–257. [Google Scholar] [CrossRef]
- Langer, R.M.; Van Buren, C.T.; Katz, S.M.; Kahan, B.D. De novo hemolytic uremic syndrome after kidney transplantation in patients treated with cyclosporine-sirolimus combination. Transplantation 2002, 73, 756–760. [Google Scholar] [CrossRef]
- Satoskar, A.A.; Pelletier, R.; Adams, P.; Nadasdy, G.M.; Brodsky, S.; Pesavento, T.; Henry, M.; Nadasdy, T. De novo thrombotic microangiopathy in renal allograft biopsies-role of antibody-mediated rejection. Am. J. Transplant. 2010, 10, 1804–1811. [Google Scholar] [CrossRef]
- Zarifian, A.; Meleg-Smith, S.; O’Donovan, R.; Tesi, R.J.; Batuman, V. Cyclosporine-associated thrombotic microangiopathy in renal allografts. Kidney Int. 1999, 55, 2457–2466. [Google Scholar] [CrossRef]
- Marks, W.H.; Mamode, N.; Montgomery, R.A.; Stegall, M.D.; Ratner, L.E.; Cornell, L.D.; Rowshani, A.T.; Colvin, R.B.; Dain, B.; Boice, J.A.; et al. Safety and efficacy of eculizumab in the prevention of antibody-mediated rejection in living-donor kidney transplant recipients requiring desensitization therapy: A randomized trial. Am. J. Transplant. 2019, 19, 2876–2888. [Google Scholar] [CrossRef]
- Zuber, J.; Le Quintrec, M.; Krid, S.; Bertoye, C.; Gueutin, V.; Lahoche, A.; Heyne, N.; Ardissino, G.; Chatelet, V.; Noel, L.H.; et al. Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am. J. Transplant. 2012, 12, 3337–3354. [Google Scholar] [CrossRef]
- Siedlecki, A.M.; Isbel, N.; Vande Walle, J.; James Eggleston, J.; Cohen, D.J.; Globala, H.U.S.R. Eculizumab Use for Kidney Transplantation in Patients with a Diagnosis of Atypical Hemolytic Uremic Syndrome. Kidney Int. Rep. 2019, 4, 434–446. [Google Scholar] [CrossRef]
- Le Clech, A.; Simon-Tillaux, N.; Provot, F.; Delmas, Y.; Vieira-Martins, P.; Limou, S.; Halimi, J.M.; Le Quintrec, M.; Lebourg, L.; Grange, S.; et al. Atypical and secondary hemolytic uremic syndromes have a distinct presentation and no common genetic risk factors. Kidney Int. 2019, 95, 1443–1452. [Google Scholar] [CrossRef]
- Fremeaux-Bacchi, V.; Sellier-Leclerc, A.L.; Vieira-Martins, P.; Limou, S.; Kwon, T.; Lahoche, A.; Novo, R.; Llanas, B.; Nobili, F.; Roussey, G.; et al. Complement Gene Variants and Shiga Toxin-Producing Escherichia coli-Associated Hemolytic Uremic Syndrome: Retrospective Genetic and Clinical Study. Clin. J. Am. Soc. Nephrol. 2019, 14, 364–377. [Google Scholar] [CrossRef]
- Ahlenstiel-Grunow, T.; Hachmeister, S.; Bange, F.C.; Wehling, C.; Kirschfink, M.; Bergmann, C.; Pape, L. Systemic complement activation and complement gene analysis in enterohaemorrhagic Escherichia coli-associated paediatric haemolytic uraemic syndrome. Nephrol. Dial. Transplant. 2016, 31, 1114–1121. [Google Scholar] [CrossRef]
- Westra, D.; Volokhina, E.B.; van der Molen, R.G.; van der Velden, T.J.; Jeronimus-Klaasen, A.; Goertz, J.; Gracchi, V.; Dorresteijn, E.M.; Bouts, A.H.; Keijzer-Veen, M.G.; et al. Serological and genetic complement alterations in infection-induced and complement-mediated hemolytic uremic syndrome. Pediatr. Nephrol. 2017, 32, 297–309. [Google Scholar] [CrossRef]
- Menne, J.; Nitschke, M.; Stingele, R.; Abu-Tair, M.; Beneke, J.; Bramstedt, J.; Bremer, J.P.; Brunkhorst, R.; Busch, V.; Dengler, R.; et al. Validation of treatment strategies for enterohaemorrhagic Escherichia coli O104:H4 induced haemolytic uraemic syndrome: Case-control study. BMJ 2012, 345, e4565. [Google Scholar] [CrossRef]
- Mody, R.K.; Gu, W.; Griffin, P.M.; Jones, T.F.; Rounds, J.; Shiferaw, B.; Tobin-D’Angelo, M.; Smith, G.; Spina, N.; Hurd, S.; et al. Postdiarrheal hemolytic uremic syndrome in United States children: Clinical spectrum and predictors of in-hospital death. J. Pediatr. 2015, 166, 1022–1029. [Google Scholar] [CrossRef]
- Kielstein, J.T.; Beutel, G.; Fleig, S.; Steinhoff, J.; Meyer, T.N.; Hafer, C.; Kuhlmann, U.; Bramstedt, J.; Panzer, U.; Vischedyk, M.; et al. Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga-toxin-producing E. coli O104:H4 induced haemolytic-uraemic syndrome: An analysis of the German STEC-HUS registry. Nephrol. Dial. Transplant. 2012, 27, 3807–3815. [Google Scholar] [CrossRef]
- Szilagyi, A.; Kiss, N.; Bereczki, C.; Talosi, G.; Racz, K.; Turi, S.; Gyorke, Z.; Simon, E.; Horvath, E.; Kelen, K.; et al. The role of complement in Streptococcus pneumoniae-associated haemolytic uraemic syndrome. Nephrol. Dial. Transplant. 2013, 28, 2237–2245. [Google Scholar] [CrossRef]
- Banerjee, R.; Hersh, A.L.; Newland, J.; Beekmann, S.E.; Polgreen, P.M.; Bender, J.; Shaw, J.; Copelovitch, L.; Kaplan, B.S.; Shah, S.S.; et al. Streptococcus pneumoniae-associated hemolytic uremic syndrome among children in North America. Pediatr. Infect. Dis. J. 2011, 30, 736–739. [Google Scholar] [CrossRef]
- Copelovitch, L.; Kaplan, B.S. Streptococcus pneumoniae--associated hemolytic uremic syndrome: Classification and the emergence of serotype 19A. Pediatrics 2010, 125, e174–e182. [Google Scholar] [CrossRef]
- Yui, J.C.; Van Keer, J.; Weiss, B.M.; Waxman, A.J.; Palmer, M.B.; D’Agati, V.D.; Kastritis, E.; Dimopoulos, M.A.; Vij, R.; Bansal, D.; et al. Proteasome inhibitor associated thrombotic microangiopathy. Am. J. Hematol. 2016, 91, E348–E352. [Google Scholar] [CrossRef]
- Cavero, T.; Rabasco, C.; Lopez, A.; Roman, E.; Avila, A.; Sevillano, A.; Huerta, A.; Rojas-Rivera, J.; Fuentes, C.; Blasco, M.; et al. Eculizumab in secondary atypical haemolytic uraemic syndrome. Nephrol. Dial. Transplant. 2017, 32, 466–474. [Google Scholar] [CrossRef]
- Izzedine, H.; Mangier, M.; Ory, V.; Zhang, S.Y.; Sendeyo, K.; Bouachi, K.; Audard, V.; Pechoux, C.; Soria, J.C.; Massard, C.; et al. Expression patterns of RelA and c-mip are associated with different glomerular diseases following anti-VEGF therapy. Kidney Int. 2014, 85, 457–470. [Google Scholar] [CrossRef]
- Page, E.E.; Little, D.J.; Vesely, S.K.; George, J.N. Quinine-Induced Thrombotic Microangiopathy: A Report of 19 Patients. Am. J. Kidney Dis. 2017, 70, 686–695. [Google Scholar] [CrossRef]
- Lechner, K.; Obermeier, H.L. Cancer-related microangiopathic hemolytic anemia: Clinical and laboratory features in 168 reported cases. Medicine 2012, 91, 195–205. [Google Scholar] [CrossRef]
- Timmermans, S.; Damoiseaux, J.; Reutelingsperger, C.P.; van Paassen, P. More About Complement in the Antiphospholipid Syndrome. Blood 2020. [Google Scholar] [CrossRef]
- Barrera-Vargas, A.; Rosado-Canto, R.; Merayo-Chalico, J.; Arreola-Guerra, J.M.; Mejia-Vilet, J.M.; Correa-Rotter, R.; Gomez-Martin, D.; Alcocer-Varela, J. Renal Thrombotic Microangiopathy in Proliferative Lupus Nephritis: Risk Factors and Clinical Outcomes: A Case-Control Study. J. Clin. Rheumatol. 2016, 22, 235–240. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Braunstein, E.M.; Yuan, X.; Yu, J.; Alexander, A.; Chen, H.; Gavriilaki, E.; Alluri, R.; Streiff, M.B.; Petri, M.; et al. Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS. Blood 2020, 135, 239–251. [Google Scholar] [CrossRef]
- Park, M.H.; Caselman, N.; Ulmer, S.; Weitz, I.C. Complement-mediated thrombotic microangiopathy associated with lupus nephritis. Blood Adv. 2018, 2, 2090–2094. [Google Scholar] [CrossRef]
- Glezerman, I.G.; Jhaveri, K.D.; Watson, T.H.; Edwards, A.M.; Papadopoulos, E.B.; Young, J.W.; Flombaum, C.D.; Jakubowski, A.A. Chronic kidney disease, thrombotic microangiopathy, and hypertension following T cell-depleted hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2010, 16, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Sartain, S.; Shubert, S.; Wu, M.F.; Srivaths, P.; Teruya, J.; Krance, R.; Martinez, C. Therapeutic Plasma Exchange does not Improve Renal Function in Hematopoietic Stem Cell Transplantation-Associated Thrombotic Microangiopathy: An Institutional Experience. Biol. Blood Marrow Transplant. 2019, 25, 157–162. [Google Scholar] [CrossRef]
- Jodele, S.; Davies, S.M.; Lane, A.; Khoury, J.; Dandoy, C.; Goebel, J.; Myers, K.; Grimley, M.; Bleesing, J.; El-Bietar, J.; et al. Diagnostic and risk criteria for HSCT-associated thrombotic microangiopathy: A study in children and young adults. Blood 2014, 124, 645–653. [Google Scholar] [CrossRef]
- Jodele, S.; Dandoy, C.E.; Lane, A.; Laskin, B.L.; Teusink-Cross, A.; Myers, K.C.; Wallace, G.; Nelson, A.; Bleesing, J.; Chima, R.S.; et al. Complement blockade for TA-TMA: Lessons learned from a large pediatric cohort treated with eculizumab. Blood 2020, 135, 1049–1057. [Google Scholar] [CrossRef]
- Jodele, S.; Zhang, K.; Zou, F.; Laskin, B.; Dandoy, C.E.; Myers, K.C.; Lane, A.; Meller, J.; Medvedovic, M.; Chen, J.; et al. The genetic fingerprint of susceptibility for transplant-associated thrombotic microangiopathy. Blood 2016, 127, 989–996. [Google Scholar] [CrossRef]
- Azukaitis, K.; Simkova, E.; Majid, M.A.; Galiano, M.; Benz, K.; Amann, K.; Bockmeyer, C.; Gajjar, R.; Meyers, K.E.; Cheong, H.I.; et al. The Phenotypic Spectrum of Nephropathies Associated with Mutations in Diacylglycerol Kinase epsilon. J. Am. Soc. Nephrol. 2017, 28, 3066–3075. [Google Scholar] [CrossRef]
- Sanchez Chinchilla, D.; Pinto, S.; Hoppe, B.; Adragna, M.; Lopez, L.; Justa Roldan, M.L.; Pena, A.; Lopez Trascasa, M.; Sanchez-Corral, P.; Rodriguez de Cordoba, S. Complement mutations in diacylglycerol kinase-epsilon-associated atypical hemolytic uremic syndrome. Clin. J. Am. Soc. Nephrol. 2014, 9, 1611–1619. [Google Scholar] [CrossRef] [PubMed]
- Brocklebank, V.; Kumar, G.; Howie, A.J.; Chandar, J.; Milford, D.V.; Craze, J.; Evans, J.; Finlay, E.; Freundlich, M.; Gale, D.P.; et al. Long-term outcomes and response to treatment in diacylglycerol kinase epsilon nephropathy. Kidney Int. 2020, 97, 1260–1274. [Google Scholar] [CrossRef]
- Lemaire, M.; Fremeaux-Bacchi, V.; Schaefer, F.; Choi, M.; Tang, W.H.; Le Quintrec, M.; Fakhouri, F.; Taque, S.; Nobili, F.; Martinez, F.; et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat. Genet. 2013, 45, 531–536. [Google Scholar] [CrossRef]
- Beck, B.B.; van Spronsen, F.; Diepstra, A.; Berger, R.M.; Komhoff, M. Renal thrombotic microangiopathy in patients with cblC defect: Review of an under-recognized entity. Pediatr. Nephrol. 2017, 32, 733–741. [Google Scholar] [CrossRef]
- Ravindran, A.; Go, R.S.; Fervenza, F.C.; Sethi, S. Thrombotic microangiopathy associated with monoclonal gammopathy. Kidney Int. 2017, 91, 691–698. [Google Scholar] [CrossRef]
- Ozaki, M.; Kang, Y.; Tan, Y.S.; Pavlov, V.I.; Liu, B.; Boyle, D.C.; Kushak, R.I.; Skjoedt, M.O.; Grabowski, E.F.; Taira, Y.; et al. Human mannose-binding lectin inhibitor prevents Shiga toxin-induced renal injury. Kidney Int. 2016, 90, 774–782. [Google Scholar] [CrossRef]
- Morigi, M.; Galbusera, M.; Gastoldi, S.; Locatelli, M.; Buelli, S.; Pezzotta, A.; Pagani, C.; Noris, M.; Gobbi, M.; Stravalaci, M.; et al. Alternative pathway activation of complement by Shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J. Immunol. 2011, 187, 172–180. [Google Scholar] [CrossRef]
- Rosales, A.; Hofer, J.; Zimmerhackl, L.B.; Jungraithmayr, T.C.; Riedl, M.; Giner, T.; Strasak, A.; Orth-Holler, D.; Wurzner, R.; Karch, H.; et al. Need for long-term follow-up in enterohemorrhagic Escherichia coli-associated hemolytic uremic syndrome due to late-emerging sequelae. Clin. Infect. Dis. 2012, 54, 1413–1421. [Google Scholar] [CrossRef]
- Alberti, M.; Valoti, E.; Piras, R.; Bresin, E.; Galbusera, M.; Tripodo, C.; Thaiss, F.; Remuzzi, G.; Noris, M. Two patients with history of STEC-HUS, posttransplant recurrence and complement gene mutations. Am. J. Transplant. 2013, 13, 2201–2206. [Google Scholar] [CrossRef]
- Frank, C.; Werber, D.; Cramer, J.P.; Askar, M.; Faber, M.; an der Heiden, M.; Bernard, H.; Fruth, A.; Prager, R.; Spode, A.; et al. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N. Engl. J. Med. 2011, 365, 1771–1780. [Google Scholar] [CrossRef]
- Nitschke, M.; Sayk, F.; Hartel, C.; Roseland, R.T.; Hauswaldt, S.; Steinhoff, J.; Fellermann, K.; Derad, I.; Wellhoner, P.; Buning, J.; et al. Association between azithromycin therapy and duration of bacterial shedding among patients with Shiga toxin-producing enteroaggregative Escherichia coli O104:H4. JAMA 2012, 307, 1046–1052. [Google Scholar] [CrossRef]
- Reese, J.A.; Bougie, D.W.; Curtis, B.R.; Terrell, D.R.; Vesely, S.K.; Aster, R.H.; George, J.N. Drug-induced thrombotic microangiopathy: Experience of the Oklahoma Registry and the BloodCenter of Wisconsin. Am. J. Hematol. 2015, 90, 406–410. [Google Scholar] [CrossRef]
- Saleem, R.; Reese, J.A.; George, J.N. Drug-induced thrombotic microangiopathy: An updated systematic review, 2014–2018. Am. J. Hematol. 2018, 93, E241–E243. [Google Scholar] [CrossRef]
- Glynne, P.; Salama, A.; Chaudhry, A.; Swirsky, D.; Lightstone, L. Quinine-induced immune thrombocytopenic purpura followed by hemolytic uremic syndrome. Am. J. Kidney Dis. 1999, 33, 133–137. [Google Scholar] [CrossRef]
- Eremina, V.; Jefferson, J.A.; Kowalewska, J.; Hochster, H.; Haas, M.; Weisstuch, J.; Richardson, C.; Kopp, J.B.; Kabir, M.G.; Backx, P.H.; et al. VEGF inhibition and renal thrombotic microangiopathy. N. Engl. J. Med. 2008, 358, 1129–1136. [Google Scholar] [CrossRef]
- Keir, L.S.; Firth, R.; Aponik, L.; Feitelberg, D.; Sakimoto, S.; Aguilar, E.; Welsh, G.I.; Richards, A.; Usui, Y.; Satchell, S.C.; et al. VEGF regulates local inhibitory complement proteins in the eye and kidney. J. Clin. Investig. 2017, 127, 199–214. [Google Scholar] [CrossRef]
- Brain, M.C.; Azzopardi, J.G.; Baker, L.R.; Pineo, G.F.; Roberts, P.D.; Dacie, J.V. Microangiopathic haemolytic anaemia and mucin-forming adenocarcinoma. Br. J. Haematol. 1970, 18, 183–193. [Google Scholar] [CrossRef]
- Moroni, G.; Radice, A.; Giammarresi, G.; Quaglini, S.; Gallelli, B.; Leoni, A.; Li Vecchi, M.; Messa, P.; Sinico, R.A. Are laboratory tests useful for monitoring the activity of lupus nephritis? A 6-year prospective study in a cohort of 228 patients with lupus nephritis. Ann. Rheum Dis. 2009, 68, 234–237. [Google Scholar] [CrossRef]
- Tektonidou, M.G.; Sotsiou, F.; Nakopoulou, L.; Vlachoyiannopoulos, P.G.; Moutsopoulos, H.M. Antiphospholipid syndrome nephropathy in patients with systemic lupus erythematosus and antiphospholipid antibodies: Prevalence, clinical associations, and long-term outcome. Arthritis Rheum. 2004, 50, 2569–2579. [Google Scholar] [CrossRef]
- Jonsen, A.; Nilsson, S.C.; Ahlqvist, E.; Svenungsson, E.; Gunnarsson, I.; Eriksson, K.G.; Bengtsson, A.; Zickert, A.; Eloranta, M.L.; Truedsson, L.; et al. Mutations in genes encoding complement inhibitors CD46 and CFH affect the age at nephritis onset in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2011, 13, R206. [Google Scholar] [CrossRef]
- Sharif, M.O.; Leavis, H.L.; van Paassen, P.; van Rhenen, A.; Timmermans, S.; Ton, E.; van Laar, J.M.; Spierings, J. Severe thrombotic microangiopathy after autologous stem cell transplantation in systemic sclerosis: A case report. Rheumatology 2021, e1–e3. [Google Scholar] [CrossRef]
- Jodele, S.; Licht, C.; Goebel, J.; Dixon, B.P.; Zhang, K.; Sivakumaran, T.A.; Davies, S.M.; Pluthero, F.G.; Lu, L.; Laskin, B.L. Abnormalities in the alternative pathway of complement in children with hematopoietic stem cell transplant-associated thrombotic microangiopathy. Blood 2013, 122, 2003–2007. [Google Scholar] [CrossRef]
- Gavriilaki, E.; Chrysanthopoulou, A.; Sakellari, I.; Batsis, I.; Mallouri, D.; Touloumenidou, T.; Papalexandri, A.; Mitsios, A.; Arampatzioglou, A.; Ritis, K.; et al. Linking Complement Activation, Coagulation, and Neutrophils in Transplant-Associated Thrombotic Microangiopathy. Thromb. Haemost. 2019, 119, 1433–1440. [Google Scholar] [CrossRef]
- Busch, M.; Timmermans, S.A.; Nagy, M.; Visser, M.; Huckriede, J.; Aendekerk, J.P.; De Vries, F.; Potjewijd, J.; Jallah, B.; Ysermans, R.; et al. Neutrophils and Contact Activation of Coagulation as Potential Drivers of COVID-19. Circulation 2020, 142, 1787–1790. [Google Scholar] [CrossRef]
- Sridharan, M.; Hook, C.C.; Leung, N.; Winters, J.L.; Go, R.S.; Mayo Clinic Complement Alternative Pathway‐Thrombotic Microangiopathy Disease-Oriented Group. Postsurgical thrombotic microangiopathy: Case series and review of the literature. Eur. J. Haematol. 2019, 103, 307–318. [Google Scholar] [CrossRef]
- Timmermans, S.; Abdul-Hamid, M.H.; van Paassen, P.; Limburg Renal, R. Chronic thrombotic microangiopathy in patients with a C3 gain of function protein. Nephrol. Dial. Transplant. 2020, 35, 1449–1451. [Google Scholar] [CrossRef]
- Noris, M.; Galbusera, M.; Gastoldi, S.; Macor, P.; Banterla, F.; Bresin, E.; Tripodo, C.; Bettoni, S.; Donadelli, R.; Valoti, E.; et al. Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood 2014, 124, 1715–1726. [Google Scholar] [CrossRef]
- Zuber, J.; Frimat, M.; Caillard, S.; Kamar, N.; Gatault, P.; Petitprez, F.; Couzi, L.; Jourde-Chiche, N.; Chatelet, V.; Gaisne, R.; et al. Use of Highly Individualized Complement Blockade Has Revolutionized Clinical Outcomes after Kidney Transplantation and Renal Epidemiology of Atypical Hemolytic Uremic Syndrome. J. Am. Soc. Nephrol. 2019, 30, 2449–2463. [Google Scholar] [CrossRef]
- Gavriilaki, E.; Yuan, X.; Ye, Z.; Ambinder, A.J.; Shanbhag, S.P.; Streiff, M.B.; Kickler, T.S.; Moliterno, A.R.; Sperati, C.J.; Brodsky, R.A. Modified Ham test for atypical hemolytic uremic syndrome. Blood 2015, 125, 3637–3646. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}



| Genetics/FHAA a | Treatment | |||||
|---|---|---|---|---|---|---|
| Condition Ref. (Patients) | Rare Variants b | Pathogenic | Dialysis at Presentation | Kidney Outcome c | Standard of Care (SoC) | Eculizumab |
| STEC-HUS | 9/125, 7% | 3/125, 2% | >50% | 1–7% ESKD; no relapse d | Kidney response is common | Efficacy unproven |
| Ref. [67] (79) | 6/75 | 3/75 | 56% (median, 9 d) | 1% ESKD | Kidney response is common | Similar to SoC (n = 12) |
| Ref. [68] (25) | 1/25 | 0/25 | 80% (mean, 7 d) | - | - | - |
| Ref. [69] (26) | 2/25 | 0/25 | 65% (average, 16 d) | - | - | - |
| Ref. [70] (298) | - | - | 54% (mean, 10 d) | 1% ESKD | Kidney response is common | Similar to SoC (n = 67) |
| Ref. [71] (770) | - | - | 57% | 7% RRT at discharge | Kidney response is common | - |
| Ref. [72] (491) | - | - | 57% | 4% RRT at discharge | Kidney response is common | Similar to SoC (n = 193) |
| Post-diarrheal HUS | ||||||
| Ref. [67] (33) | 2/23, 9% | 1/33, 3% | 41% (median, 9 d) | 0% ESKD; no relape | Kidney response is common | Efficayc unproven; similar to SoC (n = 3) |
| Pneumococcal HUS | 3/5, 60% | 2/5, 40% | >50% | 14–23% ESKD; no relapse | Kidney response is common | Efficacy unproven |
| Ref. [73] (5) | 3/5 | 2/5 | 80% | - | - | - |
| Ref. [74] (37) | – | – | 73% (median, 15 d) | 23% ESKD | Kidney response is common | - |
| Ref. [75] (14) | - | - | 57% | 14% RRT at discharge | Kidney response is common | - |
| Drug-induced TMA | 2/62, 3% | 1/62; 2% | Variable | <16% ESKD; no relapse d | Kidney response is common | Efficacy unproven |
| Ref. [66] (32) | 2/32 | 1/32 | 22% | 16% ESKD | Kidney response is common | Similar to SoC (n = 13) |
| Ref. [76] (11) | 0/2 | 0/2 | 45% | - | Kidney response is common | - |
| Ref. [77] (15) | 0/15 | 0/15 | 33% | No ESKD | - | Kidney response is common |
| Ref. [78] (14) | 0/13 | 0/13 | - | No ESKD | Kidney response is common | - |
| Ref. [79] (19) | - | - | 90% | 17% ESKD | Kidney response is common | - |
| Cancer | 2/11, 18% | 1/11, 9% | Variable | ESKD competes with survival; no relapse | Kidney response is common | Efficacy unproven |
| Ref. [66] (11) | 2/11 | 1/11 | 55% | 30% ESKD | Kidney response is common | Similar to SoC (n = 8) |
| Ref. [80] (154) | - | - | 17% | - | - | - |
| Autoimmunity | 2/34, 6% | 0/34, 0% | Variable | Variable: ~10% (i.e., CAPS) [81] to >50% ESKD [82]; no relapse | Poor prognosis | Efficacy unproven |
| Ref. [66] (26) | 1/26 | 0/26 | 52% | 61% ESKD | Poor prognosis | No response (n/N = 8/9) |
| Ref. [77] (8) | 1/8 | 0/8 | 63% | 57% ESKD | Poor prognosis | No response (n/N = 6/7) |
| Ref. [83] (10; CAPS) | 1/10 | 0/10 | - | - | - | - |
| Ref. [84] (11; SLE) | 2/10 | 0/10 | 73% | 27% ESKD | - | No response (n/N = 4/11) |
| HSCT-TMA | 7/64, 11% | 0/64, 0% | 23% | Most surviving patients had CKD G3-4 [85,86]; no relapse | Poor prognosis with 1-year survival of 17% [87] | May be considered; 1-year survival of 66% [88] |
| Ref. [89] (34) | 3/34 | 0/34 | - | - | - | - |
| Ref. [88] (30) | 4/30 | 0/30 | 23% | - | - | - |
| DGKE-HUS | 3/72, 4% | 0/72, 0% | 50–69% | “late” ESKD; relapse is common | Kidney response is common | Efficacy unproven |
| Refs. [90,91] (44) | 3/44 | 0/44 | 52% | 23% ESKD (~12 yr); 70% relapse | Kidney response is common | Similar to SoC (n = 3) |
| Ref. [92] (15) | 0/15 | 0/15 | 50% (i.e., <3 Wk) | 13% ESKD (>20 yr); 53% relapse | Kidney response is common | Similar to SoC (n = 6) |
| Ref. [93] (13) | 0/13 | 0/13 | 69% | 31% ESKD (>10 yr); 77% relapse | Kidney response is common | Similar to SoC (n = 1) |
| Cobalamine C deficiency | ||||||
| Ref. [94] (36) | 2 e/15, 13% | 0/15, 0% | 22% | High mortality; no relapse | Kidney response is common | Efficacy unproven; no response (n/N = 4/5) |
| Monoclonal gammopathy | ||||||
| Ref. [95] (20) | - | - | 55% | 50% ESKD; no relapse | Kidney response is common | Efficacy unproven; no response (n/N = 1/1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timmermans, S.A.M.E.G.; van Paassen, P. The Syndromes of Thrombotic Microangiopathy: A Critical Appraisal on Complement Dysregulation. J. Clin. Med. 2021, 10, 3034. https://doi.org/10.3390/jcm10143034
Timmermans SAMEG, van Paassen P. The Syndromes of Thrombotic Microangiopathy: A Critical Appraisal on Complement Dysregulation. Journal of Clinical Medicine. 2021; 10(14):3034. https://doi.org/10.3390/jcm10143034
Chicago/Turabian StyleTimmermans, Sjoerd A. M. E. G., and Pieter van Paassen. 2021. "The Syndromes of Thrombotic Microangiopathy: A Critical Appraisal on Complement Dysregulation" Journal of Clinical Medicine 10, no. 14: 3034. https://doi.org/10.3390/jcm10143034
APA StyleTimmermans, S. A. M. E. G., & van Paassen, P. (2021). The Syndromes of Thrombotic Microangiopathy: A Critical Appraisal on Complement Dysregulation. Journal of Clinical Medicine, 10(14), 3034. https://doi.org/10.3390/jcm10143034