An Attenuated Zika Virus Encoding Non-Glycosylated Envelope (E) and Non-Structural Protein 1 (NS1) Confers Complete Protection against Lethal Challenge in a Mouse Model

 , , , ,
, , , ,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Cells and Viruses
2.3. Reagents and Antibodies
2.4. qRT-PCR and Plaque Assay
2.5. Generation of Mutant Viruses
2.6. Mouse Experiments
2.7. Plaque Reduction Neutralization Test
2.8. Western Blotting
2.9. Acetone Precipitation of Proteins
2.10. Antibody Isotyping by ELISA
2.11. ELISPOT Assay
2.12. Statistical Analysis
3. Results
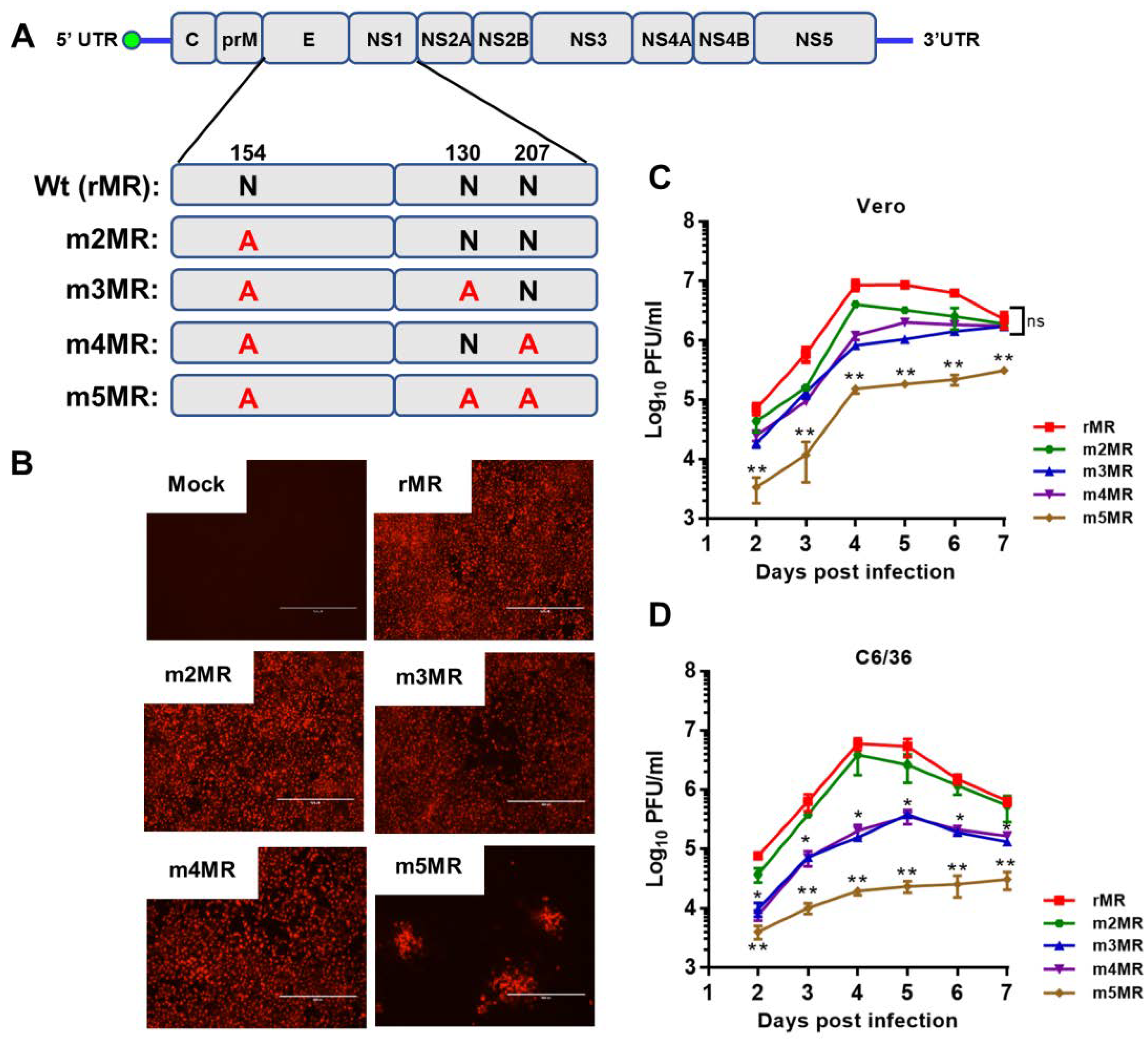
3.1. Recovery and Characterization of E/NS1 Glycosylation Mutant Viruses
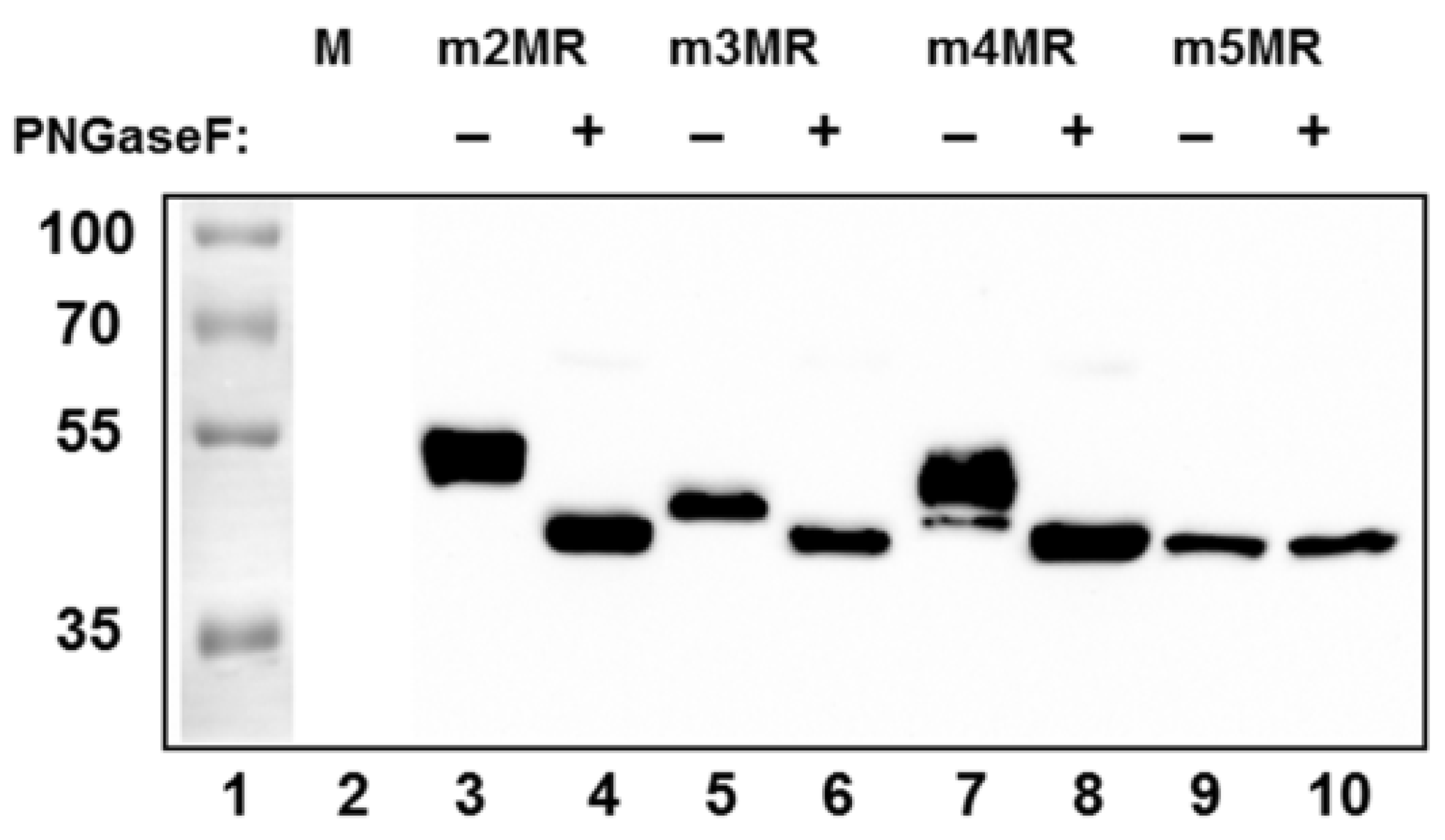
3.2. Glycosylation Status and Secretion of NS1 Protein from Mutant Virus-Infected Cells
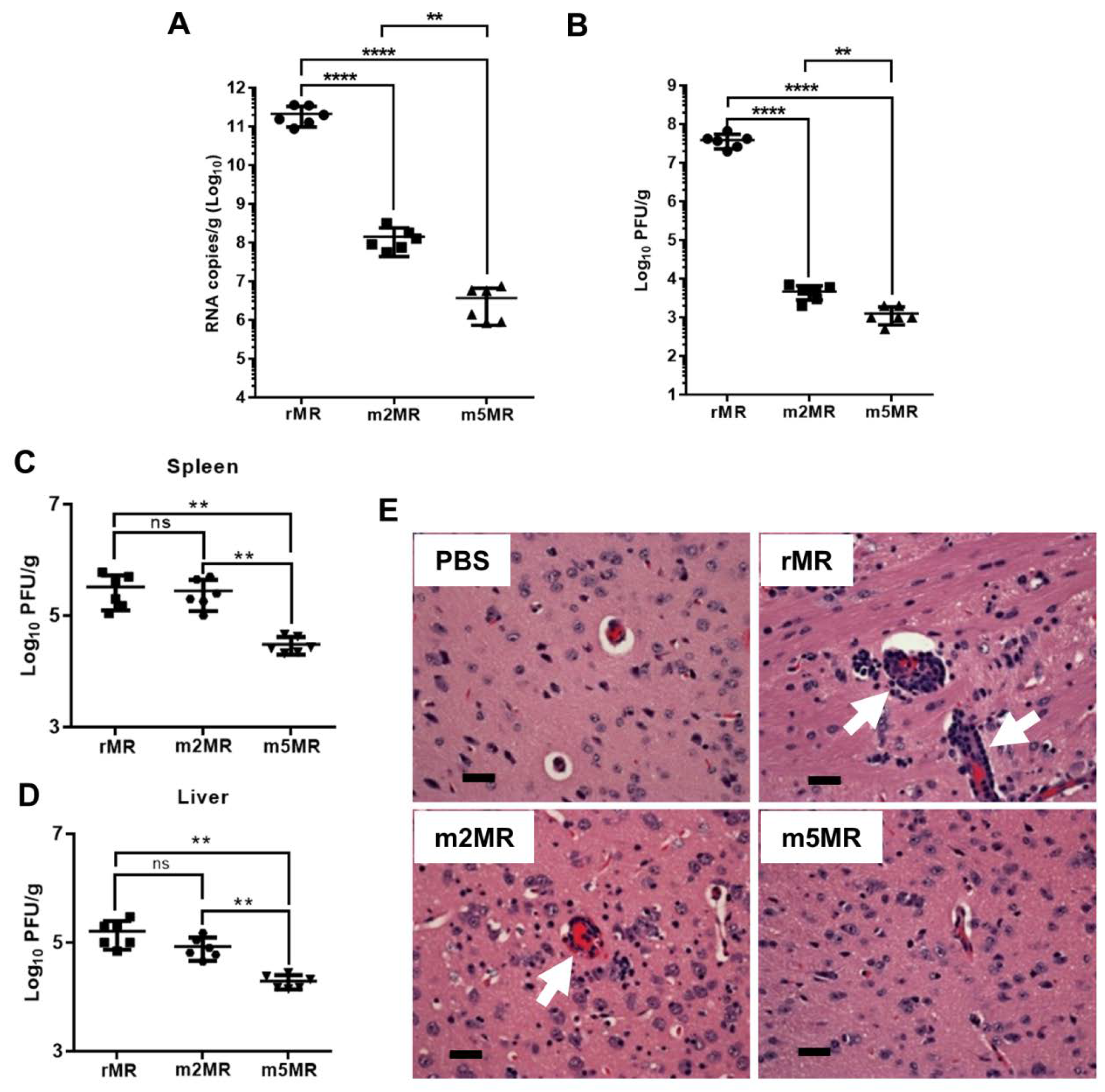
3.3. m5MR Virus Encoding both Non-Glycosylated E and NS1 Proteins is Highly Attenuated
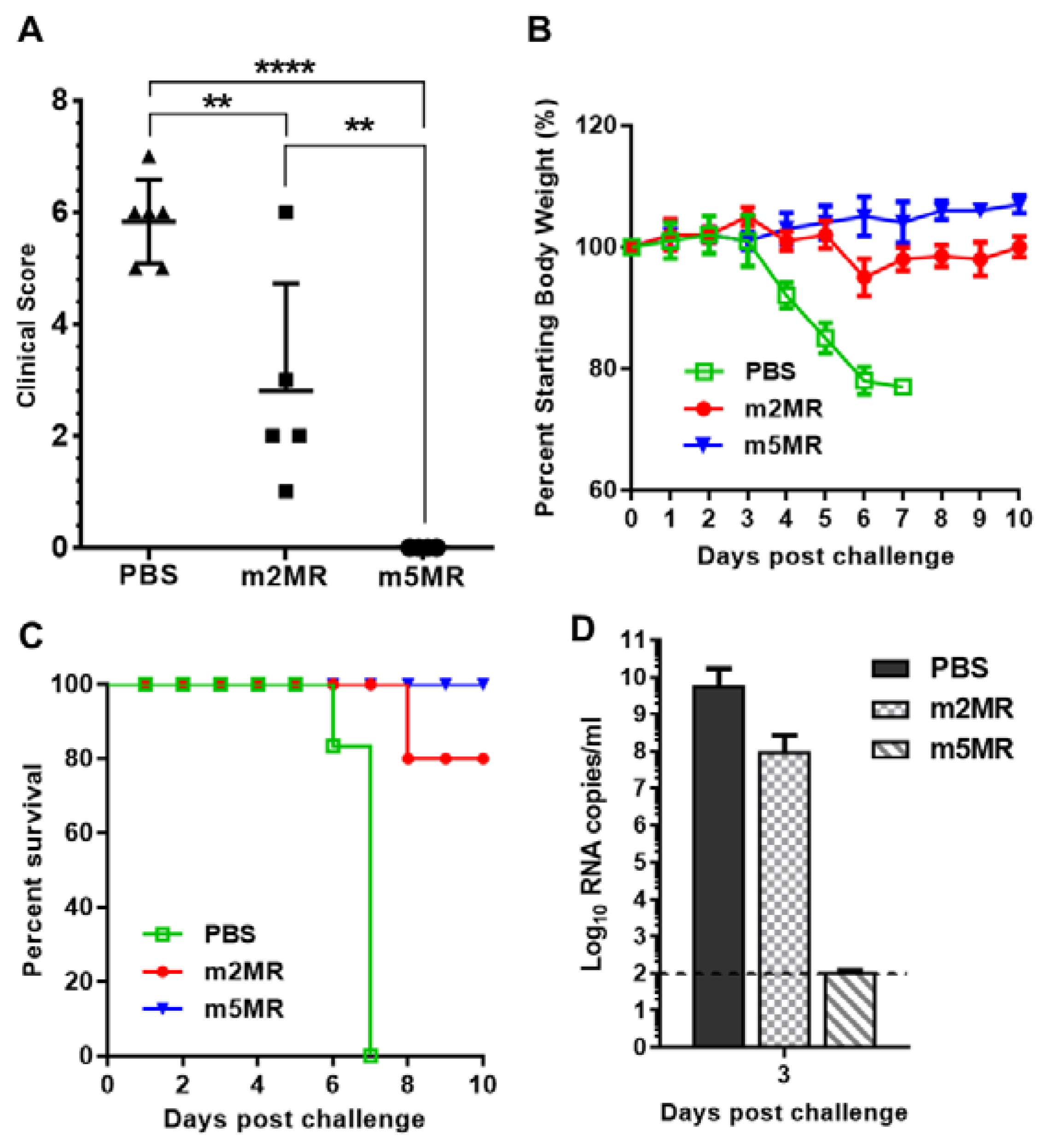
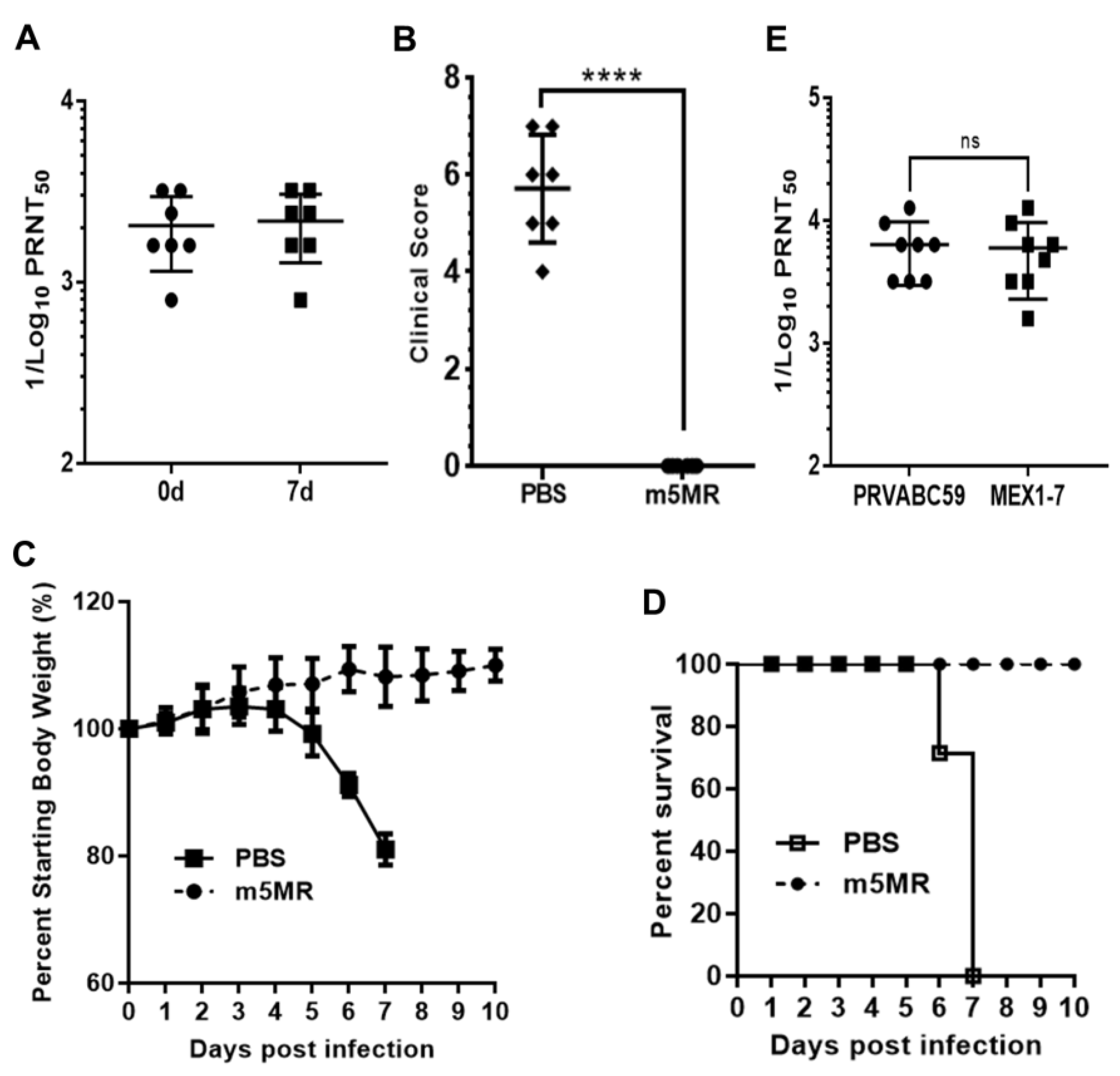
3.4. m5MR Virus Protects Immunocompromised Mice from Lethal Challenge
3.5. Protection from Challenge is Conferred by both Humoral and Cell-Mediated Immune Responses
3.6. Passive Transfer of Antibodies from Animals Infected with m5MR Virus Protects Mice from Lethal Challenge
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hills, S.L.; Russell, K.; Hennessey, M.; Williams, C.; Oster, A.M.; Fischer, M.; Mead, P. Transmission of Zika Virus Through Sexual Contact with Travelers to Areas of Ongoing Transmission—Continental United States, 2016. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 215–216. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Gubler, D.J. Zika Virus. Clin. Microbiol. Rev. 2016, 29, 487–524. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Lal, S.K. Zika Virus: Transmission, Detection, Control, and Prevention. Front. Microbiol. 2017, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.J.; Duehr, J.; Dulin, H.; Broecker, F.; Brown, J.A.; Arumemi, F.O.; Bermudez Gonzalez, M.C.; Leyva-Grado, V.H.; Evans, M.J.; Simon, V.; et al. Human antibodies targeting Zika virus NS1 provide protection against disease in a mouse model. Nat. Commun. 2018, 9, 4560. [Google Scholar] [CrossRef]
- Bogoch, I.I.; Brady, O.J.; Kraemer, M.U.G.; German, M.; Creatore, M.I.; Kulkarni, M.A.; Brownstein, J.S.; Mekaru, S.R.; Hay, S.I.; Groot, E.; et al. Anticipating the international spread of Zika virus from Brazil. Lancet 2016, 387, 335–336. [Google Scholar] [CrossRef]
- Ladhani, S.N.; O’Connor, C.; Kirkbride, H.; Brooks, T.; Morgan, D. Outbreak of Zika virus disease in the Americas and the association with microcephaly, congenital malformations and Guillain-Barre syndrome. Arch. Dis. Child. 2016, 101, 600–602. [Google Scholar] [CrossRef]
- Carod-Artal, F.J. Neurological complications of Zika virus infection. Expert Rev. Anti Infect. Ther. 2018, 16, 399–410. [Google Scholar] [CrossRef]
- Coyne, C.B.; Lazear, H.M. Zika virus—Reigniting the TORCH. Nat. Rev. Microbiol. 2016, 14, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Van Hemert, F.; Berkhout, B. Nucleotide composition of the Zika virus RNA genome and its codon usage. Virol. J. 2016, 13, 95. [Google Scholar] [CrossRef]
- Kuno, G.; Chang, G.J. Full-length sequencing and genomic characterization of Bagaza, Kedougou, and Zika viruses. Arch. Virol. 2007, 152, 687–696. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Murray, C.L.; Thiel, R.J.; Rice, C.M. Flaviviridae, 6th ed.; Knipe, D.M., Howley, P.M., Cohen, J.I., Grifin, D.E., Lamb, R.A., Martin, M.A., Recaniello, V.R., Roizman, B., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Diamond, M.S.; Pierson, T.C. Molecular Insight into Dengue Virus Pathogenesis and Its Implications for Disease Control. Cell 2015, 162, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sanchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016, 17, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Wang, Q.; Song, H.; Gao, G.F. Zika Virus Envelope Protein and Antibody Complexes. Subcell Biochem. 2018, 88, 147–168. [Google Scholar] [PubMed]
- Heinz, F.X.; Stiasny, K. The Antigenic Structure of Zika Virus and Its Relation to Other Flaviviruses: Implications for Infection and Immunoprophylaxis. Microbiol. Mol. Biol. Rev. MMBR 2017, 81, e00055-16. [Google Scholar] [CrossRef] [PubMed]
- Barba-Spaeth, G.; Dejnirattisai, W.; Rouvinski, A.; Vaney, M.C.; Medits, I.; Sharma, A.; Simon-Loriere, E.; Sakuntabhai, A.; Cao-Lormeau, V.M.; Haouz, A.; et al. Structural basis of potent Zika-dengue virus antibody cross-neutralization. Nature 2016, 536, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Stettler, K.; Beltramello, M.; Espinosa, D.A.; Graham, V.; Cassotta, A.; Bianchi, S.; Vanzetta, F.; Minola, A.; Jaconi, S.; Mele, F.; et al. Specificity, cross-reactivity, and function of antibodies elicited by Zika virus infection. Science 2016, 353, 823–826. [Google Scholar] [CrossRef]
- Schlesinger, J.J.; Foltzer, M.; Chapman, S. The Fc portion of antibody to yellow fever virus NS1 is a determinant of protection against YF encephalitis in mice. Virology 1993, 192, 132–141. [Google Scholar] [CrossRef]
- Chung, K.M.; Thompson, B.S.; Fremont, D.H.; Diamond, M.S. Antibody recognition of cell surface-associated NS1 triggers Fc-gamma receptor-mediated phagocytosis and clearance of West Nile Virus-infected cells. J. Virol. 2007, 81, 9551–9555. [Google Scholar] [CrossRef]
- Chung, K.M.; Liszewski, M.K.; Nybakken, G.; Davis, A.E.; Townsend, R.R.; Fremont, D.H.; Atkinson, J.P.; Diamond, M.S. West Nile virus nonstructural protein NS1 inhibits complement activation by binding the regulatory protein factor H. Proc. Natl. Acad. Sci. USA 2006, 103, 19111–19116. [Google Scholar] [CrossRef]
- Li, A.; Yu, J.; Lu, M.; Ma, Y.; Attia, Z.; Shan, C.; Xue, M.; Liang, X.; Craig, K.; Makadiya, N.; et al. A Zika virus vaccine expressing premembrane-envelope-NS1 polyprotein. Nat. Commun. 2018, 9, 3067. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.J.; Broecker, F.; Duehr, J.; Arumemi, F.; Krammer, F.; Palese, P.; Tan, G.S. Antibodies Elicited by an NS1-Based Vaccine Protect Mice against Zika Virus. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Tan, K.H.; Rathore, A.P.; Rozen-Gagnon, K.; Shuai, W.; Ruedl, C.; Vasudevan, S.G. The magnitude of dengue virus NS1 protein secretion is strain dependent and does not correlate with severe pathologies in the mouse infection model. J. Virol. 2012, 86, 5508–5514. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, M.; Sharma, N.; Singh, S.K. Flavivirus NS1: A multifaceted enigmatic viral protein. Virol. J. 2016, 13, 131. [Google Scholar] [CrossRef] [PubMed]
- Watterson, D.; Modhiran, N.; Young, P.R. The many faces of the flavivirus NS1 protein offer a multitude of options for inhibitor design. Antivir. Res. 2016, 130, 7–18. [Google Scholar] [CrossRef]
- Westaway, E.G.; Goodman, M.R. Variation in distribution of the three flavivirus-specified glycoproteins detected by immunofluorescence in infected Vero cells. Arch. Virol. 1987, 94, 215–228. [Google Scholar] [CrossRef]
- Flamand, M.; Megret, F.; Mathieu, M.; Lepault, J.; Rey, F.A.; Deubel, V. Dengue virus type 1 nonstructural glycoprotein NS1 is secreted from mammalian cells as a soluble hexamer in a glycosylation-dependent fashion. J. Virol. 1999, 73, 6104–6110. [Google Scholar] [PubMed]
- Muller, D.A.; Young, P.R. The flavivirus NS1 protein: Molecular and structural biology, immunology, role in pathogenesis and application as a diagnostic biomarker. Antivir. Res. 2013, 98, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.M.; Jones, M.K.; Young, P.R. Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology 1996, 220, 232–240. [Google Scholar] [CrossRef]
- Westaway, E.G.; Mackenzie, J.M.; Kenney, M.T.; Jones, M.K.; Khromykh, A.A. Ultrastructure of Kunjin virus-infected cells: Colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J. Virol. 1997, 71, 6650–6661. [Google Scholar] [PubMed]
- Yap, S.S.L.; Nguyen-Khuong, T.; Rudd, P.M.; Alonso, S. Dengue Virus Glycosylation: What Do We Know? Front. Microbiol. 2017, 8, 1415. [Google Scholar] [CrossRef] [PubMed]
- Crooks, A.J.; Lee, J.M.; Dowsett, A.B.; Stephenson, J.R. Purification and analysis of infectious virions and native non-structural antigens from cells infected with tick-borne encephalitis virus. J. Chromatogr. 1990, 502, 59–68. [Google Scholar] [CrossRef]
- Avirutnan, P.; Fuchs, A.; Hauhart, R.E.; Somnuke, P.; Youn, S.; Diamond, M.S.; Atkinson, J.P. Antagonism of the complement component C4 by flavivirus nonstructural protein NS1. J. Exp. Med. 2010, 207, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Avirutnan, P.; Punyadee, N.; Noisakran, S.; Komoltri, C.; Thiemmeca, S.; Auethavornanan, K.; Jairungsri, A.; Kanlaya, R.; Tangthawornchaikul, N.; Puttikhunt, C.; et al. Vascular leakage in severe dengue virus infections: A potential role for the nonstructural viral protein NS1 and complement. J. Infect. Dis. 2006, 193, 1078–1088. [Google Scholar] [CrossRef] [PubMed]
- Dalgarno, L.; Trent, D.W.; Strauss, J.H.; Rice, C.M. Partial nucleotide sequence of the Murray Valley encephalitis virus genome. Comparison of the encoded polypeptides with yellow fever virus structural and non-structural proteins. J. Mol. Biol. 1986, 187, 309–323. [Google Scholar] [CrossRef]
- Naik, N.G.; Wu, H.N. Mutation of Putative N-Glycosylation Sites on Dengue Virus NS4B Decreases RNA Replication. J. Virol. 2015, 89, 6746–6760. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Yun, S.I.; Song, B.H.; Hahn, Y.S.; Lee, C.H.; Oh, H.W.; Lee, Y.M. A single N-linked glycosylation site in the Japanese encephalitis virus prM protein is critical for cell type-specific prM protein biogenesis, virus particle release, and pathogenicity in mice. J. Virol. 2008, 82, 7846–7862. [Google Scholar] [CrossRef]
- Somnuke, P.; Hauhart, R.E.; Atkinson, J.P.; Diamond, M.S.; Avirutnan, P. N-linked glycosylation of dengue virus NS1 protein modulates secretion, cell-surface expression, hexamer stability, and interactions with human complement. Virology 2011, 413, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Pryor, M.J.; Wright, P.J. Glycosylation mutants of dengue virus NS1 protein. J. Gen. Virol. 1994, 75 Pt 5, 1183–1187. [Google Scholar] [CrossRef]
- Flamand, M.; Deubel, V.; Girard, M. Expression and secretion of Japanese encephalitis virus nonstructural protein NS1 by insect cells using a recombinant baculovirus. Virology 1992, 191, 826–836. [Google Scholar] [CrossRef]
- Whiteman, M.C.; Li, L.; Wicker, J.A.; Kinney, R.M.; Huang, C.; Beasley, D.W.; Chung, K.M.; Diamond, M.S.; Solomon, T.; Barrett, A.D. Development and characterization of non-glycosylated E and NS1 mutant viruses as a potential candidate vaccine for West Nile virus. Vaccine 2010, 28, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Sirohi, D.; Chen, Z.; Sun, L.; Klose, T.; Pierson, T.C.; Rossmann, M.G.; Kuhn, R.J. The 3.8 A resolution cryo-EM structure of Zika virus. Science 2016, 352, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Song, H.; Qi, J.; Liu, Y.; Wang, H.; Su, C.; Shi, Y.; Gao, G.F. Contribution of intertwined loop to membrane association revealed by Zika virus full-length NS1 structure. EMBO J. 2016, 35, 2170–2178. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, M.B.; Kinney, R.M.; Miller, B.R. Deglycosylation of the NS1 protein of dengue 2 virus, strain 16681: Construction and characterization of mutant viruses. Arch. Virol. 2005, 150, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Pletnev, A.G.; Bray, M.; Lai, C.J. Chimeric tick-borne encephalitis and dengue type 4 viruses: Effects of mutations on neurovirulence in mice. J. Virol. 1993, 67, 4956–4963. [Google Scholar] [PubMed]
- Pryor, M.J.; Gualano, R.C.; Lin, B.; Davidson, A.D.; Wright, P.J. Growth restriction of dengue virus type 2 by site-specific mutagenesis of virus-encoded glycoproteins. J. Gen. Virol. 1998, 79, 2631–2639. [Google Scholar] [CrossRef] [PubMed]
- Muylaert, I.R.; Chambers, T.J.; Galler, R.; Rice, C.M. Mutagenesis of the N-linked glycosylation sites of the yellow fever virus NS1 protein: Effects on virus replication and mouse neurovirulence. Virology 1996, 222, 159–168. [Google Scholar] [CrossRef]
- Annamalai, A.S.; Pattnaik, A.; Sahoo, B.R.; Muthukrishnan, E.; Natarajan, S.K.; Steffen, D.; Vu, H.L.X.; Delhon, G.; Osorio, F.A.; Petro, T.M.; et al. Zika Virus Encoding Non-Glycosylated Envelope Protein is Attenuated and Defective in Neuroinvasion. J. Virol. 2017, 91, e01348-17. [Google Scholar] [CrossRef]
- Pattnaik, A.; Palermo, N.; Sahoo, B.R.; Yuan, Z.; Hu, D.; Annamalai, A.S.; Vu, H.L.X.; Correas, I.; Prathipati, P.K.; Destache, C.J.; et al. Discovery of a non-nucleoside RNA polymerase inhibitor for blocking Zika virus replication through in silico screening. Antivir. Res. 2018, 151, 78–86. [Google Scholar] [CrossRef]
- Bullard, B.L.; Corder, B.N.; Gorman, M.J.; Diamond, M.S.; Weaver, E.A. Efficacy of a T Cell-Biased Adenovirus Vector as a Zika Virus Vaccine. Sci. Rep. 2018, 8, 18017. [Google Scholar] [CrossRef]
- Rossi, S.L.; Tesh, R.B.; Azar, S.R.; Muruato, A.E.; Hanley, K.A.; Auguste, A.J.; Langsjoen, R.M.; Paessler, S.; Vasilakis, N.; Weaver, S.C. Characterization of a Novel Murine Model to Study Zika Virus. Am. J. Trop. Med. Hyg. 2016, 94, 1362–1369. [Google Scholar] [CrossRef]
- Morrison, T.E.; Diamond, M.S. Animal Models of Zika Virus Infection, Pathogenesis, and Immunity. J. Virol. 2017, 91, e00009-17. [Google Scholar] [CrossRef]
- Shan, C.; Xie, X.; Barrett, A.D.; Garcia-Blanco, M.A.; Tesh, R.B.; Vasconcelos, P.F.; Vasilakis, N.; Weaver, S.C.; Shi, P.Y. Zika Virus: Diagnosis, Therapeutics, and Vaccine. ACS Infect. Dis. 2016, 2, 170–172. [Google Scholar] [CrossRef]
- Khou, C.; Pardigon, N. Identifying Attenuating Mutations: Tools for a New Vaccine Design against Flaviviruses. Intervirology 2017, 60, 8–18. [Google Scholar] [CrossRef]
- Heinz, F.X.; Stiasny, K. Flaviviruses and flavivirus vaccines. Vaccine 2012, 30, 4301–4306. [Google Scholar] [CrossRef]
- Barzon, L.; Palu, G. Current views on Zika virus vaccine development. Expert Opin. Biol. Ther. 2017, 17, 1185–1192. [Google Scholar] [CrossRef][Green Version]
- Larocca, R.A.; Abbink, P.; Peron, J.P.; Zanotto, P.M.; Iampietro, M.J.; Badamchi-Zadeh, A.; Boyd, M.; Ng’ang’a, D.; Kirilova, M.; Nityanandam, R.; et al. Vaccine protection against Zika virus from Brazil. Nature 2016, 536, 474–478. [Google Scholar] [CrossRef]
- Abbink, P.; Larocca, R.A.; De La Barrera, R.A.; Bricault, C.A.; Moseley, E.T.; Boyd, M.; Kirilova, M.; Li, Z.; Ng’ang’a, D.; Nanayakkara, O.; et al. Protective efficacy of multiple vaccine platforms against Zika virus challenge in rhesus monkeys. Science 2016, 353, 1129–1132. [Google Scholar] [CrossRef]
- Dowd, K.A.; Ko, S.Y.; Morabito, K.M.; Yang, E.S.; Pelc, R.S.; DeMaso, C.R.; Castilho, L.R.; Abbink, P.; Boyd, M.; Nityanandam, R.; et al. Rapid development of a DNA vaccine for Zika virus. Science 2016, 354, 237–240. [Google Scholar] [CrossRef]
- Chambers, T.J.; Nestorowicz, A.; Mason, P.W.; Rice, C.M. Yellow fever/Japanese encephalitis chimeric viruses: Construction and biological properties. J. Virol. 1999, 73, 3095–3101. [Google Scholar]
- Arroyo, J.; Miller, C.; Catalan, J.; Myers, G.A.; Ratterree, M.S.; Trent, D.W.; Monath, T.P. ChimeriVax-West Nile virus live-attenuated vaccine: Preclinical evaluation of safety, immunogenicity, and efficacy. J. Virol. 2004, 78, 12497–12507. [Google Scholar] [CrossRef]
- Richner, J.M.; Jagger, B.W.; Shan, C.; Fontes, C.R.; Dowd, K.A.; Cao, B.; Himansu, S.; Caine, E.A.; Nunes, B.T.D.; Medeiros, D.B.A.; et al. Vaccine Mediated Protection Against Zika Virus-Induced Congenital Disease. Cell 2017, 170, 273–283. [Google Scholar] [CrossRef]
- Fontes-Garfias, C.R.; Shan, C.; Luo, H.; Muruato, A.E.; Medeiros, D.B.A.; Mays, E.; Xie, X.; Zou, J.; Roundy, C.M.; Wakamiya, M.; et al. Functional Analysis of Glycosylation of Zika Virus Envelope Protein. Cell Rep. 2017, 21, 1180–1190. [Google Scholar] [CrossRef]
- Carbaugh, D.L.; Baric, R.S.; Lazear, H.M. Envelope Protein Glycosylation Mediates Zika Virus Pathogenesis. J. Virol. 2019, 93, e00113-19. [Google Scholar] [CrossRef]
- Duggal, N.K.; McDonald, E.M.; Weger-Lucarelli, J.; Hawks, S.A.; Ritter, J.M.; Romo, H.; Ebel, G.D.; Brault, A.C. Mutations present in a low-passage Zika virus isolate result in attenuated pathogenesis in mice. Virology 2019, 530, 19–26. [Google Scholar] [CrossRef]
- Hilgenfeld, R. Zika virus NS1, a pathogenicity factor with many faces. EMBO J. 2016, 35, 2631–2633. [Google Scholar] [CrossRef]
- Chen, H.W.; Huang, H.W.; Hu, H.M.; Chung, H.H.; Wu, S.H.; Chong, P.; Tao, M.H.; Pan, C.H. A poorly neutralizing IgG2a/c response elicited by a DNA vaccine protects mice against Japanese encephalitis virus. J. Gen. Virol. 2014, 95, 1983–1990. [Google Scholar] [CrossRef]
- Brault, A.C.; Domi, A.; McDonald, E.M.; Talmi-Frank, D.; McCurley, N.; Basu, R.; Robinson, H.L.; Hellerstein, M.; Duggal, N.K.; Bowen, R.A.; et al. A Zika Vaccine Targeting NS1 Protein Protects Immunocompetent Adult Mice in a Lethal Challenge Model. Sci. Rep. 2017, 7, 14769. [Google Scholar] [CrossRef]
- Diamond, M.S.; Shrestha, B.; Marri, A.; Mahan, D.; Engle, M. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J. Virol. 2003, 77, 2578–2586. [Google Scholar] [CrossRef]
- Shrestha, B.; Diamond, M.S. Role of CD8+ T cells in control of West Nile virus infection. J. Virol. 2004, 78, 8312–8321. [Google Scholar] [CrossRef]
- Scott, J.M.; Lebratti, T.J.; Richner, J.M.; Jiang, X.; Fernandez, E.; Zhao, H.; Fremont, D.H.; Diamond, M.S.; Shin, H. Cellular and Humoral Immunity Protect against Vaginal Zika Virus Infection in Mice. J. Virol. 2018, 92, e00038-18. [Google Scholar] [CrossRef]
- Barrett, P.N.; Mundt, W.; Kistner, O.; Howard, M.K. Vero cell platform in vaccine production: Moving towards cell culture-based viral vaccines. Expert Rev. Vaccines 2009, 8, 607–618. [Google Scholar] [CrossRef]
- Genzel, Y. Designing cell lines for viral vaccine production: Where do we stand? Biotechnol. J. 2015, 10, 728–740. [Google Scholar] [CrossRef]
- Shan, C.; Muruato, A.E.; Nunes, B.T.D.; Luo, H.; Xie, X.; Medeiros, D.B.A.; Wakamiya, M.; Tesh, R.B.; Barrett, A.D.; Wang, T.; et al. A live-attenuated Zika virus vaccine candidate induces sterilizing immunity in mouse models. Nat. Med. 2017, 23, 763–767. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer and Name | Sequence (5’ to 3’) * | Application | Nucleotide Change |
|---|---|---|---|
| 1. MR-701F | GTGTACGGAACCTGTCATC | Upstream primer to generate PCR product for cloning | |
| 2. NS1-130-N to A-2864F | GCGGCAAAGACCGCCAACAGTTTTGTTGTC | N130A glycosylation mutation | AAC to GCC |
| 3. NS1-207-N to A-3090F | GGATTGAAAGTGAAAAGGCTGACACATGGAG | Single (N207A) glycosylation mutation | AAT to GCT |
| 4. NS1-207-N to A-3090R | CTCCATGTGTCAGCCTTTTCACTTTCAATCC | Double (N130A/N207A) glycosylation mutation | AAC to GCC and AAT to GCT in reverse |
| 5. MR-3873R | GCCAGGGCTAGCAGCATGCTCTCCCGCGGTGTCCAATTGGC | Downstream primer to generate PCR product for cloning |
| Peptide | Sequence * | Amino Acid Position in E (1–504) |
|---|---|---|
| 1 | IRCIGVSNRDFVEGM | 1–15 |
| 2 | IGVSNRDFVEGMSGG | 4–18 |
| 3 | SNRDFVEGMSGGTWV | 7–21 |
| 6 | SGGTWVDVVLEHGGC | 16–30 |
| 8 | DVVLEHGGCVTVMAQ | 22–36 |
| 19 | EVRSYCYEASISDMA | 55–69 |
| 21 | YEASISDMASDSRCP | 61–75 |
| 24 | SDSRCPTQGEAYLDK | 70–84 |
| 25 | RCPTQGEAYLDKQSD | 73–87 |
| 102 | SYSLCTAAFTFTKIP | 304–318 |
| 103 | LCTAAFTFTKIPAET | 307–321 |
| 104 | AAFTFTKIPAETLHG | 310–324 |
| 119 | VGRLITANPVITEST | 355–369 |
| 120 | LITANPVITESTENS | 358–372 |
| 123 | ESTENSKMMLELDPP | 367–381 |
| 124 | ENSKMMLELDPPFGD | 370–384 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Annamalai, A.S.; Pattnaik, A.; Sahoo, B.R.; Guinn, Z.P.; Bullard, B.L.; Weaver, E.A.; Steffen, D.; Natarajan, S.K.; Petro, T.M.; Pattnaik, A.K. An Attenuated Zika Virus Encoding Non-Glycosylated Envelope (E) and Non-Structural Protein 1 (NS1) Confers Complete Protection against Lethal Challenge in a Mouse Model. Vaccines 2019, 7, 112. https://doi.org/10.3390/vaccines7030112
Annamalai AS, Pattnaik A, Sahoo BR, Guinn ZP, Bullard BL, Weaver EA, Steffen D, Natarajan SK, Petro TM, Pattnaik AK. An Attenuated Zika Virus Encoding Non-Glycosylated Envelope (E) and Non-Structural Protein 1 (NS1) Confers Complete Protection against Lethal Challenge in a Mouse Model. Vaccines. 2019; 7(3):112. https://doi.org/10.3390/vaccines7030112
Chicago/Turabian StyleAnnamalai, Arun S., Aryamav Pattnaik, Bikash R. Sahoo, Zack P. Guinn, Brianna L. Bullard, Eric A. Weaver, David Steffen, Sathish Kumar Natarajan, Thomas M. Petro, and Asit K. Pattnaik. 2019. "An Attenuated Zika Virus Encoding Non-Glycosylated Envelope (E) and Non-Structural Protein 1 (NS1) Confers Complete Protection against Lethal Challenge in a Mouse Model" Vaccines 7, no. 3: 112. https://doi.org/10.3390/vaccines7030112
APA StyleAnnamalai, A. S., Pattnaik, A., Sahoo, B. R., Guinn, Z. P., Bullard, B. L., Weaver, E. A., Steffen, D., Natarajan, S. K., Petro, T. M., & Pattnaik, A. K. (2019). An Attenuated Zika Virus Encoding Non-Glycosylated Envelope (E) and Non-Structural Protein 1 (NS1) Confers Complete Protection against Lethal Challenge in a Mouse Model. Vaccines, 7(3), 112. https://doi.org/10.3390/vaccines7030112