
Preclinical Establishment of a Divalent Vaccine against SARS-CoV-2

 , , , , , , and
, , , , , , and 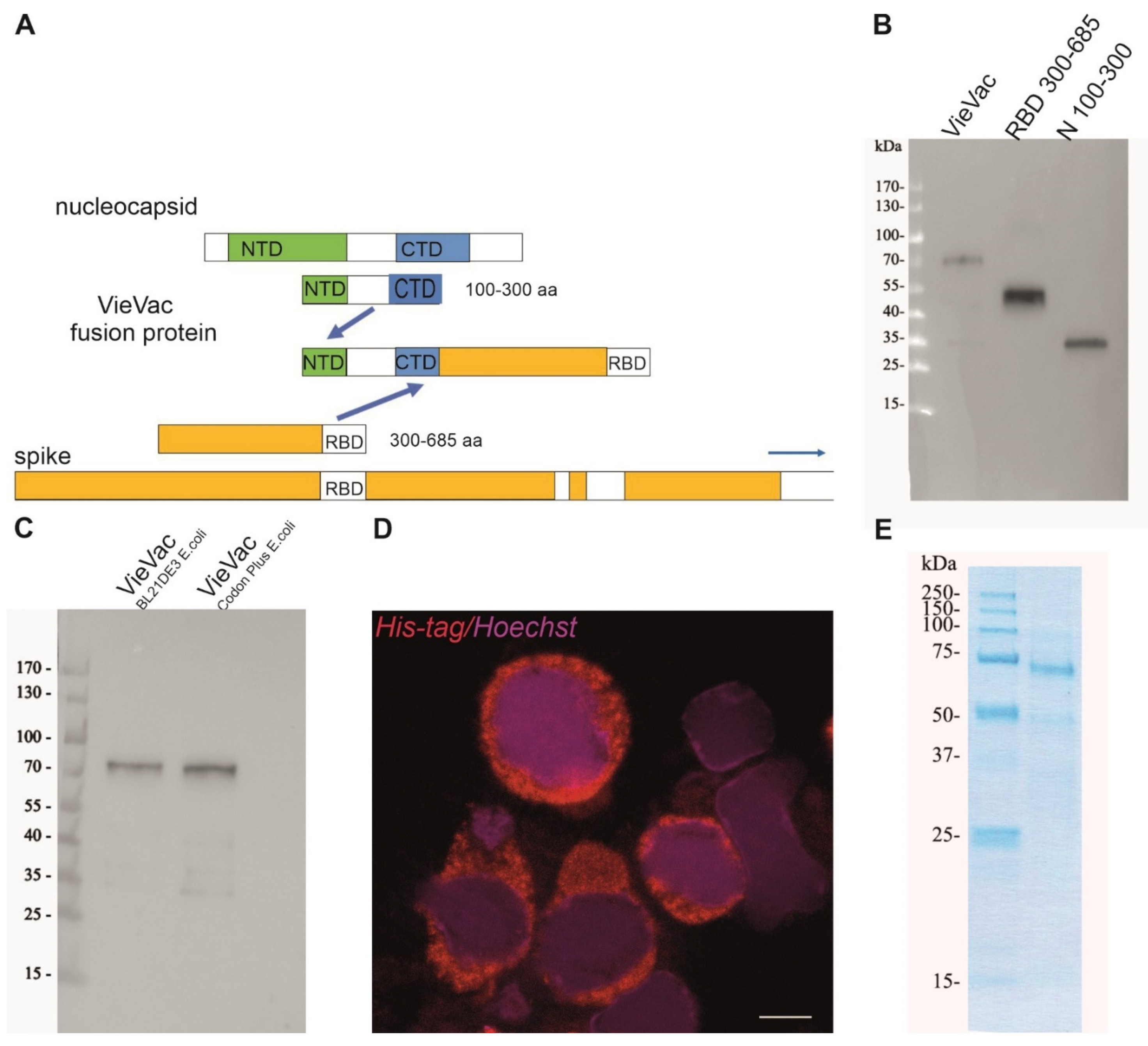{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Rationale
2.2. Animals, Blood Sampling and Tissue Processing
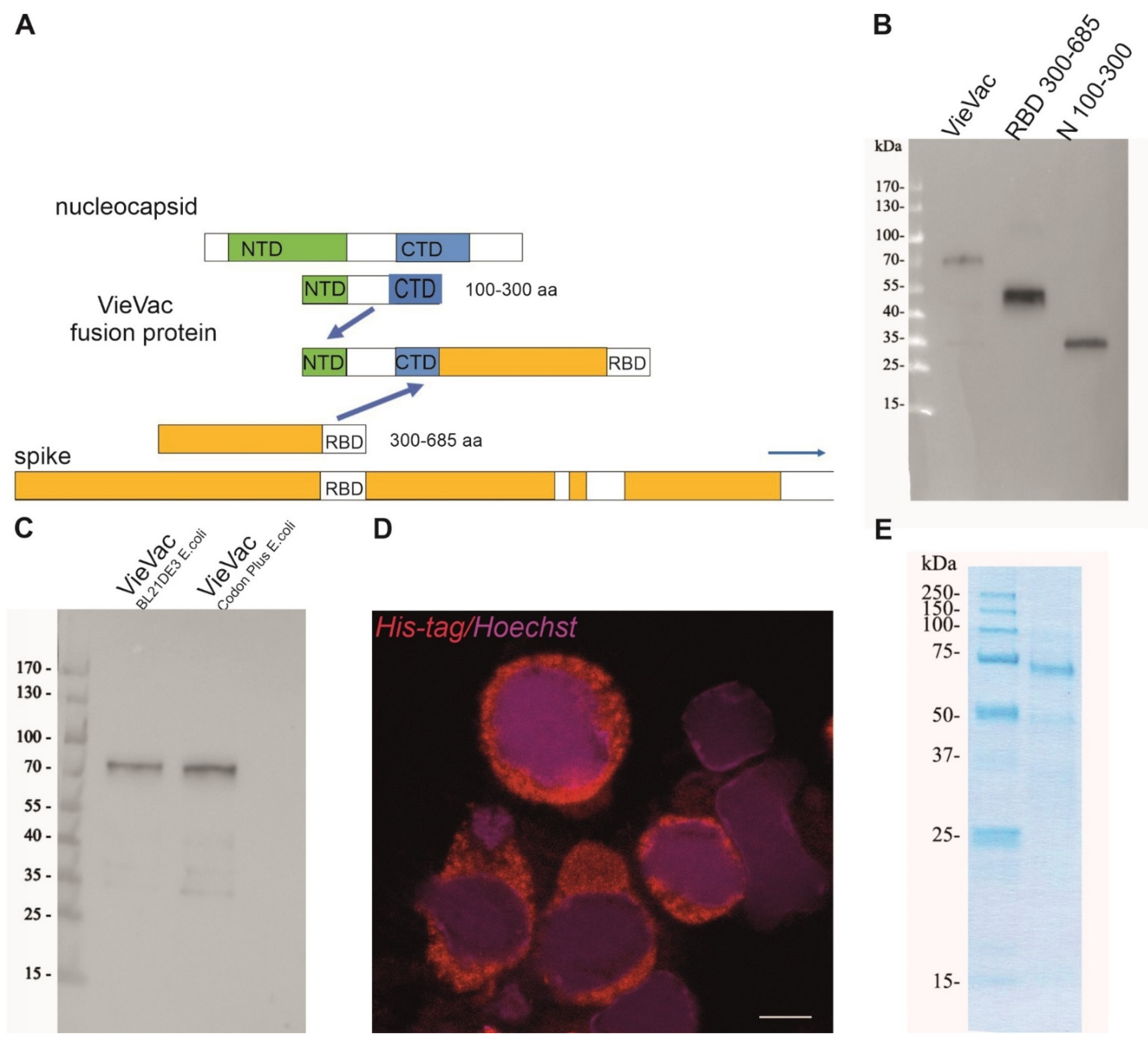
2.3. Construction and Heterologous Expression of the Fusion Protein
2.4. Directional Cloning into the Gateway pEntry/D-TOPO Vector
2.5. Insertion into Baculovirus and Amplification in Insect Cells
2.6. ‘VieVac’ Purification from Insect Cells
2.7. Evaluation of Protein Expression by Immunofluorescence
2.8. Protein Production and Purification from E. coli
2.9. Immunoblotting of ‘VieVac’ Protein with Immune Sera
2.10. Protein Adsorption to Imject™ Alum Adjuvant
2.11. Mouse Immunization
2.12. ELISA
2.13. RNA Isolation from Blood and Spleen
2.14. Quantitative PCR
2.15. Ex Vivo T-Cell Stimulation with N- and S-Specific Peptides and CD44 Evaluation by Cell Surface Flow Cytometry
2.16. Immunohistochemistry
2.17. Statistical Analysis
3. Results
3.1. Protein Production, Characterization and Purification
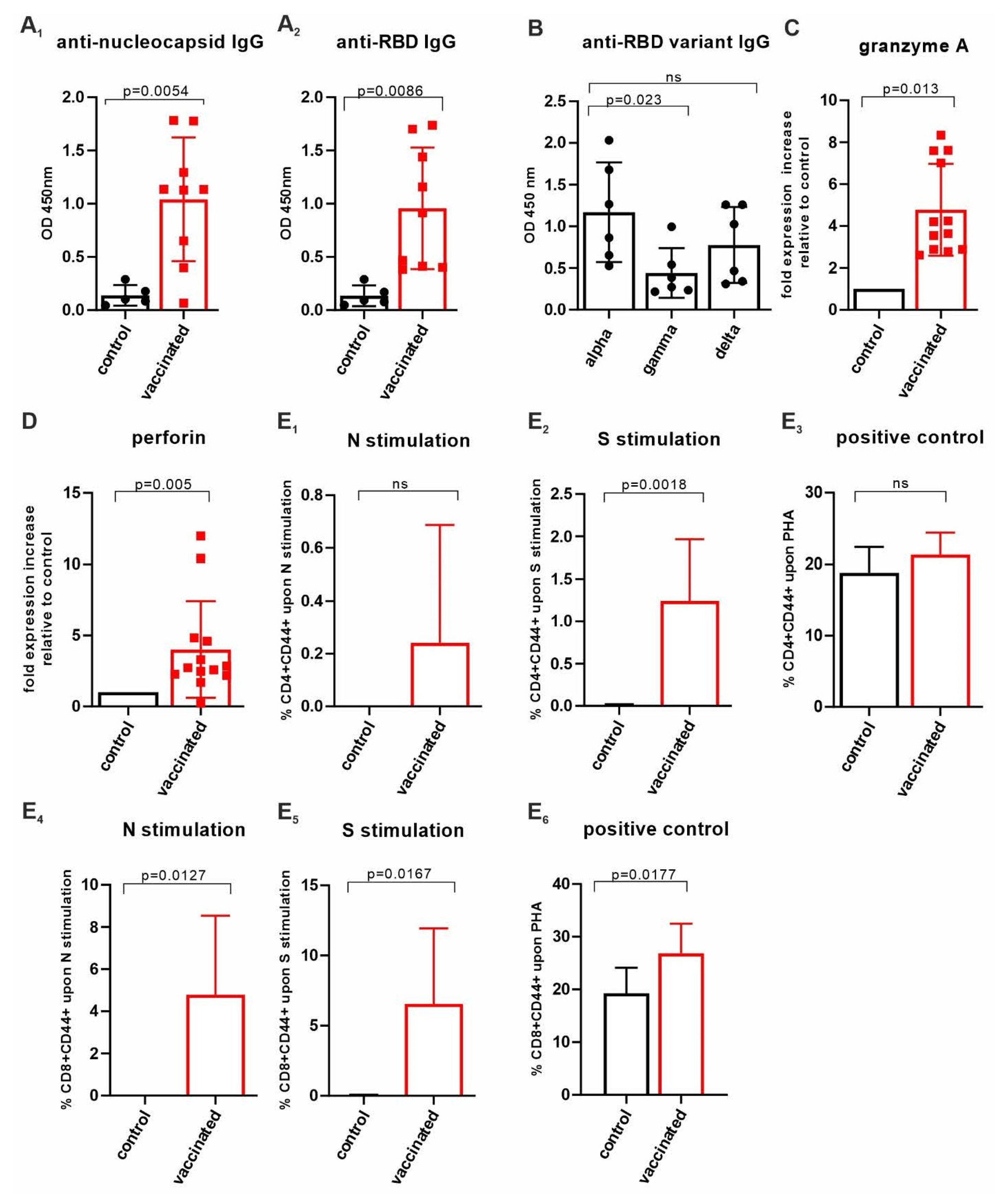
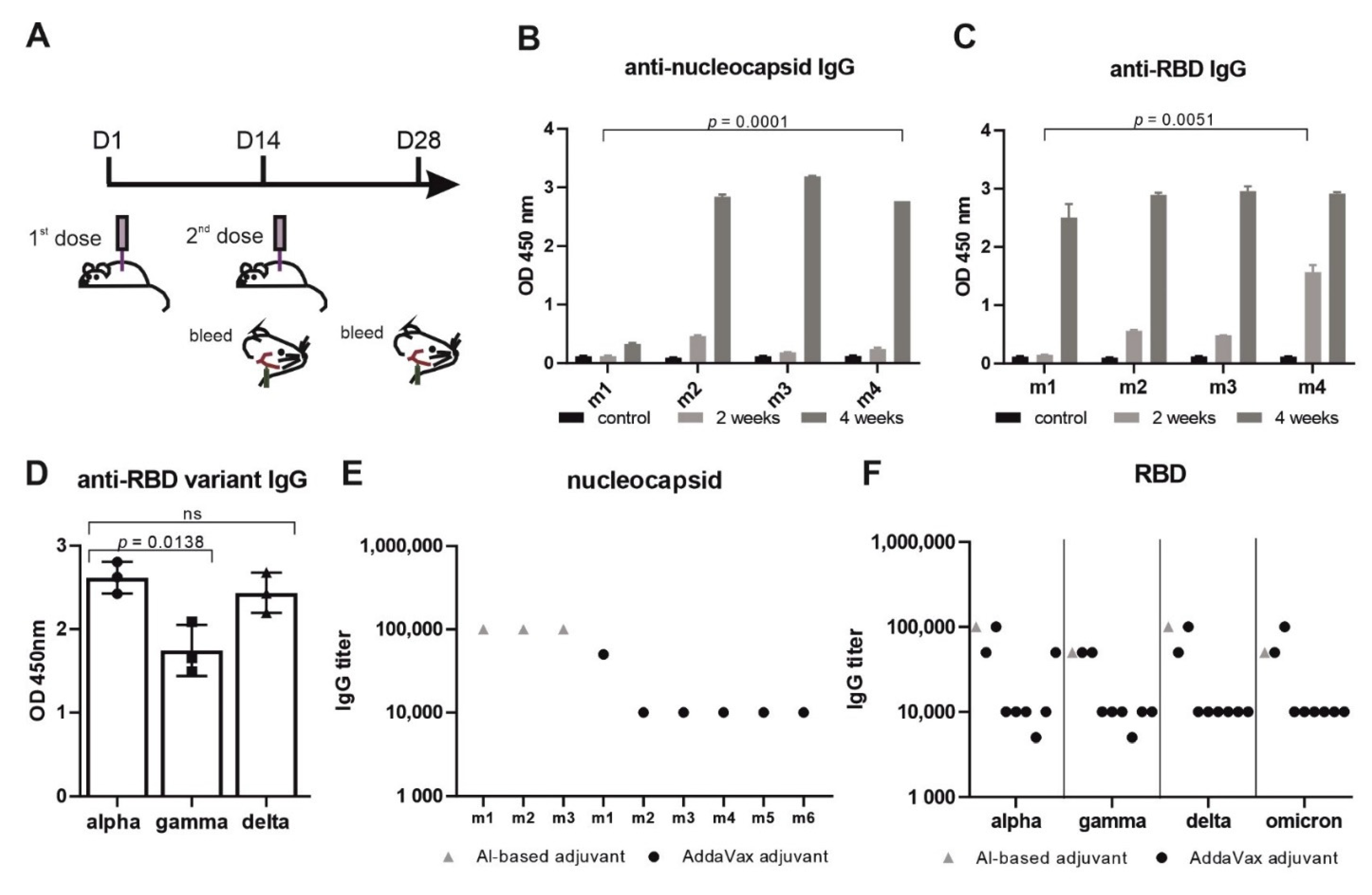
3.2. Imject™ Alum/’VieVac’ Induces IgGs to Both Nucleocapsid and RBD
3.3. AddaVax™ with ‘VieVac’ Primes IgG Production to Both Nucleocapsid and RBD
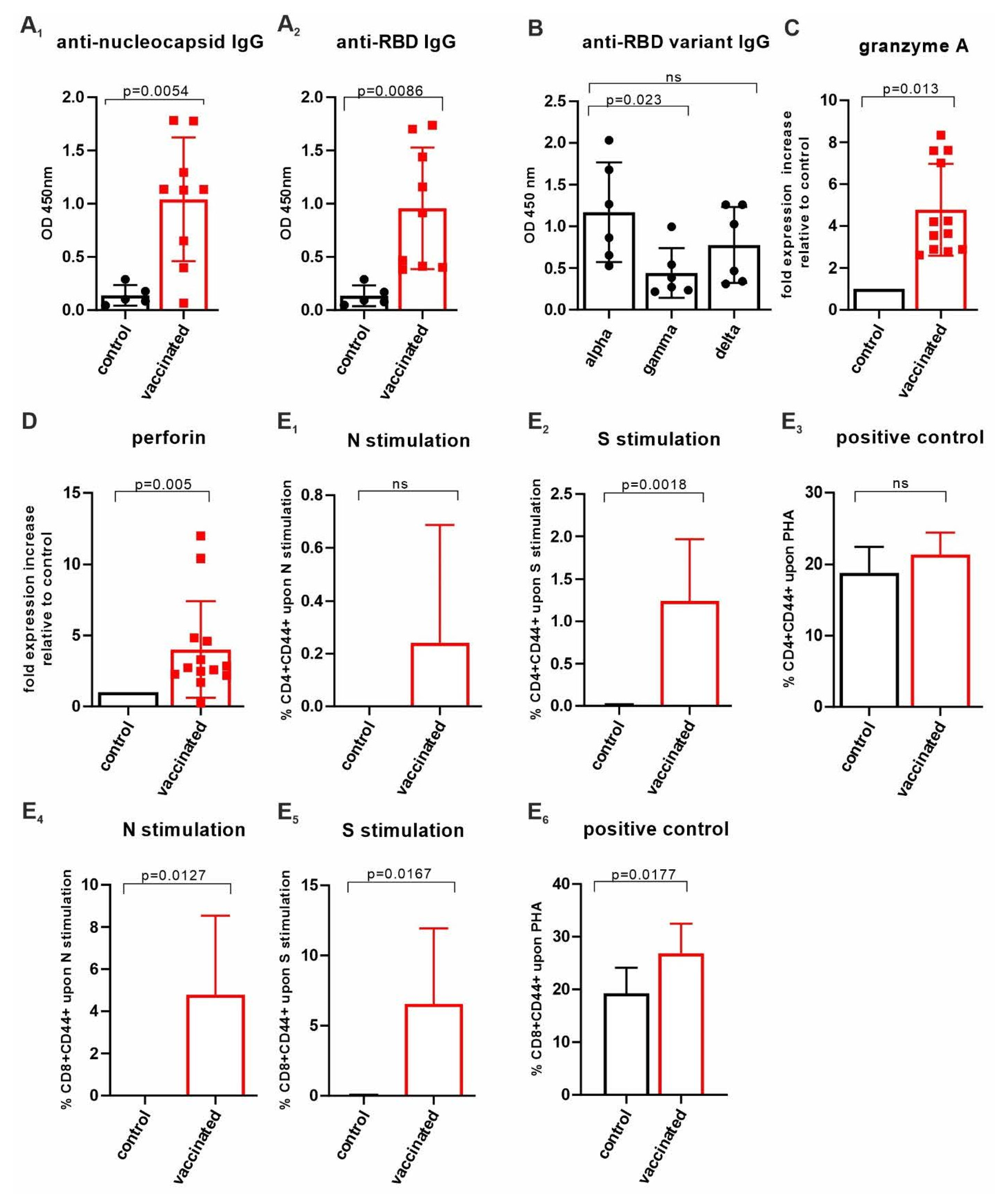
3.4. Insect-Cell-Produced AddaVax™/VieVac Generates Effector Cell Immunoreactivity
3.5. CD4+ and CD8+ T-Cell Responses Evaluated by CD44 Cell Surface Upregulation in Ex Vivo-Stimulated Spleen Cells of AddaVax™-/VieVac-Challenged Mice
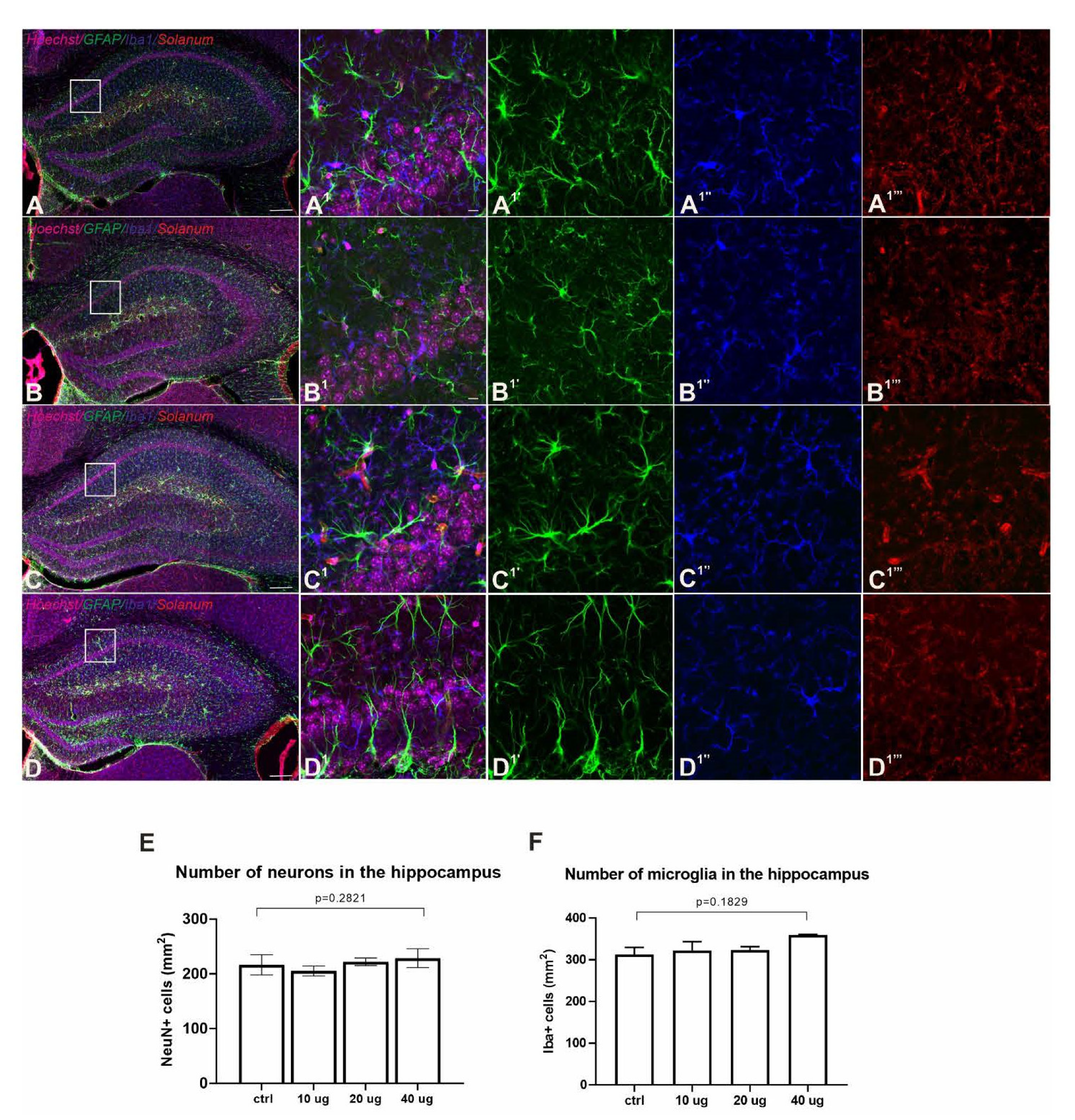
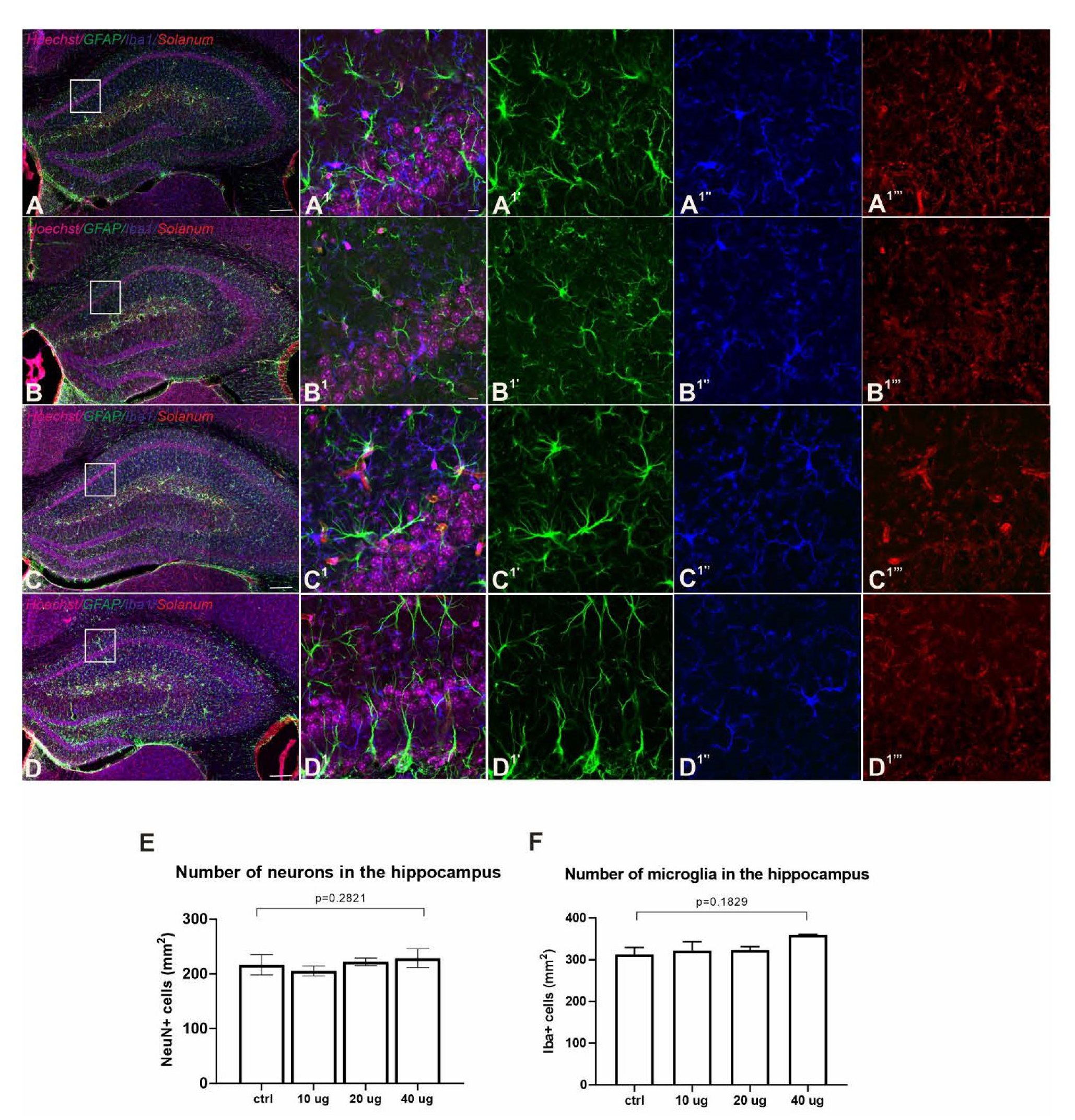
3.6. Lack of Adverse Neuropathology in Addavax™-/VieVac-Injected Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Engl. J. Med. 2020, 383, 1920–1931. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, M.J.; Lyke, K.E.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Raabe, V.; Bailey, R.; Swanson, K.A.; et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 2020, 586, 589–593. [Google Scholar] [CrossRef]
- Guebre-Xabier, M.; Patel, N.; Tian, J.-H.; Zhou, B.; Maciejewski, S.; Lam, K.; Portnoff, A.D.; Massare, M.J.; Frieman, M.B.; Piedra, P.A.; et al. NVX-CoV2373 vaccine protects cynomolgus macaque upper and lower airways against SARS-CoV-2 challenge. Vaccine 2020, 38, 7892–7896. [Google Scholar] [CrossRef] [PubMed]
- Watterson, D.; Wijesundara, D.K.; Modhiran, N.; Mordant, F.L.; Li, Z.; Avumegah, M.S.; McMillan, C.L.; Lackenby, J.; Guilfoyle, K.; van Amerongen, G.; et al. Preclinical development of a molecular clamp-stabilised subunit vaccine for severe acute respiratory syndrome coronavirus 2. Clin. Transl. Immunol. 2021, 10, e1269. [Google Scholar] [CrossRef] [PubMed]
- Mostaghimi, D.; Valdez, C.N.; Larson, H.T.; Kalinich, C.C.; Iwasaki, A. Prevention of host-to-host transmission by SARS-CoV-2 vaccines. Lancet Infect. Dis. 2022, 22, e52–e58. [Google Scholar] [CrossRef]
- Puhach, O.; Adea, K.; Hulo, N.; Sattonnet, P.; Genecand, C.; Iten, A.; Bausch, F.J.; Kaiser, L.; Vetter, P.; Eckerle, I.; et al. Infectious viral load in unvaccinated and vaccinated patients infected with SARS-CoV-2 WT, Delta and Omicron. medRxiv 2022. [Google Scholar] [CrossRef]
- Li, T.; Wang, L.; Wang, H.; Li, X.; Zhang, S.; Xu, Y.; Wei, W. Serum SARS-COV-2 Nucleocapsid Protein: A Sensitivity and Specificity Early Diagnostic Marker for SARS-COV-2 Infection. Front. Cell. Infect. Microbiol. 2020, 10, 470. [Google Scholar] [CrossRef]
- Dutta, N.K.; Mazumdar, K.; Gordy, J.T. The Nucleocapsid Protein of SARS-CoV-2: A Target for Vaccine Development. J. Virol. 2020, 94, e00647-20. [Google Scholar] [CrossRef] [PubMed]
- Ahlen, G.; Frelin, L.; Nikouyan, N.; Weber, F.; Hoglund, U.; Larsson, O.; Westman, M.; Tuvesson, O.; Gidlund, E.K.; Cadossi, M.; et al. The SARS-CoV-2 N Protein Is a Good Component in a Vaccine. J. Virol. 2020, 94, e01279-20. [Google Scholar] [CrossRef] [PubMed]
- Diao, B.; Wen, K.; Chen, J.; Liu, Y.; Yuan, Z.; Han, C.; Chen, J.; Pan, Y.; Chen, L.; Dan, Y.; et al. Diagnosis of Acute Respiratory Syndrome Coronavirus 2 Infection by Detection of Nucleocapsid Protein. medRxiv 2020. [Google Scholar] [CrossRef]
- Burbelo, P.D.; Riedo, F.X.; Morishima, C.; Rawlings, S.; Smith, D.; Das, S.; Strich, J.R.; Chertow, D.S.; Davey, R.T., Jr.; Cohen, J.I. Detection of nucleocapsid antibody to SARS-CoV-2 is more sensitive than antibody to spike protein in COVID-19 patients. medRxiv 2020. [Google Scholar] [CrossRef]
- Guo, L.; Ren, L.; Yang, S.; Xiao, M.; Chang, D.; Yang, F.; Dela Cruz, C.S.; Wang, Y.; Wu, C.; Xiao, Y.; et al. Profiling Early Humoral Response to Diagnose Novel Coronavirus Disease (COVID-19). Clin. Infect. Dis. 2020, 71, 778–785. [Google Scholar] [CrossRef] [Green Version]
- Smits, V.A.J.; Hernández-Carralero, E.; Paz-Cabrera, M.C.; Cabrera, E.; Hernández-Reyes, Y.; Hernández-Fernaud, J.R.; Gillespie, D.A.; Salido, E.; Hernández-Porto, M.; Freire, R. The Nucleocapsid protein triggers the main humoral immune response in COVID-19 patients. Biochem. Biophys. Res. Commun. 2021, 543, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Pajenda, S.; Kapps, S.; Reiter, T.; Freire, R.; Smits, V.A.J.; Wagner, L.; Gerges, D.; Winnicki, W.; Sunder-Plassmann, G.; Schmidt, A. Antibody Response against the SARS-CoV-2 Nucleocapsid Protein and Its Subdomains—Identification of Pre-Immunization Status by Human Coronaviruses with Multipanel Nucleocapsid Fragment Immunoblotting. COVID 2021, 1, 105–114. [Google Scholar] [CrossRef]
- Ladner, J.T.; Henson, S.N.; Boyle, A.S.; Engelbrektson, A.L.; Fink, Z.W.; Rahee, F.; D’Ambrozio, J.; Schaecher, K.E.; Stone, M.; Dong, W.; et al. Epitope-resolved profiling of the SARS-CoV-2 antibody response identifies cross-reactivity with endemic human coronaviruses. Cell Rep. Med. 2021, 2, 100189. [Google Scholar] [CrossRef]
- Nakanaga, K.; Yamanouchi, K.; Fujiwara, K. Protective effect of monoclonal antibodies on lethal mouse hepatitis virus infection in mice. J. Virol. 1986, 59, 168–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Q.; Bielefeldt-Ohmann, H.; Maison, R.M.; Masleša-Galić, S.; Cooper, S.K.; Bowen, R.A.; Horwitz, M.A. Replicating bacterium-vectored vaccine expressing SARS-CoV-2 Membrane and Nucleocapsid proteins protects against severe COVID-19-like disease in hamsters. npj Vaccines 2021, 6, 47. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, F.; Shen, C.; Peng, W.; Li, D.; Zhao, C.; Li, Z.; Li, S.; Bi, Y.; Yang, Y.; et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science 2020, 368, 1274–1278. [Google Scholar] [CrossRef]
- Premkumar, L.; Segovia-Chumbez, B.; Jadi, R.; Martinez, D.R.; Raut, R.; Markmann, A.; Cornaby, C.; Bartelt, L.; Weiss, S.; Park, Y.; et al. The receptor binding domain of the viral spike protein is an immunodominant and highly specific target of antibodies in SARS-CoV-2 patients. Sci. Immunol. 2020, 5, eabc8413. [Google Scholar] [CrossRef] [PubMed]
- Zuhorn, F.; Graf, T.; Klingebiel, R.; Schabitz, W.R.; Rogalewski, A. Postvaccinal Encephalitis after ChAdOx1 nCov-19. Ann. Neurol. 2021, 90, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Rzymski, P.; Perek, B.; Flisiak, R. Thrombotic Thrombocytopenia after COVID-19 Vaccination: In Search of the Underlying Mechanism. Vaccines 2021, 9, 559. [Google Scholar] [CrossRef]
- Greinacher, A.; Thiele, T.; Warkentin, T.E.; Weisser, K.; Kyrle, P.A.; Eichinger, S. Thrombotic Thrombocytopenia after ChAdOx1 nCov-19 Vaccination. N. Engl. J. Med. 2021, 384, 2092–2101. [Google Scholar] [CrossRef]
- Sterlin, D.; Mathian, A.; Miyara, M.; Mohr, A.; Anna, F.; Claër, L.; Quentric, P.; Fadlallah, J.; Devilliers, H.; Ghillani, P.; et al. IgA dominates the early neutralizing antibody response to SARS-CoV-2. Sci. Transl. Med. 2021, 13, eabd2223. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.-Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Hogenesch, H. Mechanism of immunopotentiation and safety of aluminum adjuvants. Front. Immunol. 2012, 3, 406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, L.; Gessl, A.; Parzer, S.B.; Base, W.; Waldhäusl, W.; Pasternack, M.S. Haptoglobin phenotyping by newly developed monoclonal antibodies. Demonstration of haptoglobin uptake into peripheral blood neutrophils and monocytes. J. Immunol. 1996, 156, 1989–1996. [Google Scholar] [PubMed]
- Mbow, M.L.; De Gregorio, E.; Valiante, N.M.; Rappuoli, R. New adjuvants for human vaccines. Curr. Opin. Immunol. 2010, 22, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Chappell, K.J.; Mordant, F.L.; Li, Z.; Wijesundara, D.K.; Ellenberg, P.; Lackenby, J.A.; Cheung, S.T.M.; Modhiran, N.; Avumegah, M.S.; Henderson, C.L.; et al. Safety and immunogenicity of an MF59-adjuvanted spike glycoprotein-clamp vaccine for SARS-CoV-2: A randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Infect. Dis 2021, 21, 1383–1394. [Google Scholar] [CrossRef]
- Chun, J.Y.; Park, S.; Jung, J.; Kim, S.-H.; Kim, T.-S.; Choi, Y.J.; Kim, H.J.; Eom, H.-S.; Hyun, J.-W. Guillain-Barré syndrome after vaccination against COVID-19. Lancet Neurol. 2022, 21, 117–119. [Google Scholar] [CrossRef]
- Sharifian-Dorche, M.; Bahmanyar, M.; Sharifian-Dorche, A.; Mohammadi, P.; Nomovi, M.; Mowla, A. Vaccine-induced immune thrombotic thrombocytopenia and cerebral venous sinus thrombosis post COVID-19 vaccination; a systematic review. J. Neurol. Sci. 2021, 428, 117607. [Google Scholar] [CrossRef]
- Stam, J. Thrombosis of the cerebral veins and sinuses. N. Engl. J. Med. 2005, 352, 1791–1798. [Google Scholar] [CrossRef] [Green Version]
- Schultz, N.H.; Sørvoll, I.H.; Michelsen, A.E.; Munthe, L.A.; Lund-Johansen, F.; Ahlen, M.T.; Wiedmann, M.; Aamodt, A.H.; Skattør, T.H.; Tjønnfjord, G.E.; et al. Thrombosis and Thrombocytopenia after ChAdOx1 nCoV-19 Vaccination. N. Engl. J. Med. 2021, 384, 2124–2130. [Google Scholar] [CrossRef]
- Härtig, W.; Reichenbach, A.; Voigt, C.; Boltze, J.; Bulavina, L.; Schuhmann, M.U.; Seeger, J.; Schusser, G.F.; Freytag, C.; Grosche, J. Triple fluorescence labelling of neuronal, glial and vascular markers revealing pathological alterations in various animal models. J. Chem. Neuroanat. 2009, 37, 128–138. [Google Scholar] [CrossRef]
- Bignami, A.; Eng, L.F.; Dahl, D.; Uyeda, C.T. Localization of the glial fibrillary acidic protein in astrocytes by immunofluorescence. Brain Res. 1972, 43, 429–435. [Google Scholar] [CrossRef]
- Sasaki, Y.; Ohsawa, K.; Kanazawa, H.; Kohsaka, S.; Imai, Y. Iba1 is an actin-cross-linking protein in macrophages/microglia. Biochem. Biophys. Res. Commun. 2001, 286, 292–297. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Boots, A.M.; Kusters, J.G.; van Noort, J.M.; Zwaagstra, K.A.; Rijke, E.; van der Zeijst, B.A.; Hensen, E.J. Localization of a T-cell epitope within the nucleocapsid protein of avian coronavirus. Immunology 1991, 74, 8–13. [Google Scholar]
- Wang, H.; Hou, X.; Wu, X.; Liang, T.; Zhang, X.; Wang, D.; Teng, F.; Dai, J.; Duan, H.; Guo, S.; et al. SARS-CoV-2 proteome microarray for mapping COVID-19 antibody interactions at amino acid resolution. bioRxiv 2020. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15. [Google Scholar] [CrossRef]
- Tian, J.H.; Patel, N.; Haupt, R.; Zhou, H.; Weston, S.; Hammond, H.; Logue, J.; Portnoff, A.D.; Norton, J.; Guebre-Xabier, M.; et al. SARS-CoV-2 spike glycoprotein vaccine candidate NVX-CoV2373 immunogenicity in baboons and protection in mice. Nat. Commun. 2021, 12, 372. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, J.; Van Rooijen, N.; Perlman, S. Evasion by Stealth: Inefficient Immune Activation Underlies Poor T Cell Response and Severe Disease in SARS-CoV-Infected Mice. PLoS Pathog. 2009, 5, e1000636. [Google Scholar] [CrossRef]
- See, R.H.; Petric, M.; Lawrence, D.J.; Mok, C.P.Y.; Rowe, T.; Zitzow, L.A.; Karunakaran, K.P.; Voss, T.G.; Brunham, R.C.; Gauldie, J.; et al. Severe acute respiratory syndrome vaccine efficacy in ferrets: Whole killed virus and adenovirus-vectored vaccines. J. Gen. Virol. 2008, 89, 2136–2146. [Google Scholar] [CrossRef]
- Shrock, E.; Fujimura, E.; Kula, T.; Timms, R.T.; Lee, I.H.; Leng, Y.; Robinson, M.L.; Sie, B.M.; Li, M.Z.; Chen, Y.; et al. Viral epitope profiling of COVID-19 patients reveals cross-reactivity and correlates of severity. Science 2020, 370, eabd4250. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hevesi, Z.; Gerges, D.A.; Kapps, S.; Freire, R.; Schmidt, S.; Pollak, D.D.; Schmetterer, K.; Frey, T.; Lang, R.; Winnicki, W.; et al. Preclinical Establishment of a Divalent Vaccine against SARS-CoV-2. Vaccines 2022, 10, 516. https://doi.org/10.3390/vaccines10040516
Hevesi Z, Gerges DA, Kapps S, Freire R, Schmidt S, Pollak DD, Schmetterer K, Frey T, Lang R, Winnicki W, et al. Preclinical Establishment of a Divalent Vaccine against SARS-CoV-2. Vaccines. 2022; 10(4):516. https://doi.org/10.3390/vaccines10040516
Chicago/Turabian StyleHevesi, Zsofia, Daniela Anna Gerges, Sebastian Kapps, Raimundo Freire, Sophie Schmidt, Daniela D. Pollak, Klaus Schmetterer, Tobias Frey, Rita Lang, Wolfgang Winnicki, and et al. 2022. "Preclinical Establishment of a Divalent Vaccine against SARS-CoV-2" Vaccines 10, no. 4: 516. https://doi.org/10.3390/vaccines10040516
APA StyleHevesi, Z., Gerges, D. A., Kapps, S., Freire, R., Schmidt, S., Pollak, D. D., Schmetterer, K., Frey, T., Lang, R., Winnicki, W., Schmidt, A., Harkany, T., & Wagner, L. (2022). Preclinical Establishment of a Divalent Vaccine against SARS-CoV-2. Vaccines, 10(4), 516. https://doi.org/10.3390/vaccines10040516