
Optimization and Validation of a Harmonized Protocol for Generating Therapeutic-Grade Dendritic Cells in a Randomized Phase II Clinical Trial, Using Two Varied Antigenic Sources
 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Plan and Patient Recruitment
2.2. Maintenance of Clean Room for GMP-Grade Production of DCs
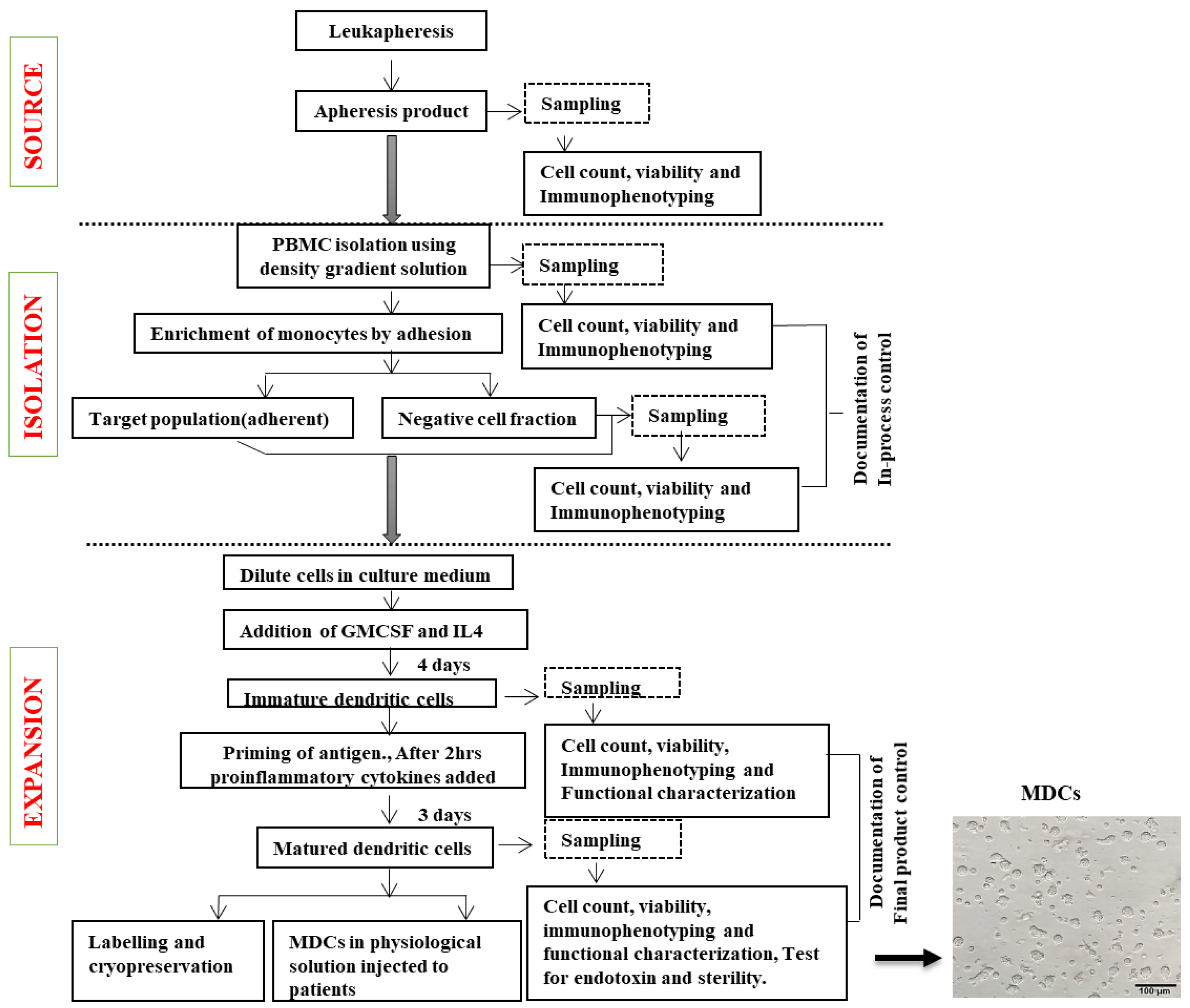
2.3. Generation of Immature Dendritic Cells (IDCs) from Monocytes
2.4. Validating Bag-Cultured Immature DCs for Antigen Uptake
2.5. Preparation of Antigenic Sources for DC Maturation
2.6. Dendritic Cell Maturation Using Two Different Antigenic Sources
2.7. Cryopreservation and Storage of MDCs
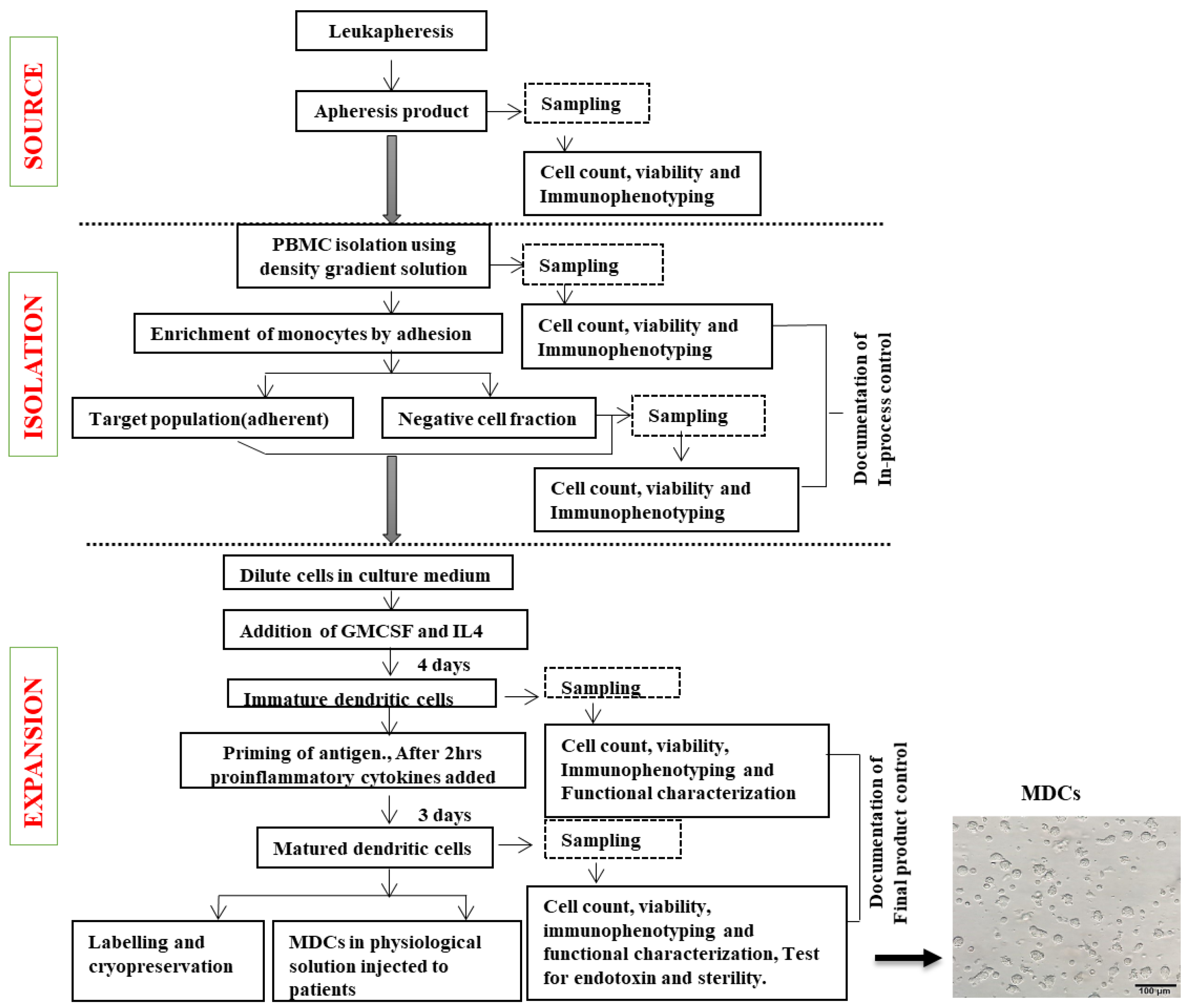
2.8. Quality Control (QC) Measures for Large-Scale Dendritic Cell Production (Table 1)
2.9. Phenotypic Expression
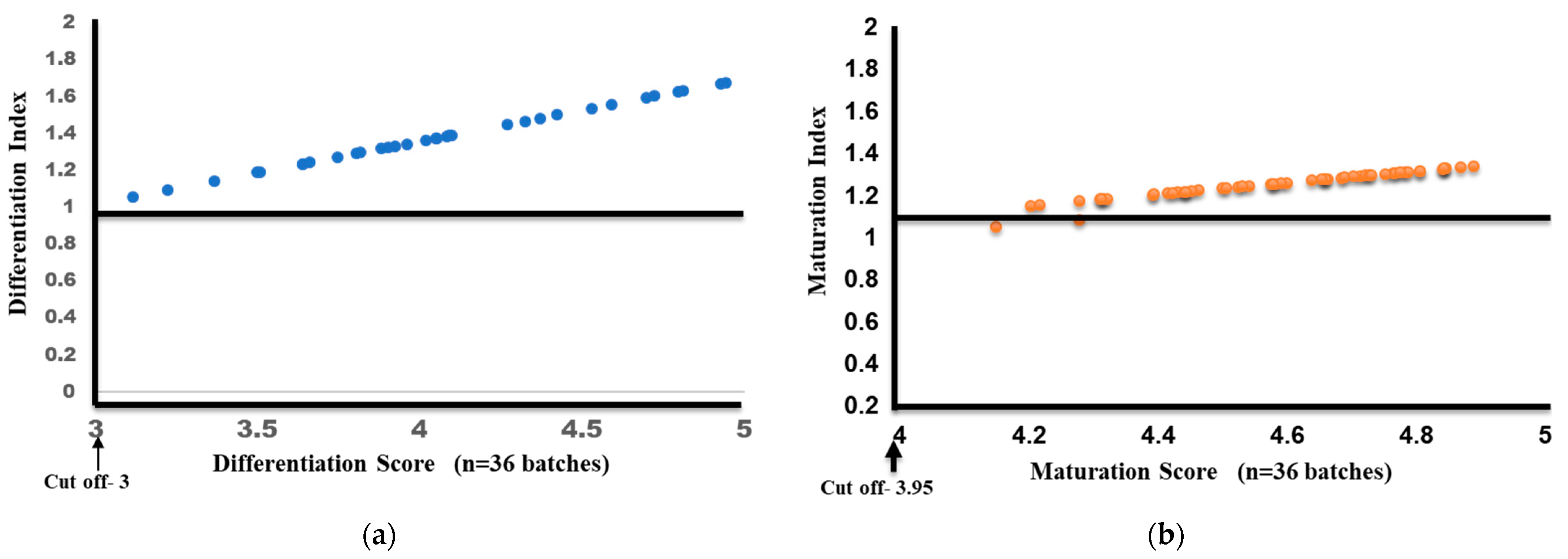
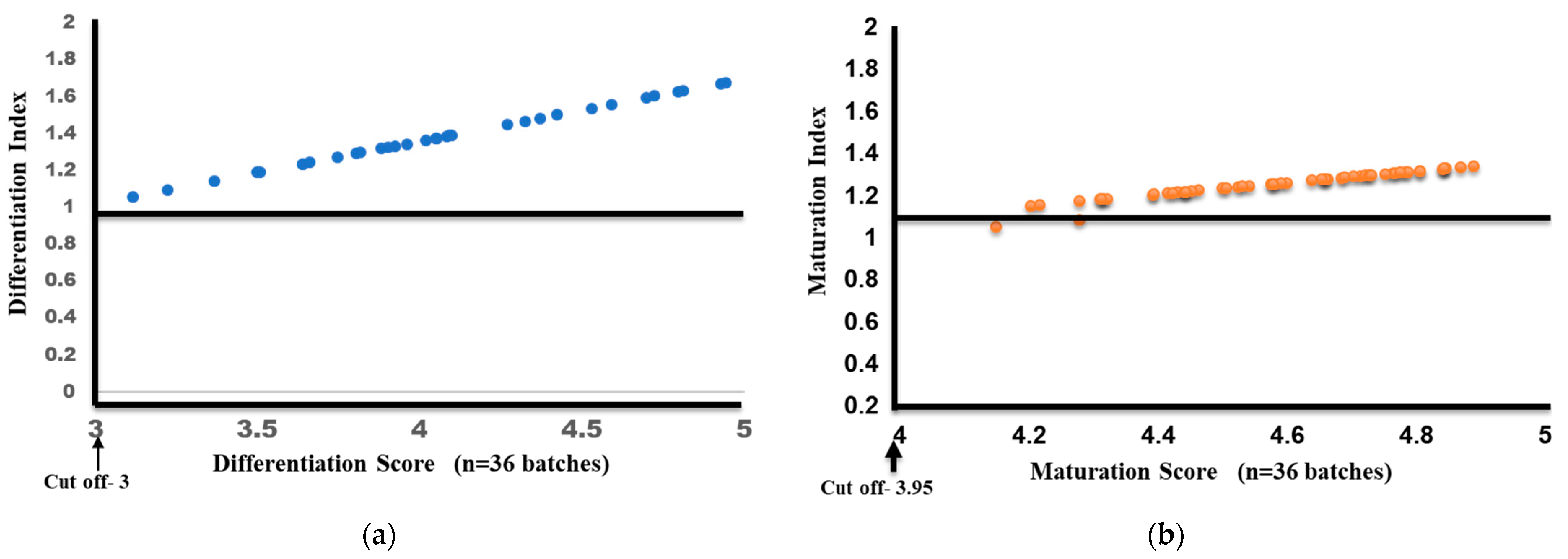
Differentiation and Maturation Index Calculation
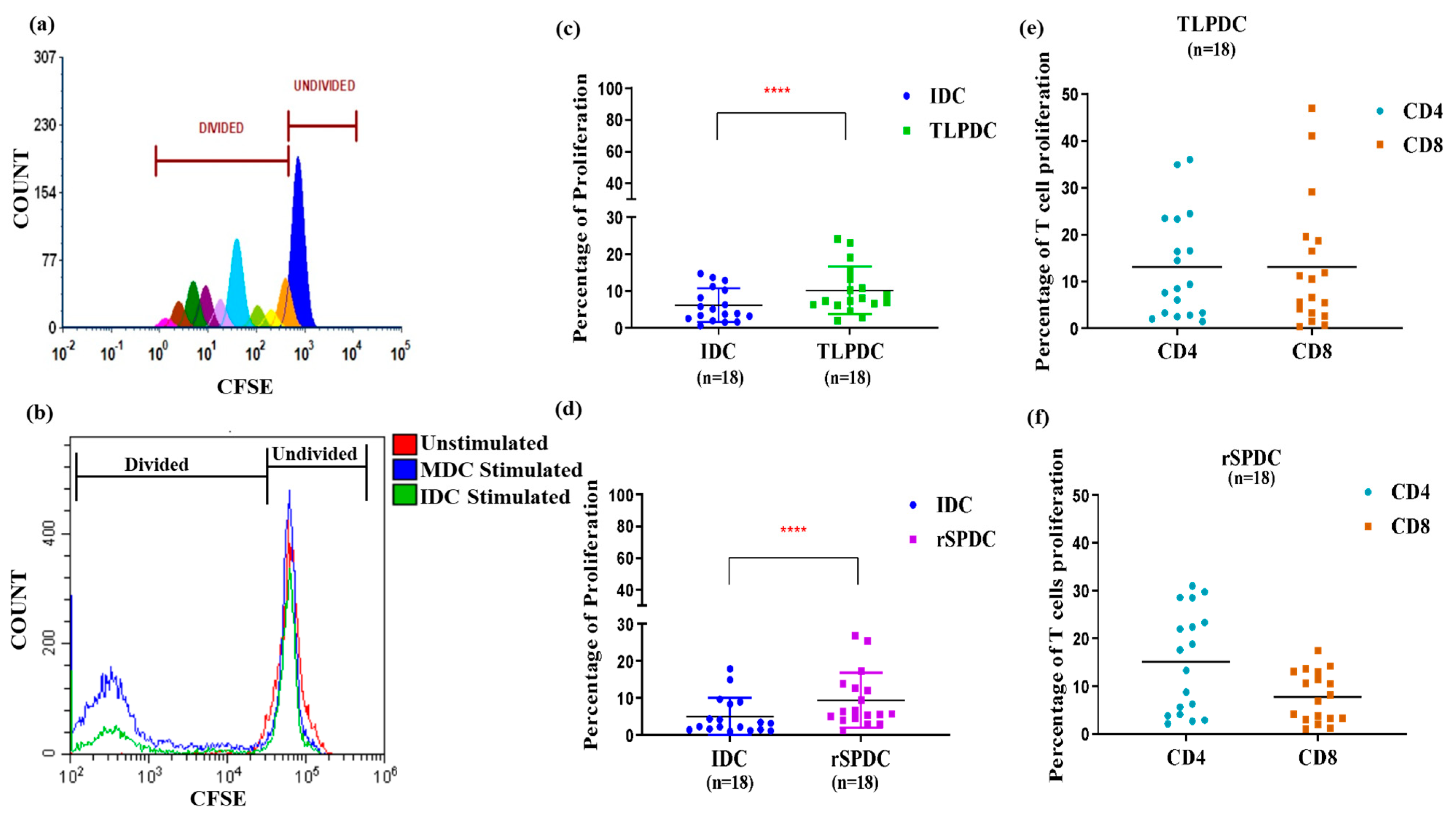
2.10. Proliferation Assay
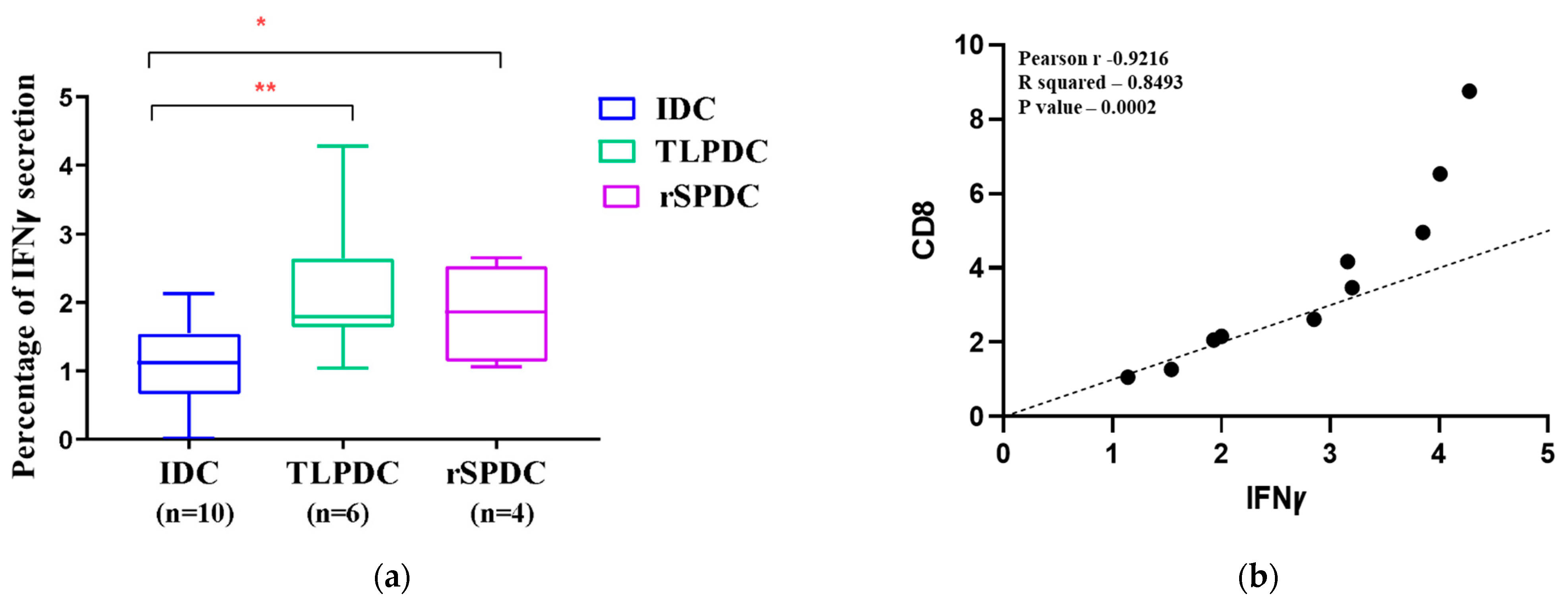
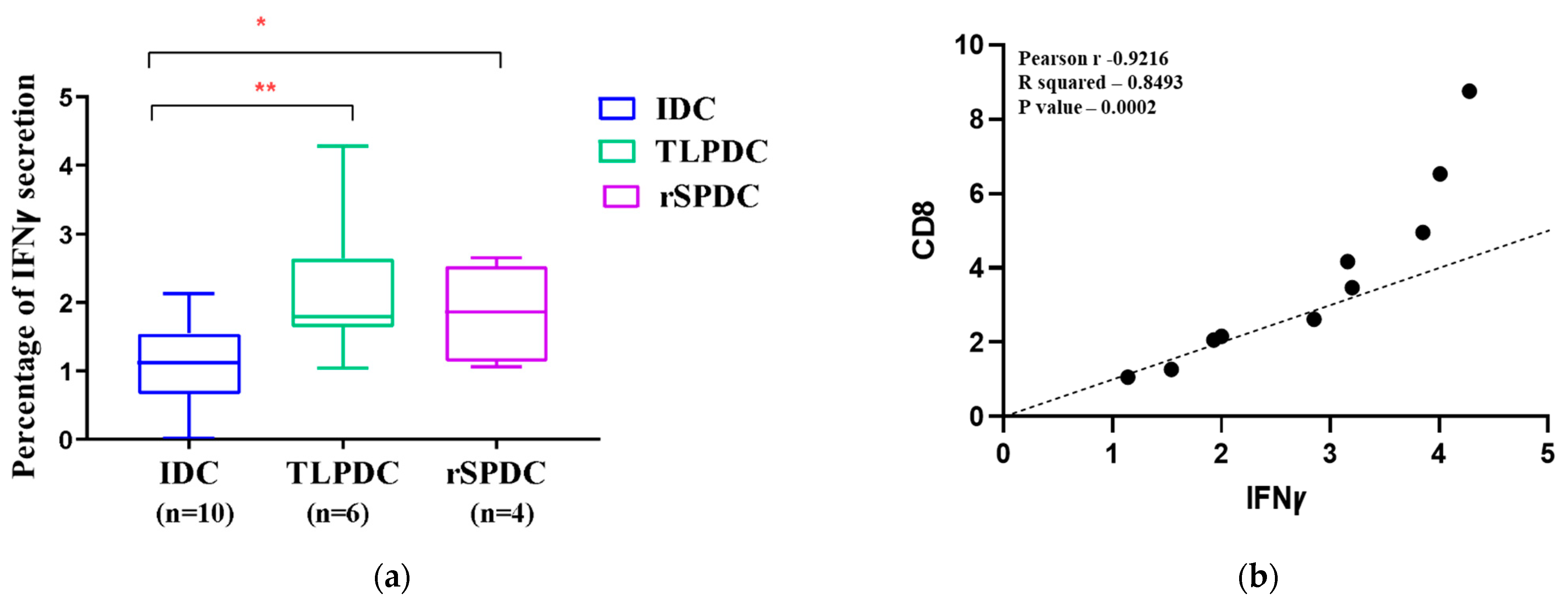
2.11. Flow Cytometric Detection of IFNγ Secretion in Allogenic Responders
2.12. Microbiological Sterility and Assessment of Bacterial Endotoxin Level
2.13. Statistical Analysis
3. Results
3.1. Screening of Patients for HPV and SPAG9 Positivity
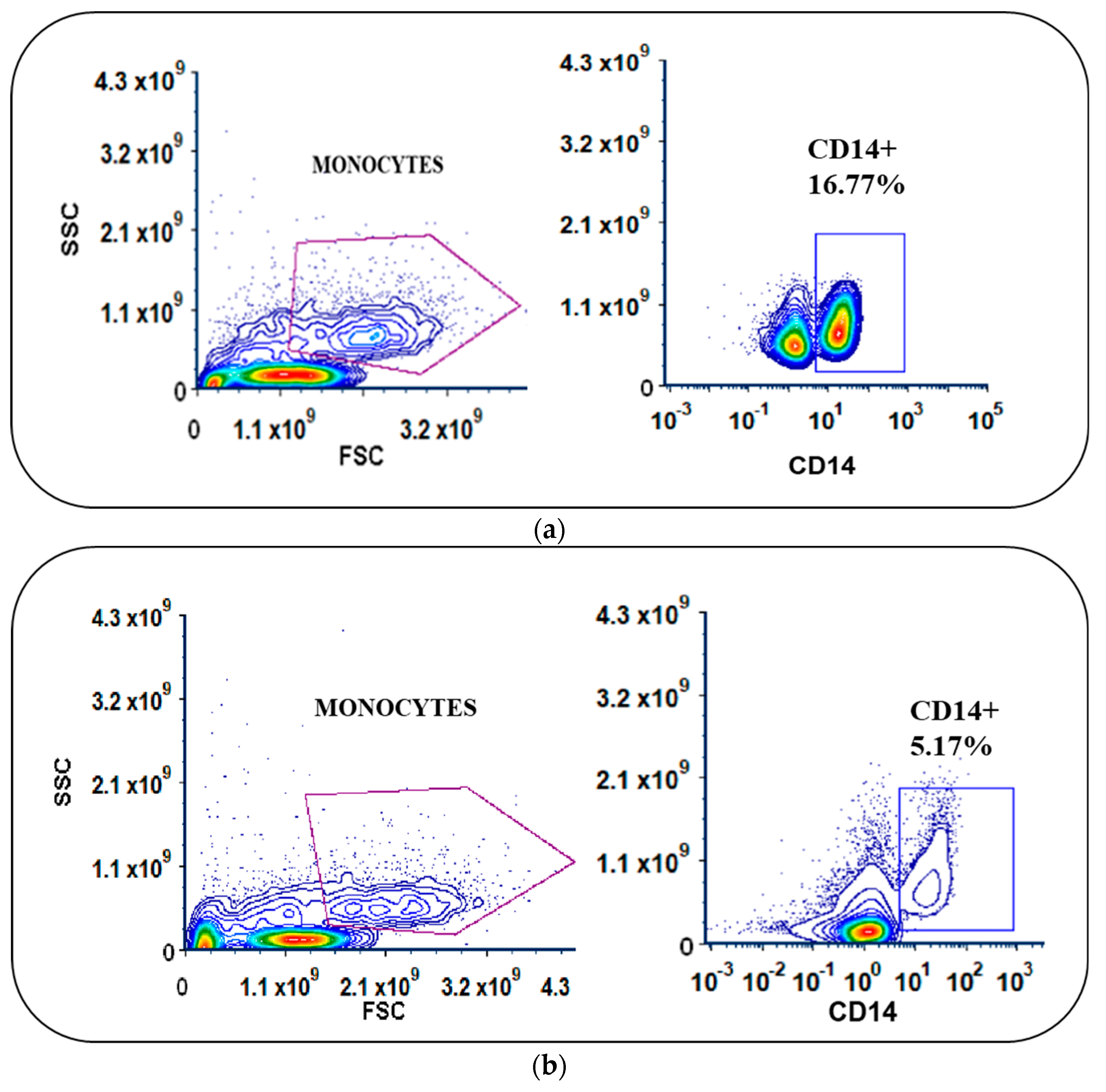
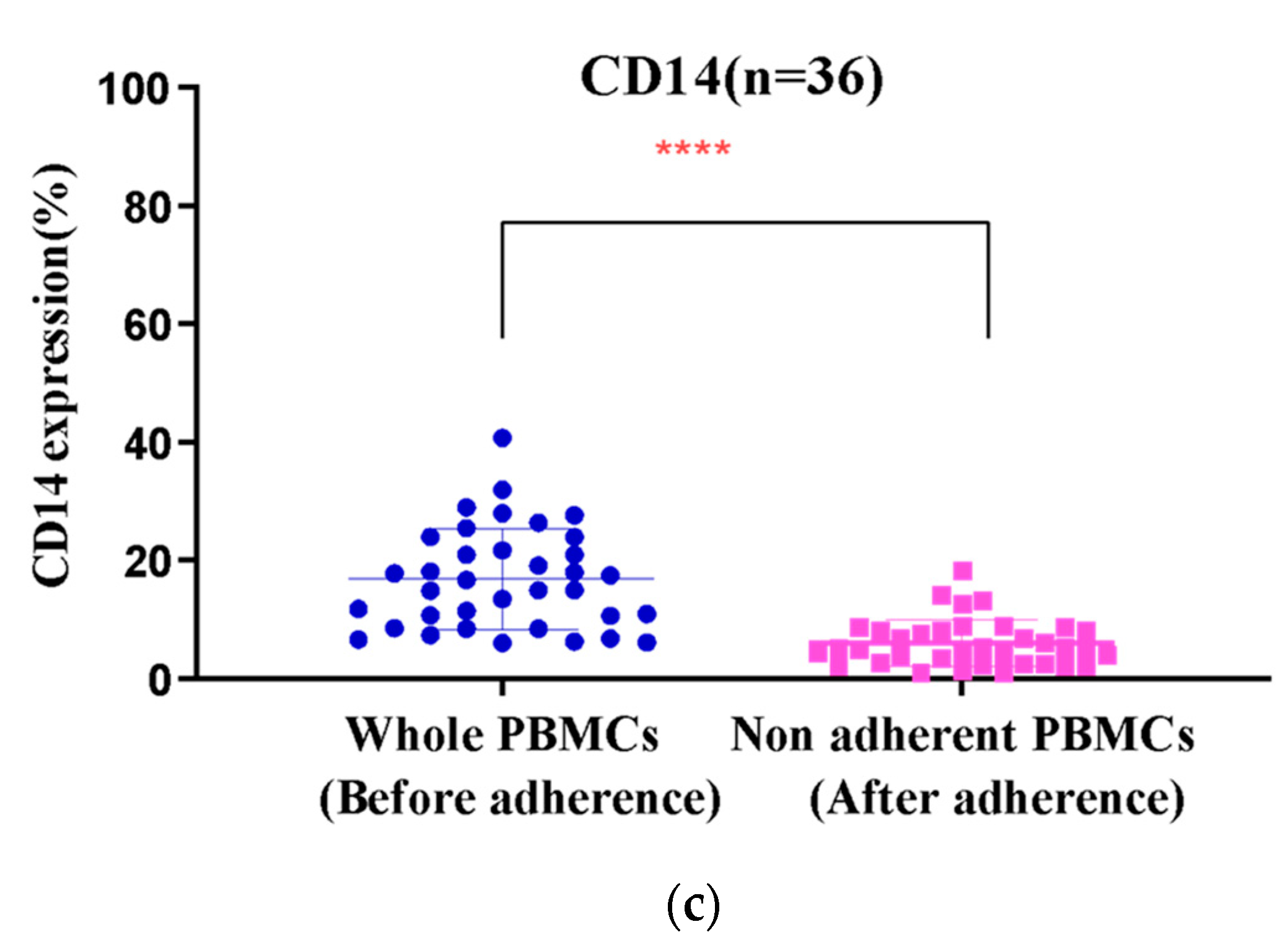
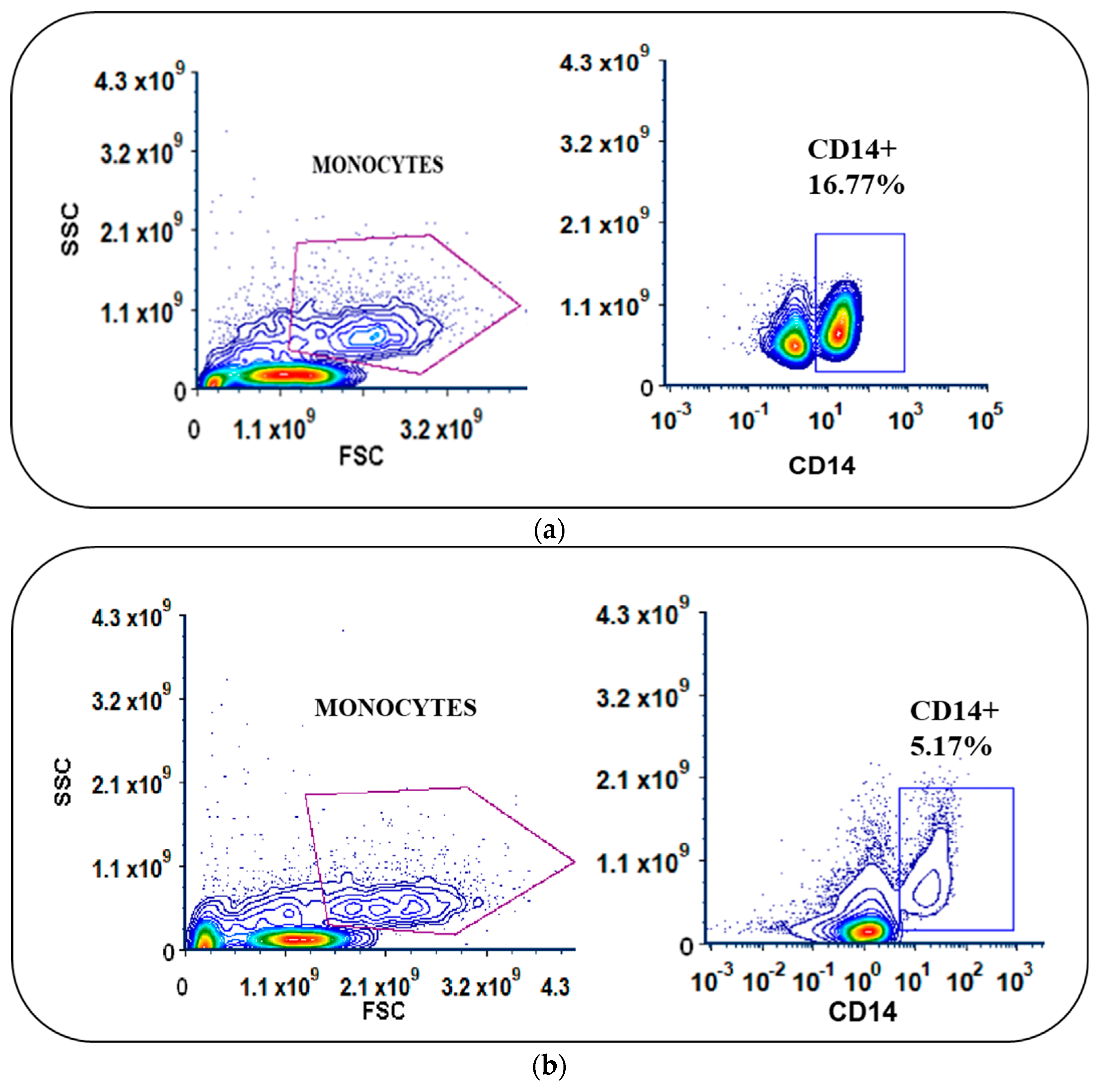
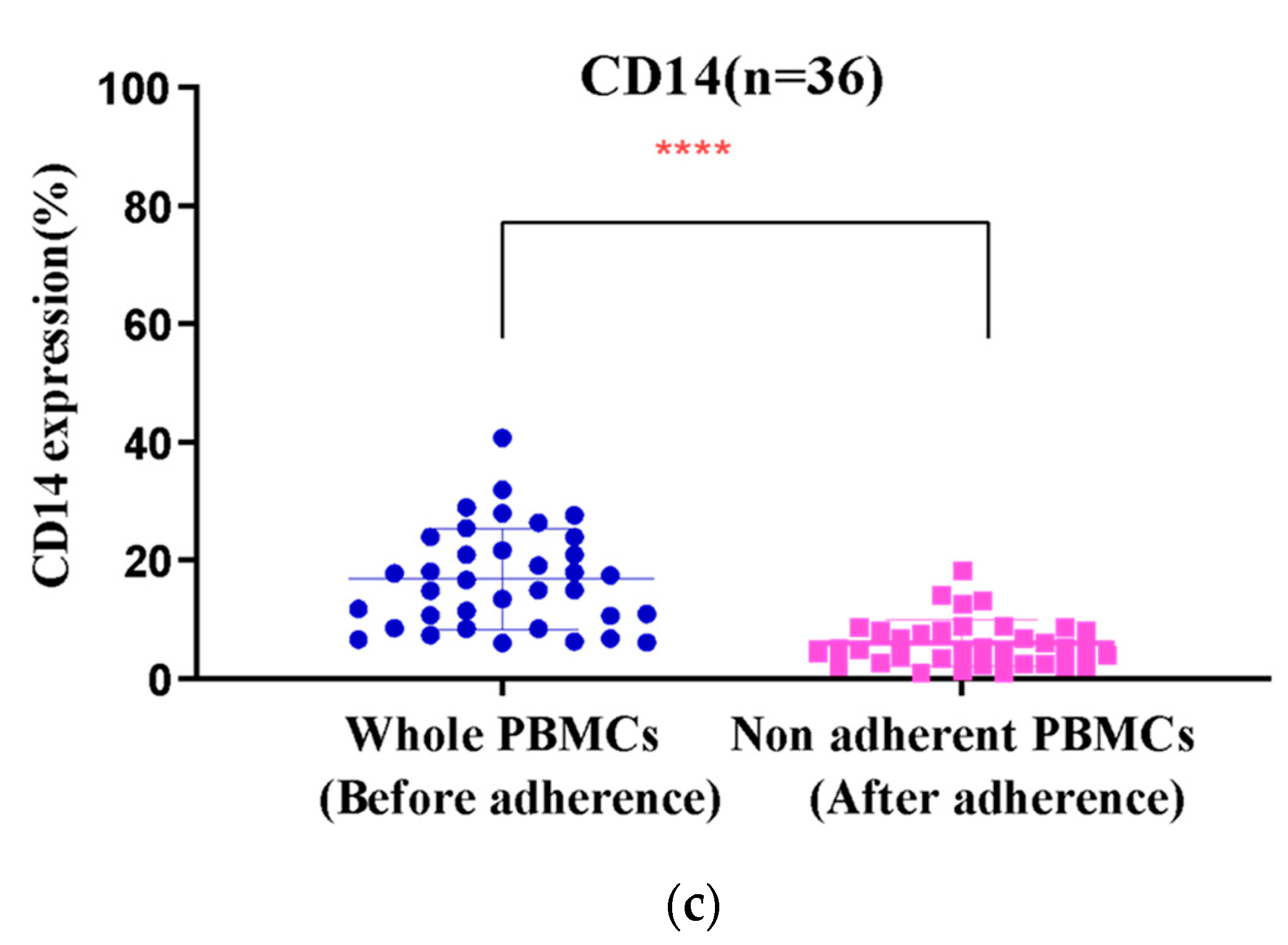
3.2. Monocyte Enrichment and Adherence Was Improved by Using Culture Bags with Coated Surfaces
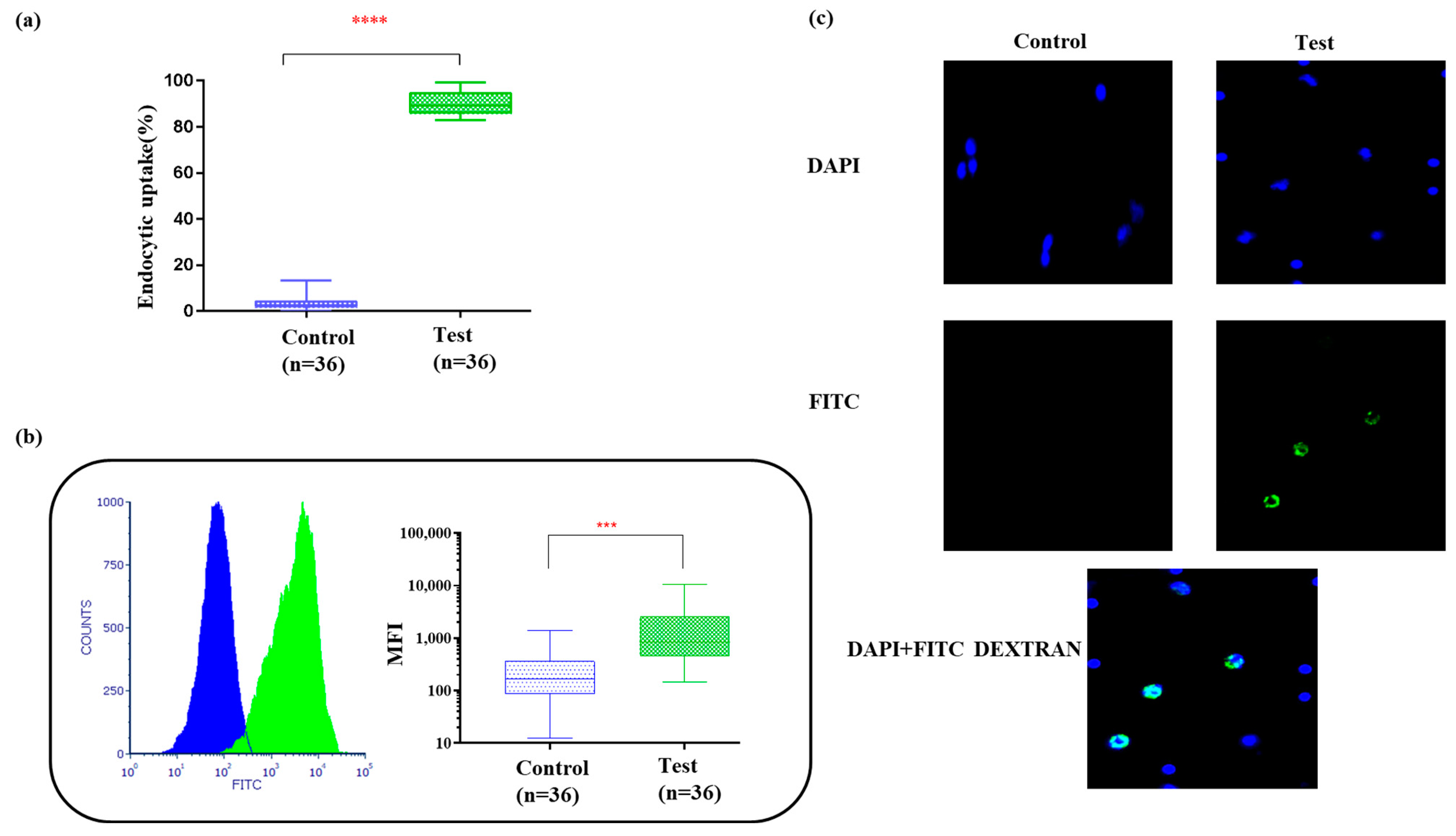
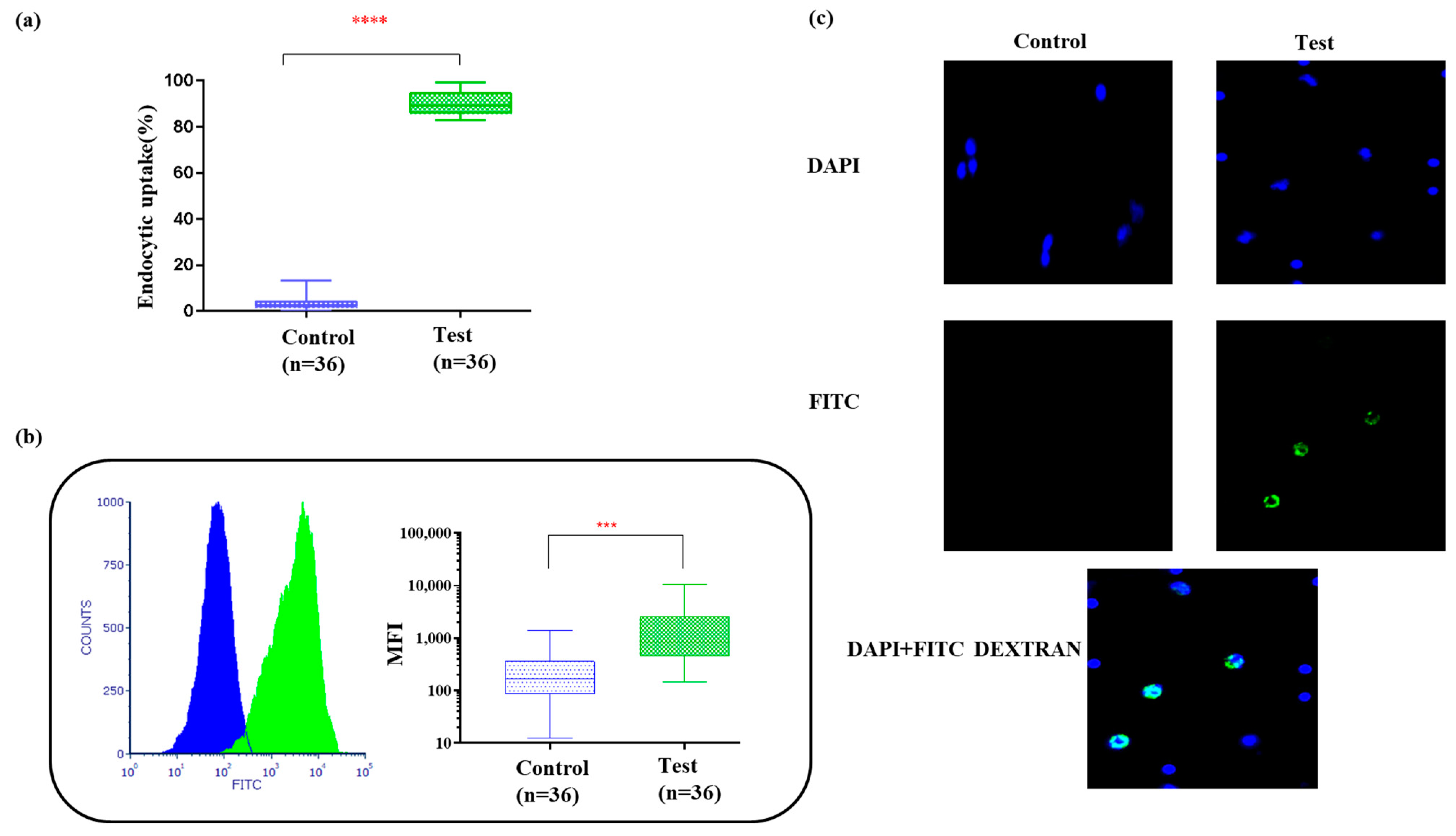
3.3. Bag-Cultured Immature DCs Showed Superior Antigen Engulfment Potential
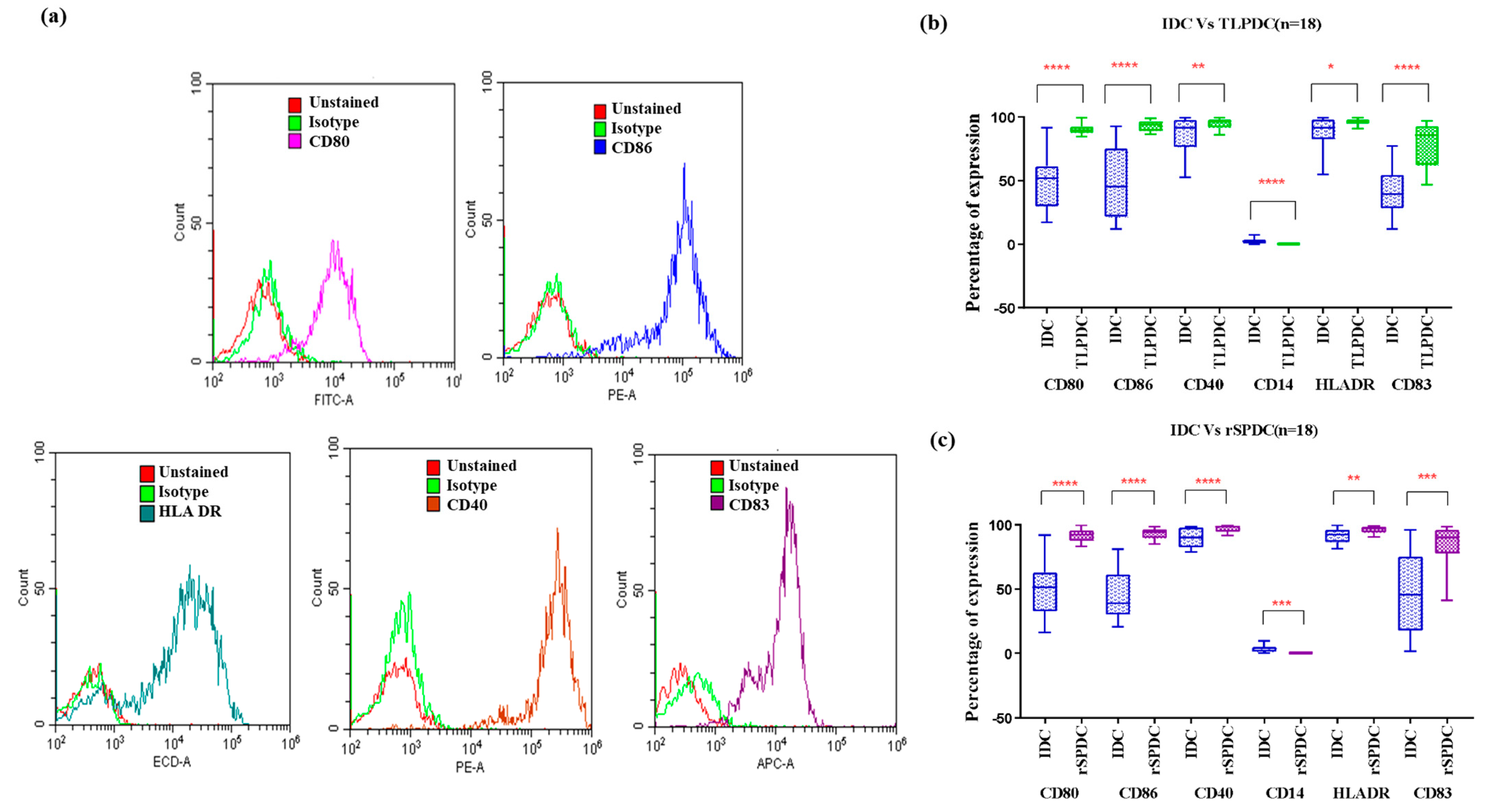
3.4. Bag Generation Minimized DC Loss to Adherence and Maintained DC Phenotypes despite Cytokine Withdrawal
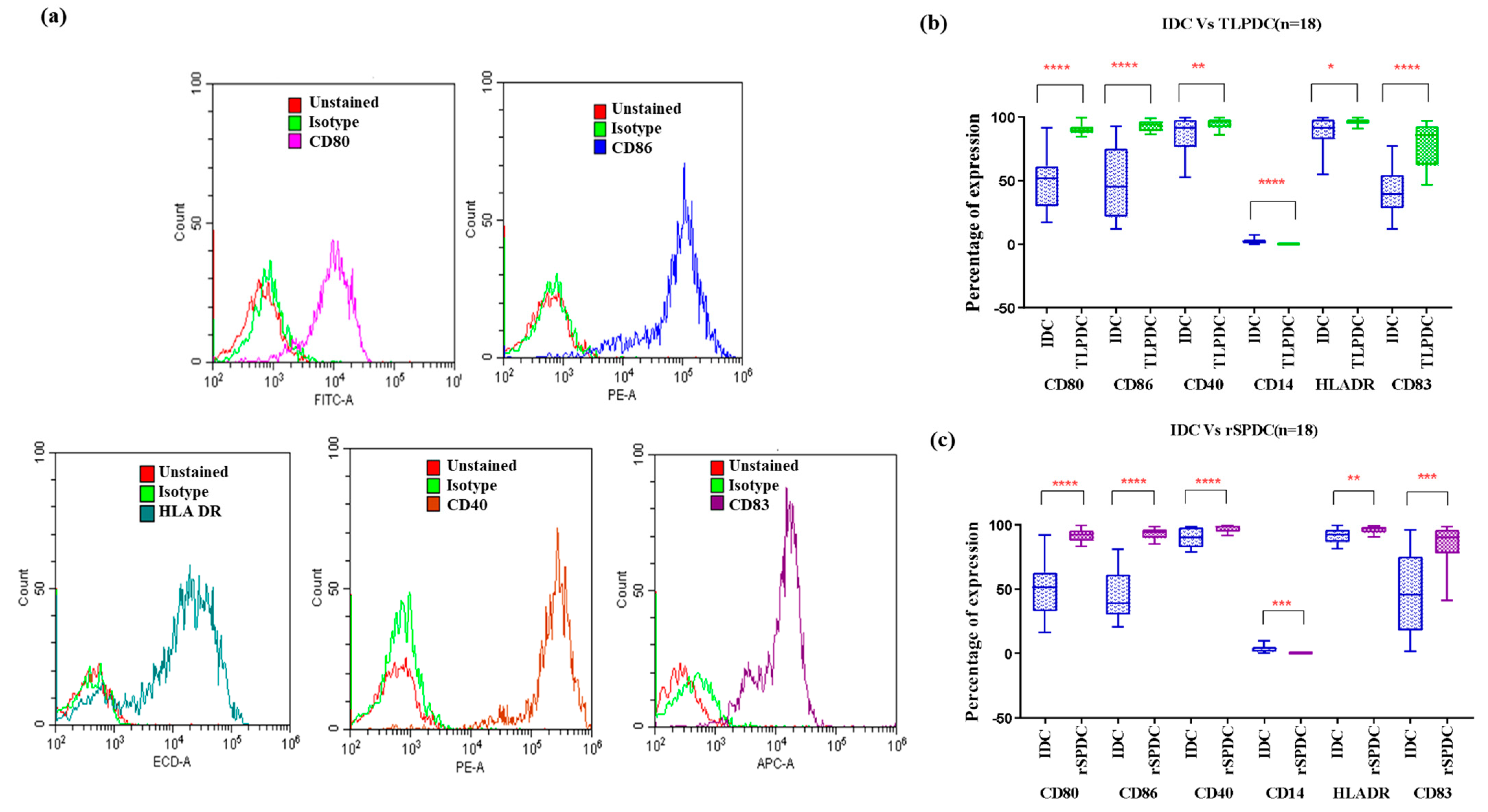
Phenotypic Expression of Batch-Produced DCs
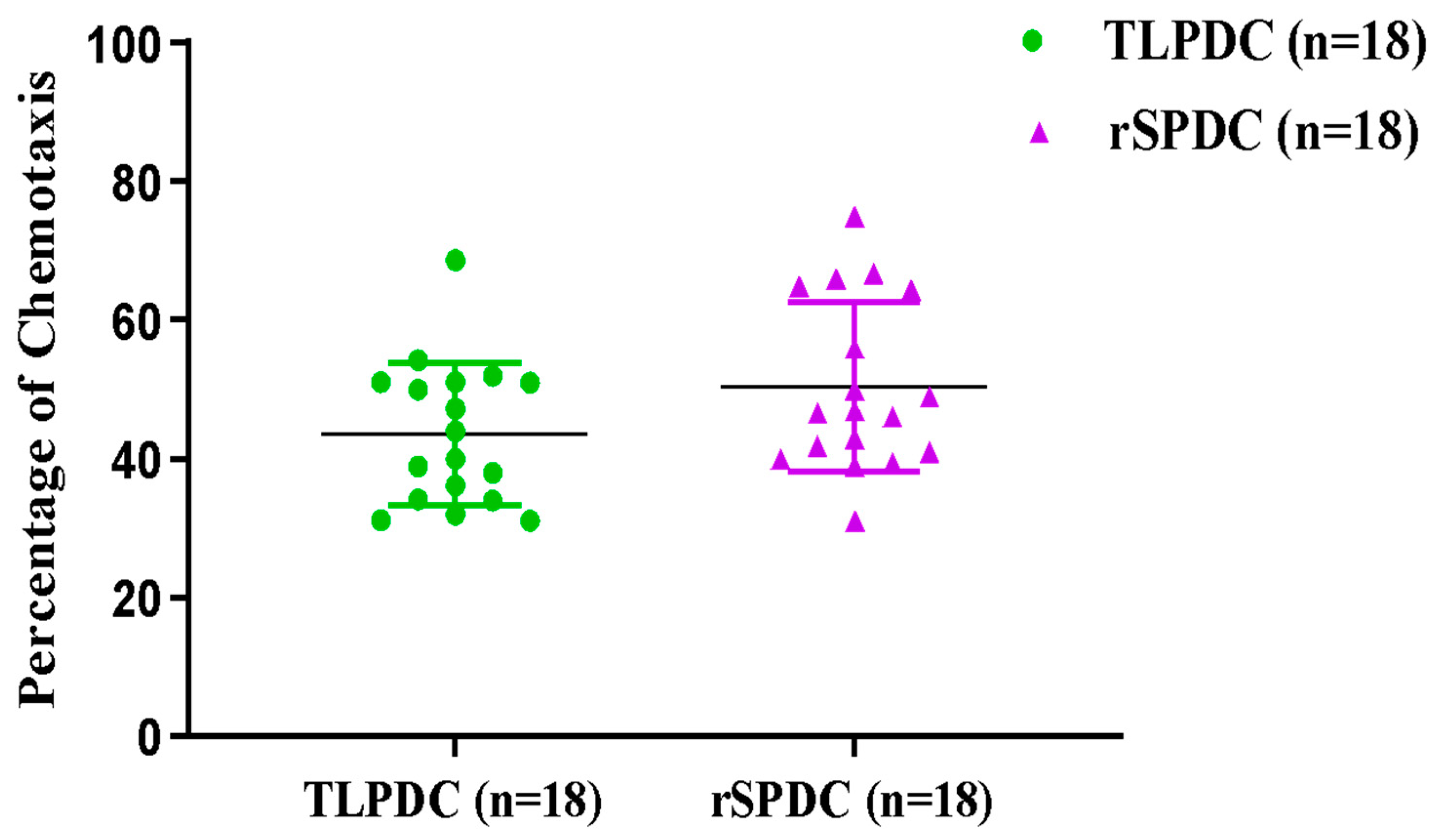
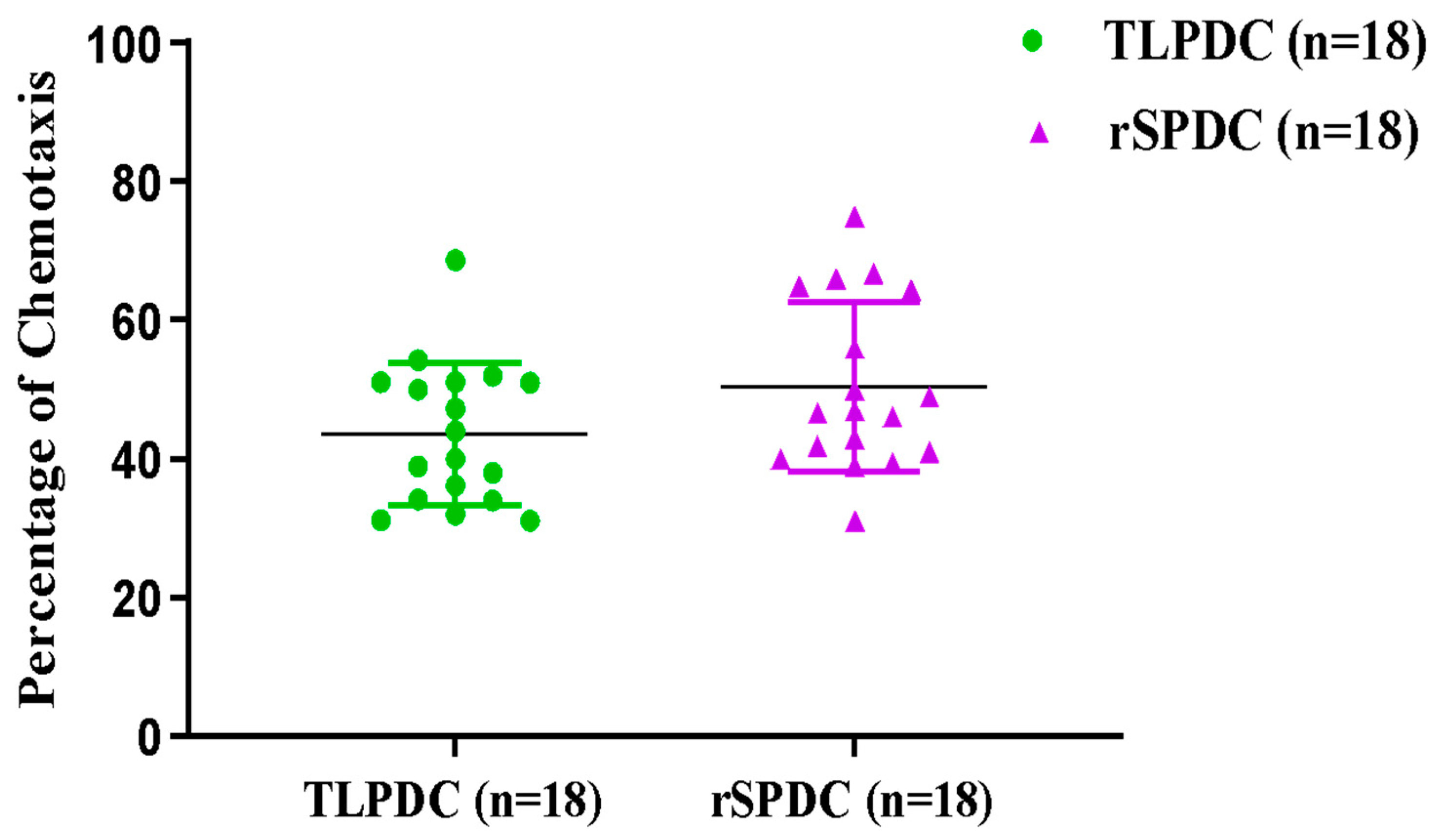
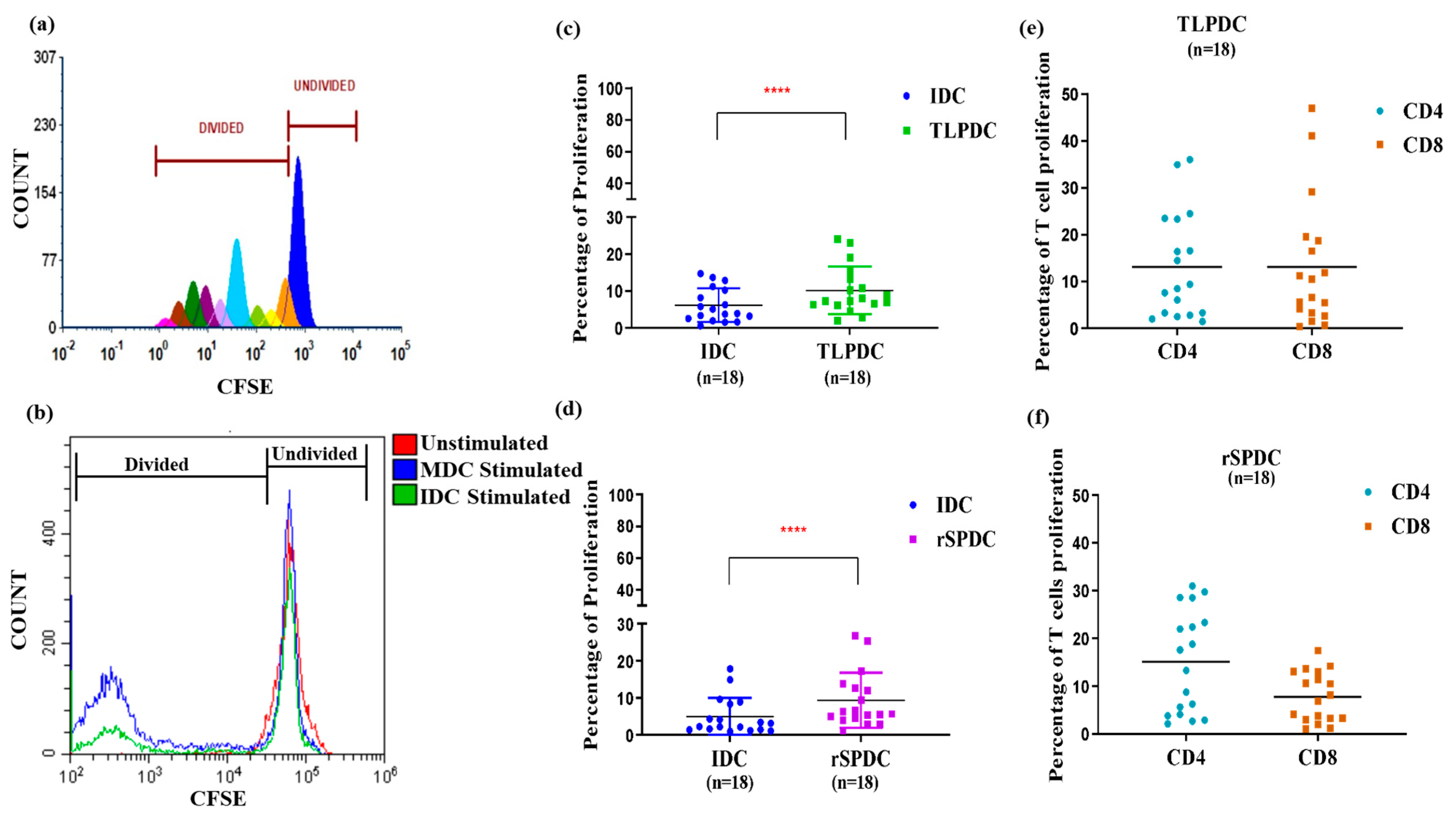
3.5. Antigen-Primed Bag-Cultured DCs Migrate Efficiently and Stimulate Allogeneic T Cells to Proliferate
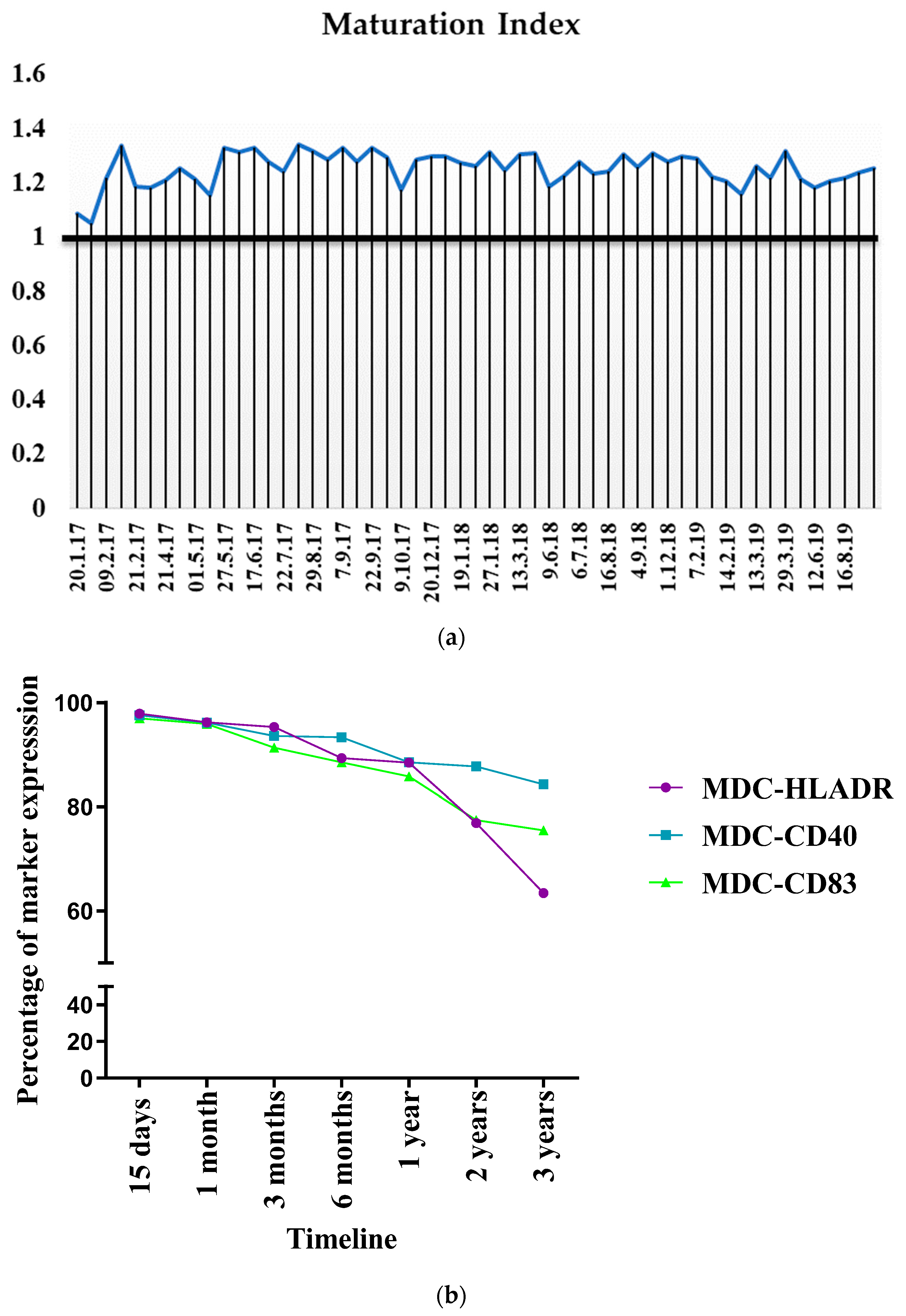
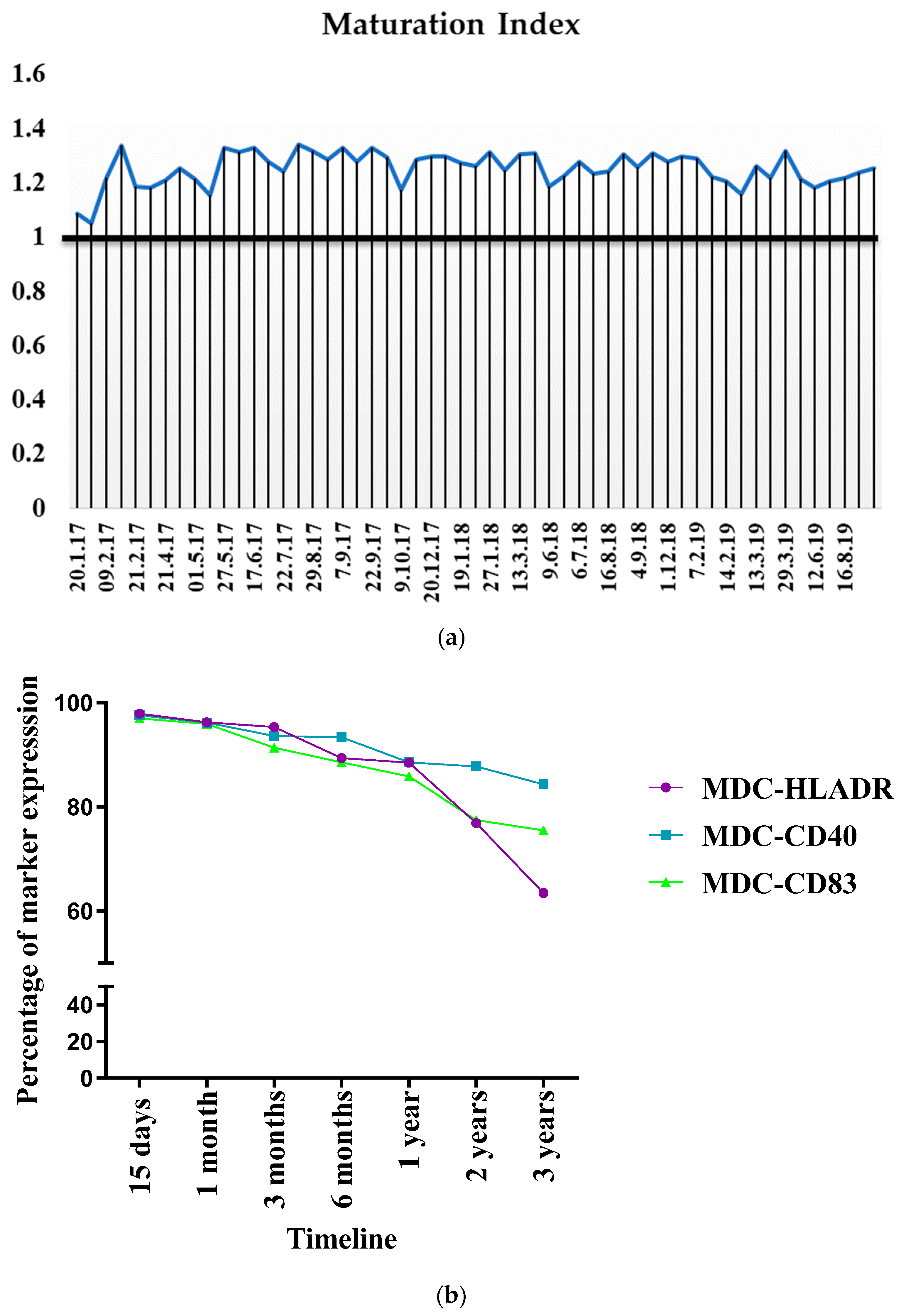
3.6. Evaluation of Inter-Batch Consistency
3.7. Assessment of Microbiological Sterility
3.8. MDCs Generated on a Large Scale Have the Capacity to Stimulate the Secretion of IFNγ in Allogenic PBMCs
3.9. Consistency in Bag Generation Ensures Stability during Short- and Long-Term Cryopreservation
3.10. Establishment of Standardized Workflow and in-Process QC for Product Release
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lubong-Sabado, R.; Bhardwaj, N. Directing dendritic cell immunotherapy towards successful cancer treatment. Immunotherapy 2010, 2, 37. [Google Scholar] [CrossRef]
- Massa, C.; Thomas, C.; Wang, E.; Marincola, F.; Seliger, B. Different maturation cocktails provide dendritic cells with different chemoattractive properties. J. Transl. Med. 2015, 13, 175. [Google Scholar] [CrossRef]
- Dhandapani, H.; Jayakumar, H.; Seetharaman, A.; Singh, S.S.; Ganeshrajah, S.; Jagadish, N.; Suri, A.; Thangarajan, R.; Ramanathan, P. Dendritic cells matured with recombinant human sperm associated antigen 9 (rhSPAG9) induce CD4+, CD8+ T cells and activate NK cells: A potential candidate molecule for immunotherapy in cervical cancer. Cancer Cell Int. 2021, 21, 473. [Google Scholar] [CrossRef]
- Ramanathan, P.; Ganeshrajah, S.; Raghanvan, R.K.; Singh, S.S.; Thangarajan, R. Development and clinical evaluation of dendritic cell vaccines for HPV related cervical cancer—A feasibility study. Asian Pac. J. Cancer Prev. 2014, 15, 5909–5916. [Google Scholar] [CrossRef]
- Ponsaerts, P.; Ponsaerts, P.; Van Tendeloo, V.F.I.; Berneman, Z.N. Cancer immunotherapy using RNA-loaded dendritic cells. Clin. Exp. Immunol. 2003, 134, 378–384. [Google Scholar] [CrossRef]
- Suri, A.; Saini, S.; Sinha, A.; Agarwal, S.; Verma, A.; Parashar, D.; Singh, S.; Gupta, N.; Jagadish, N. Cancer testis antigens: A new paradigm for cancer therapy. Oncoimmunology 2012, 1, 1194. [Google Scholar] [CrossRef]
- Permanyer, M.; Boš Njak, B.; Fö, R. Dendritic cells, T cells and lymphatics: Dialogues in migration and beyond. Curr. Opin. Immunol. 2018, 53, 173–179. [Google Scholar] [CrossRef]
- Garg, A.D.; Vara Perez, M.; Schaaf, M.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Dendritic cell-based anticancer immunotherapy. OncoImmunology 2017, 6, e1328341. [Google Scholar] [CrossRef]
- Butterfield, L.H. Dendritic cells in cancer immunotherapy clinical trials: Are we making progress? Front. Immunol. 2013, 4, 73135. [Google Scholar] [CrossRef]
- Mathur, P.; Sathishkumar, K.; Chaturvedi, M.; Das, P.; Sudarshan, K.L.; Santhappan, S.; Nallasamy, V.; John, A.; Narasimhan, S.; Roselind, F.S.; et al. Cancer Statistics, 2020: Report From National Cancer Registry Programme, India. JCO Glob. Oncol. 2020, 6, 1063–1075. [Google Scholar] [CrossRef]
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191. [Google Scholar] [CrossRef]
- Bobdey, S.; Sathwara, J.; Jain, A.; Balasubramaniam, G. Burden of cervical cancer and role of screening in India. Indian J. Med. Paediatr. Oncol. 2016, 37, 278. [Google Scholar] [CrossRef]
- Burd, E.M. Human Papillomavirus and Cervical Cancer. Clin. Microbiol. Rev. 2003, 16, 1. [Google Scholar] [CrossRef]
- Steinbach, A.; Riemer, A.B. Immune evasion mechanisms of human papillomavirus: An update. Int. J. Cancer 2018, 142, 224–229. [Google Scholar] [CrossRef]
- Chopra, S.; Gupta, M.; Mathew, A.; Mahantshetty, U.; Engineer, R.; Lavanya, G.; Gupta, S.; Ghosh, J.; Thakur, M.; Deodhar, K.; et al. Locally advanced cervical cancer: A study of 5-year outcomes. Indian J. Cancer 2018, 55, 45–49. [Google Scholar] [CrossRef]
- Friedlander, M.; Grogan, M. Guidelines for the Treatment of Recurrent and Metastatic Cervical Cancer; Guidelines for the Treatment of Recurrent and Metastatic Cervical Cancer. Oncologist 2002, 7, 342–347. [Google Scholar] [CrossRef]
- Sankaranarayanan, R.; Swaminathan, R.; Brenner, H.; Chen, K.; Chia, K.S.; Chen, J.G.; Law, S.C.; Ahn, Y.-O.; Xiang, Y.B.; Yeole, B.B.; et al. Cancer survival in Africa, Asia, and Central America: A population-based study. Lancet Oncol. 2010, 11, 165–173. [Google Scholar] [CrossRef]
- World Health Organization. Annex 6 WHO Good Manufacturing Practices for Sterile Pharmaceutical Products; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Ramanathan, P.; Dhandapani, H.; Jayakumar, H.; Ganeshrajah, S.; Thangarajan, R. Dendritic cells primed with HPV positive cervical tumor lysate are superior to unprimed DCs in migratory capacity and induce a potent Th1 response. Hum. Immunol. 2014, 75, 1216–1224. [Google Scholar] [CrossRef]
- Remmele, W.; Stegner, H.E. Recommendation for uniform definition of an immunoreactive score (IRS) for immunohistochemical estrogen receptor detection (ER-ICA) in breast cancer tissue. Pathologe 1987, 8, 138–140. [Google Scholar] [PubMed]
- Boudousquié, C.; Boand, V.; Lingre, E.; Dutoit, L.; Balint, K.; Danilo, M.; Harari, A.; Gannon, P.O.; Kandalaft, L.E. Development and Optimization of a GMP-Compliant Manufacturing Process for a Personalized Tumor Lysate Dendritic Cell Vaccine. Vaccines 2020, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, J.; Das, B.; Mishra, A.; Sahay, P.; Upadhyay, P. Open Peer Review Discuss this article (0) Comments Monocytes isolated by positive and negative magnetic sorting techniques show different molecular characteristics and immunophenotypic behaviour [version 3; referees: 2 approved]. F1000Research 2017, 6, 2045. [Google Scholar] [CrossRef]
- Figueroa, G.; Parira, T.; Laverde, A.; Casteleiro, G.; El-Mabhouh, A.; Nair, M.; Agudelo, M. Characterization of Human Monocyte-derived Dendritic Cells by Imaging Flow Cytometry: A Comparison between Two Monocyte Isolation Protocols. J. Vis. Exp. 2016, 2016, 54296. [Google Scholar] [CrossRef]
- Hopewell, E.L.; Cox, C. Manufacturing Dendritic Cells for Immunotherapy: Monocyte Enrichment. Mol. Ther. Methods Clin. Dev. 2020, 16, 155. [Google Scholar] [CrossRef]
- Stroncek, D.F.; Fellowes, V.; Pham, C.; Khuu, H.; Fowler, D.H.; Wood, L.V.; Sabatino, M. Counter-flow elutriation of clinical peripheral blood mononuclear cell concentrates for the production of dendritic and T cell therapies. J. Transl. Med. 2014, 12, 241. [Google Scholar] [CrossRef]
- Fekete, N.; Béland, A.V.; Campbell, K.; Clark, S.L.; Hoesli, C.A. Bags versus flasks: A comparison of cell culture systems for the production of dendritic cell-based immunotherapies. Transfusion 2018, 58, 1800–1813. [Google Scholar] [CrossRef] [PubMed]
- Berger, T.G.; Feuerstein, B.; Strasser, E.; Hirsch, U.; Schreiner, D.; Schuler, G.; Schuler-Thurner, B. Large-scale generation of mature monocyte-derived dendritic cells for clinical application in cell factories™. J. Immunol. Methods 2002, 268, 131–140. [Google Scholar] [CrossRef]
- Guyre, C.A.; Fisher, J.L.; Waugh, M.G.; Wallace, P.K.; Tretter, C.G.; Ernstoff, M.S.; Barth, R.J. Advantages of hydrophobic culture bags over flasks for the generation of monocyte-derived dendritic cells for clinical applications. J. Immunol. Methods 2002, 262, 85–94. [Google Scholar] [CrossRef]
- Bastien, J.-P.; Fekete, N.; Beland, A.V.; Lachambre, M.-P.; Laforte, V.; Juncker, D.; Dave, V.; Roy, D.-C.; Hoesli, C.A. Closing the system: Production of viral antigen-presenting dendritic cells eliciting specific CD8+ T cell activation in fluorinated ethylene propylene cell culture bags. J. Transl. Med. 2020, 18, 383. [Google Scholar] [CrossRef]
- Delirezh, N.; Shojaeefar, E. Phenotypic and functional comparison between flask adherent and magnetic activated cell sorted monocytes derived dendritic cells. Iran. J. Immunol. 2012, 9, 98–108. [Google Scholar]
- Elias, M.; Van Zanten, J.; Hospers, G.; Setroikromo, A.; De Jong, M.; De Leij, L.; Mulder, N. Closed system generation of dendritic cells from a single blood volume for clinical application in immunotherapy. J. Clin. Apher. 2005, 20, 197–207. [Google Scholar] [CrossRef]
- Kurlander, R.J.; Tawab, A.; Fan, Y.; Carter, C.S.; Read, E.J. A functional comparison of mature human dendritic cells prepared in fluorinated ethylene-propylene bags or polystyrene flasks. Transfusion 2006, 46, 1494–1504. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, L.; Ling, F.; Wen, S.; Luo, Y.; Liu, H.; Liu, J.; Zheng, W.; Liang, M.; Sun, J.; et al. Effect of immune tolerance induced by immature dendritic cells and CTLA4-Ig on systemic lupus erythematosus: An in vivo study. Exp. Ther. Med. 2018, 15, 2499–2506. [Google Scholar] [CrossRef]
- Fucikova, J.; Palova-Jelinkova, L.; Bartunkova, J.; Spisek, R. Induction of Tolerance and Immunity by Dendritic Cells: Mechanisms and Clinical Applications. Front. Immunol. 2019, 10, 2393. [Google Scholar] [CrossRef]
- Kvistborg, P.; Boegh, M.; Pedersen, A.; Claesson, M.; Zocca, M. Fast generation of dendritic cells. Cell Immunol. 2009, 260, 56–62. [Google Scholar] [CrossRef]
- Brabants, E.; Heyns, K.; De Smet, S.; Devreker, P.; Ingels, J.; De Cabooter, N.; Debacker, V.; Dullaers, M.; Van Meerbeeck, J.; Vandekerckhove, B.; et al. An accelerated, clinical-grade protocol to generate high yields of type 1-polarizing messenger RNAÀloaded dendritic cells for cancer vaccination. Cytotherapy 2018, 20, 1164–1181. [Google Scholar] [CrossRef]
- Lasky, J.L.; Panosyan, E.H.; Plant, A.; Davidson, T.; Yong, W.H.; Prins, R.M.; Liau, L.M.; Moore, T.B. Autologous Tumor Lysate-pulsed Dendritic Cell Immunotherapy for Pediatric Patients with Newly Diagnosed or Recurrent High-grade Gliomas. Anticancer Res. 2013, 33, 2047. [Google Scholar]
- Yu, J.S.; Liu, G.; Ying, H.; Yong, W.H.; Black, K.L.; Wheeler, C.J. Vaccination with Tumor Lysate-Pulsed Dendritic Cells Elicits Antigen-Specific, Cytotoxic T-Cells in Patients with Malignant Glioma. Cancer Res. 2004, 64, 4973–4979. [Google Scholar] [CrossRef]
- Kodack, D.P.; Farago, A.F.; Dastur, A.; Held, M.A.; Dardaei, L.; Friboulet, L.; von Flotow, F.; Damon, L.J.; Lee, D.; Parks, M.; et al. Primary Patient-Derived Cancer Cells and Their Potential for Personalized Cancer Patient Care. Cell Rep. 2017, 21, 3298. [Google Scholar] [CrossRef]
- Benteyn, D.; Van Nuffel, A.M.; Wilgenhof, S.; Bonehill, A. Single-step antigen loading and maturation of dendritic cells through mRNA electroporation of a tumor-associated antigen and a TriMix of costimulatory molecules. Methods Mol. Biol. 2014, 1139, 3–15. [Google Scholar] [CrossRef]
- Butterfield, L.H.; Palucka, A.K.; Britten, C.M.; Dhodapkar, M.V.; Håkansson, L.; Janetzki, S.; Kawakami, Y.; Kleen, T.-O.; Lee, P.P.; Maccalli, C.; et al. Recommendations from the iSBTc-SITC/FDA/NCI Workshop on Immunotherapy Biomarkers. Clin. Cancer Res. 2011, 17, 3064. [Google Scholar] [CrossRef]
- Foster, B.; Prussin, C.; Liu, F.; Whitmire, J.K.; Whitton, J.L. Detection of intracellular cytokines by flow cytometry. Curr. Protoc. Immunol. 2007, 6, 6–24. [Google Scholar] [CrossRef]
- Celluzzi, C.M.; Welbon, C. A simple cryopreservation method for dendritic cells and cells used in their derivation and functional assessment. Transfusion 2003, 43, 488–494. [Google Scholar] [CrossRef]
- Kawaguchi, H.; Sakamoto, T.; Koya, T.; Togi, M.; Date, I.; Watanabe, A.; Yoshida, K.; Kato, T.; Nakamura, Y.; Ishigaki, Y.; et al. Quality verification with a cluster−controlled manufacturing system to generate monocyte−derived dendritic cells. Vaccines 2021, 9, 533. [Google Scholar] [CrossRef]
- Pardali, E.; Schmitz, T.; Borgscheiper, A.; Iking, J.; Stegger, L.; Waltenberger, J. Cryopreservation of primary human monocytes does not negatively affect their functionality or their ability to be labelled with radionuclides: Basis for molecular imaging and cell therapy. EJNMMI Res. 2016, 6, 77. [Google Scholar] [CrossRef]
- Westermann, J.; Körner, I.J.; Kopp, J.; Kurz, S.; Zenke, M.; Dörken, B.; Pezzutto, A. Cryopreservation of mature monocyte-derived human dendritic cells for vaccination: Influence on phenotype and functional properties. Cancer Immunol. Immunother. 2003, 52, 194–198. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Attributes (Method) | Assay | IDC (Max—100%, Differentiation Score = 1 *) | MDC (Max—100%, Maturation Score = 1 #) |
|---|---|---|---|
| IDENTITY (Flow cytometry) | Immunophenotype | Cut-off | Cut-off |
| anti-CD14 PC5 (IgG1 Mouse) | <5% | 0–0.5% | |
| anti-HLA-DR ECD (IgG1 Mouse) | >80% (0.8) * | >90% (0.9) # | |
| anti-CD40 PE (IgG1 Mouse) | >75% (0.75) * | >90% (0.9) # | |
| anti-CD80 FITC (IgG1 Mouse) | >10% (0.1) * | >80% (0.8) # | |
| anti-CD86 PE (IgG1 Mouse) | 5–50% (0.5) * | >85% (0.85) # | |
| anti-CD83 APC (IgG1 Rat) | 0.5–5% (0.05) * | >20% (0.2) # | |
| POTENCY (Flow cytometry) | FITC dextran uptake | >80% (0.8) * | No data |
| Migration towards CCL19/21 in 3 hrs time | None or <1% | >30% (0.3) # | |
| Proliferation of allogeneic PBMC | Percentage of cell division (overall, CD4, CD8T cells) | Percentage of cell division (overall, CD4, CD8 T cells >IDCs) | |
| STERILITY | Clinical tests before apheresis | Blood sample negative for HIV, HBV and HCV | |
| Microbiological tests (direct inoculation and Gram stain) | Sterile upon test for bacterial and fungal culture up to 2 weeks | ||
| Test for endotoxin | Level of endotoxin less than 1.0 EU/ml | ||
| Clinicopathological Features of Screened Patients | No. of Patients |
|---|---|
| Patient classification: | |
| Median age in years (range) | 52 (30–65) |
| Locally advanced (Stage III B) | 146 |
| Metastatic (Stage IV) | 36 |
| Cell type (WHO classification): | |
| Squamous cell carcinoma | 176 |
| Papillary squamous cell carcinoma | 2 |
| Adenocarcinoma | 1 |
| Adenosquamous carcinoma | 0 |
| Poorly differentiated carcinoma | 3 |
| Histological Grade: | |
| Grade 1 | 1 |
| Grade 2 | 48 |
| Grade 3 | 133 |
| Keratinization status: | |
| LCNK | 129 |
| LCK | 53 |
| HPV status: | |
| HPV-Positive | 181 |
| HPV-Negative | 1 |
| IRS score for SPAG9: | |
| 0—Negative | 0 |
| 1 to 3—Mild positivity | 33 |
| 4 to 8—Moderate positivity | 119 |
| 9 to 12—Strong positivity | 27 |
| Tumor Percentage (range) | 72 (50–90%) |
| Total number of patients recruited for trial | 54/182 |
| Duration | Percentage of Viability Post-Thaw * | |
|---|---|---|
| TLPDCs (n = 5) | rSPDCs (n = 5) | |
| 0 | 97 ± 1.3 | 96 ± 2 |
| 30 min | 97 ± 0.7 | 96.4 ± 00.8 |
| 1 h | 95 ± 2 | 96 ± 1.2 |
| 2 h | 95 ± 2 | 96 ± 1 |
| 3 h | 94 ± 2 | 95 ± 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seetharaman, A.; Christopher, V.; Dhandapani, H.; Jayakumar, H.; Dhanushkodi, M.; Bhaskaran, N.; Rajaraman, S.; Ranganathan, R.; Sunder Singh, S.; Vijayakumar, V.; et al. Optimization and Validation of a Harmonized Protocol for Generating Therapeutic-Grade Dendritic Cells in a Randomized Phase II Clinical Trial, Using Two Varied Antigenic Sources. Vaccines 2024, 12, 112. https://doi.org/10.3390/vaccines12020112
Seetharaman A, Christopher V, Dhandapani H, Jayakumar H, Dhanushkodi M, Bhaskaran N, Rajaraman S, Ranganathan R, Sunder Singh S, Vijayakumar V, et al. Optimization and Validation of a Harmonized Protocol for Generating Therapeutic-Grade Dendritic Cells in a Randomized Phase II Clinical Trial, Using Two Varied Antigenic Sources. Vaccines. 2024; 12(2):112. https://doi.org/10.3390/vaccines12020112
Chicago/Turabian StyleSeetharaman, Abirami, Vasanth Christopher, Hemavathi Dhandapani, Hascitha Jayakumar, Manikandan Dhanushkodi, Narmadha Bhaskaran, Swaminathan Rajaraman, Rama Ranganathan, Shirley Sunder Singh, Varalakshmi Vijayakumar, and et al. 2024. "Optimization and Validation of a Harmonized Protocol for Generating Therapeutic-Grade Dendritic Cells in a Randomized Phase II Clinical Trial, Using Two Varied Antigenic Sources" Vaccines 12, no. 2: 112. https://doi.org/10.3390/vaccines12020112
APA StyleSeetharaman, A., Christopher, V., Dhandapani, H., Jayakumar, H., Dhanushkodi, M., Bhaskaran, N., Rajaraman, S., Ranganathan, R., Sunder Singh, S., Vijayakumar, V., Rajamanickam, A., Suri, A., Jagadish, N., Rajkumar, T., & Ramanathan, P. (2024). Optimization and Validation of a Harmonized Protocol for Generating Therapeutic-Grade Dendritic Cells in a Randomized Phase II Clinical Trial, Using Two Varied Antigenic Sources. Vaccines, 12(2), 112. https://doi.org/10.3390/vaccines12020112