Transcriptome Analyses of lncRNAs in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa

 ,
,  ,
, 


Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. MTT Assay
2.3. Total RNA Sequencing
2.4. Quality Assessment and Read Alignment
2.5. Gene Expression Quantification and Normalization
2.6. Filtering and Annotation of Non-Coding RNAs
2.7. Long Non-Coding RNAs Alignment-Free Algorithms of Analysis
2.8. Specific Circular RNAs Detection Pipelines
2.9. Differential lncRNAs Expression and Statistical Analysis
2.10. lncRNAs Validation by qRT-PCR
2.11. lncRNA Host and Target Genes Pathway Analysis
2.12. Pathway Analysis of microRNA Targeting to Most Altered RPE Expressed lncRNAs
3. Results
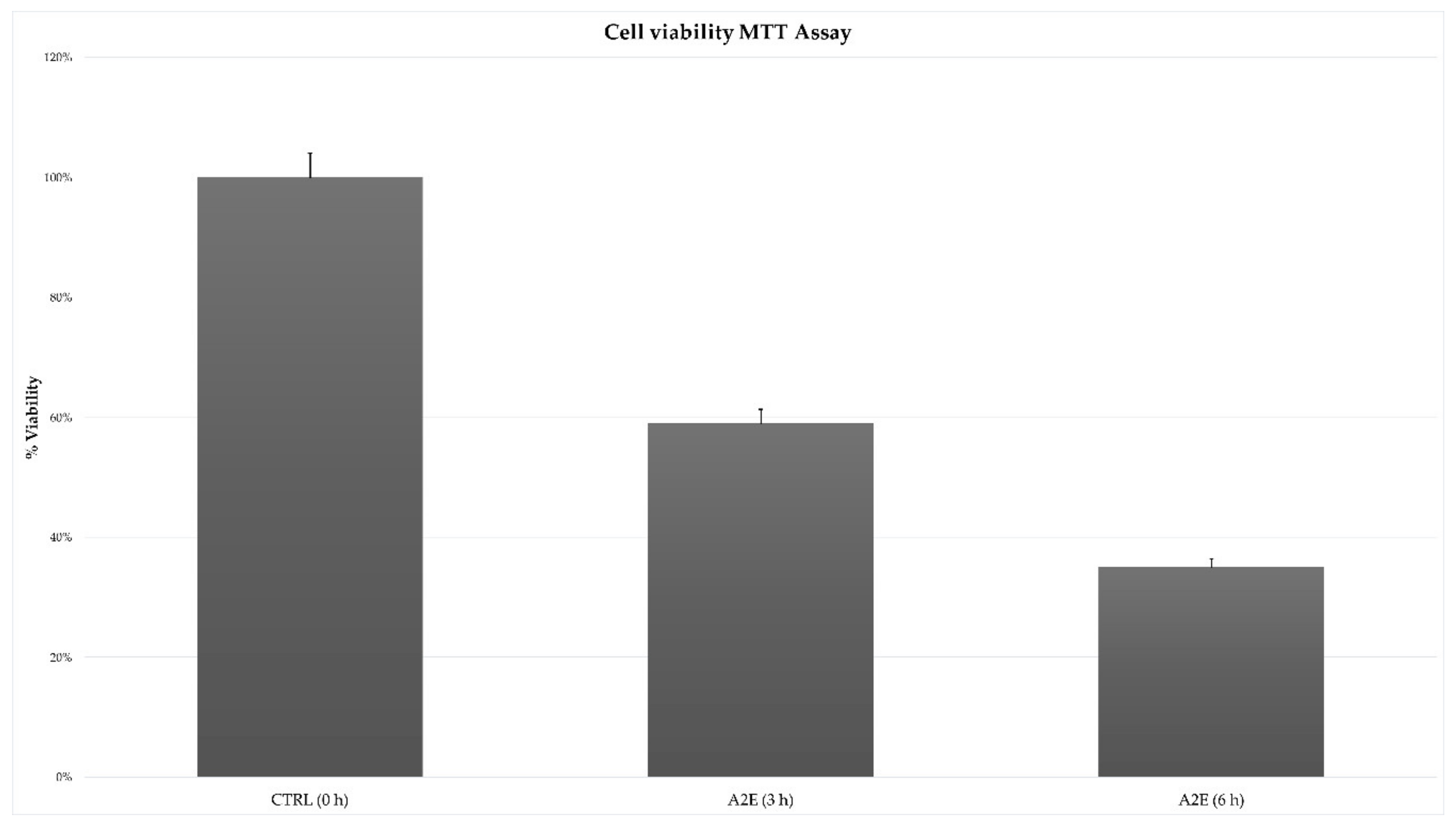
3.1. MTT Cell Viability Assay Showed an Exposure Time-Related Increased Death
3.2. Sequencing and Differential Expression Analyses Highlighted a Prevalence of Down-Regulated lncRNAs upon Up-Regulated Ones
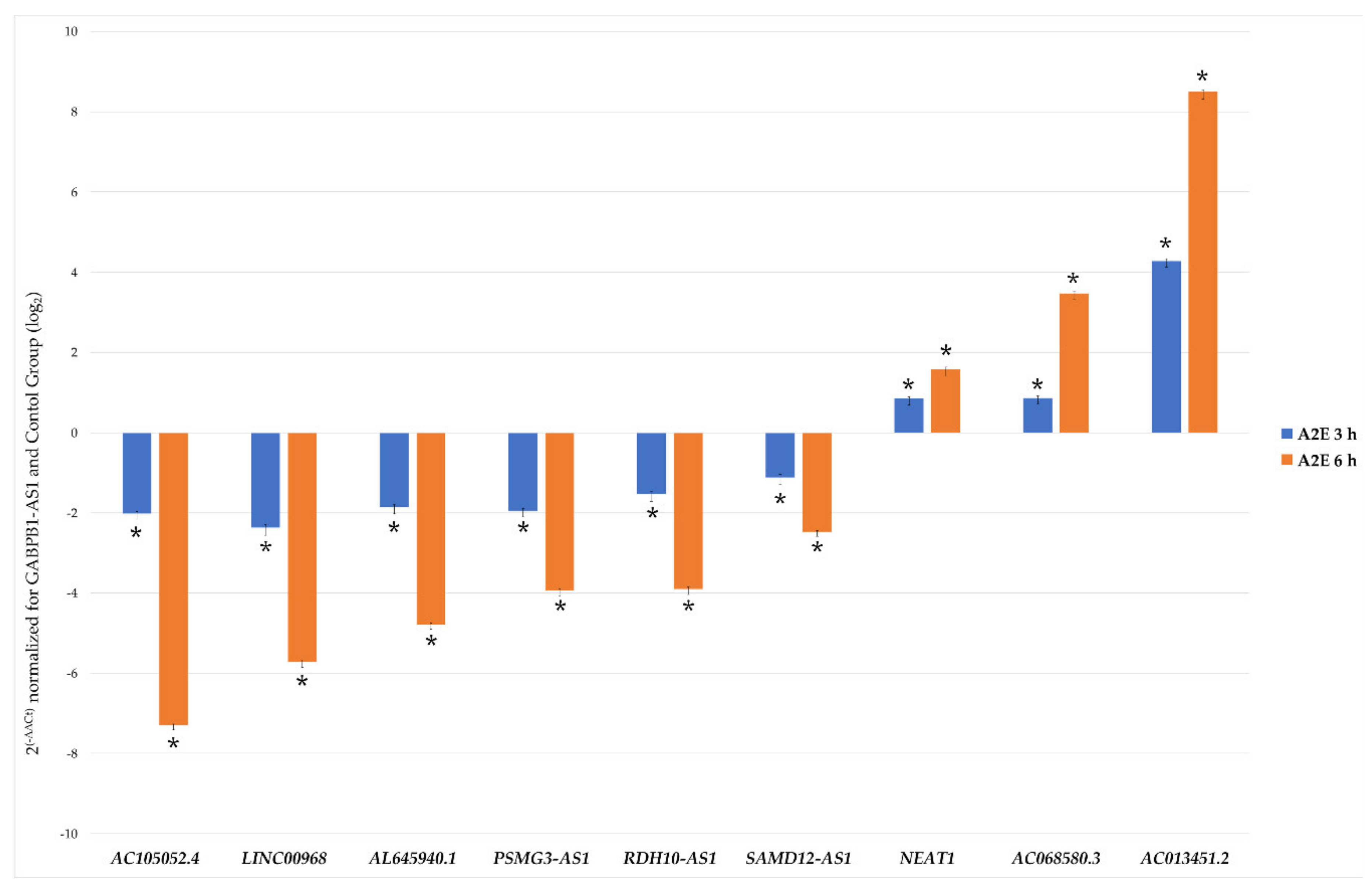
3.3. Lnc-RNAs Validation by qRT-PCR
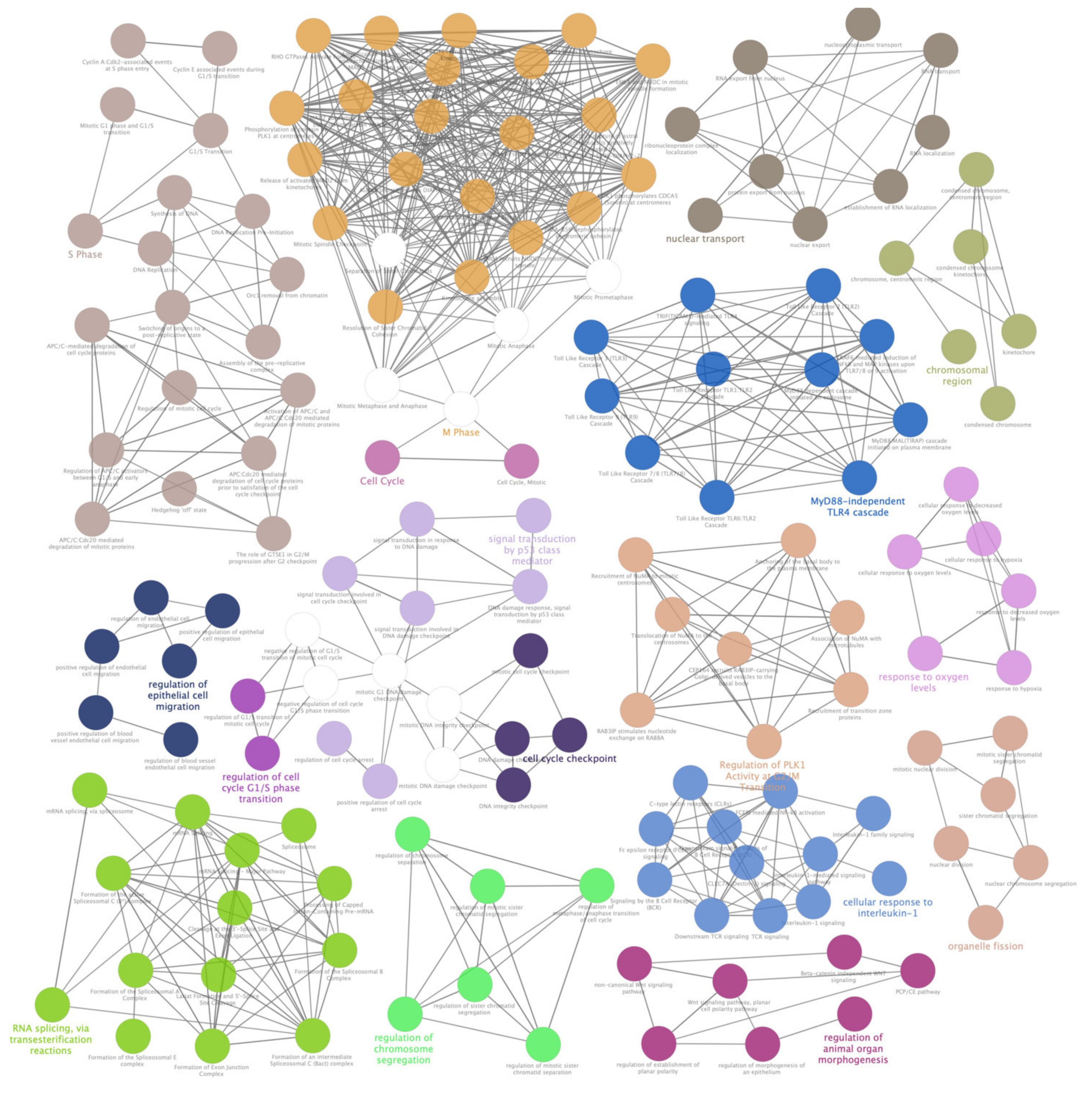
3.4. Pathway Analysis of Selected lncRNAs Target and Host Genes Shed Light on Metabolic Pathways Impaired by Induced Oxidative Stress
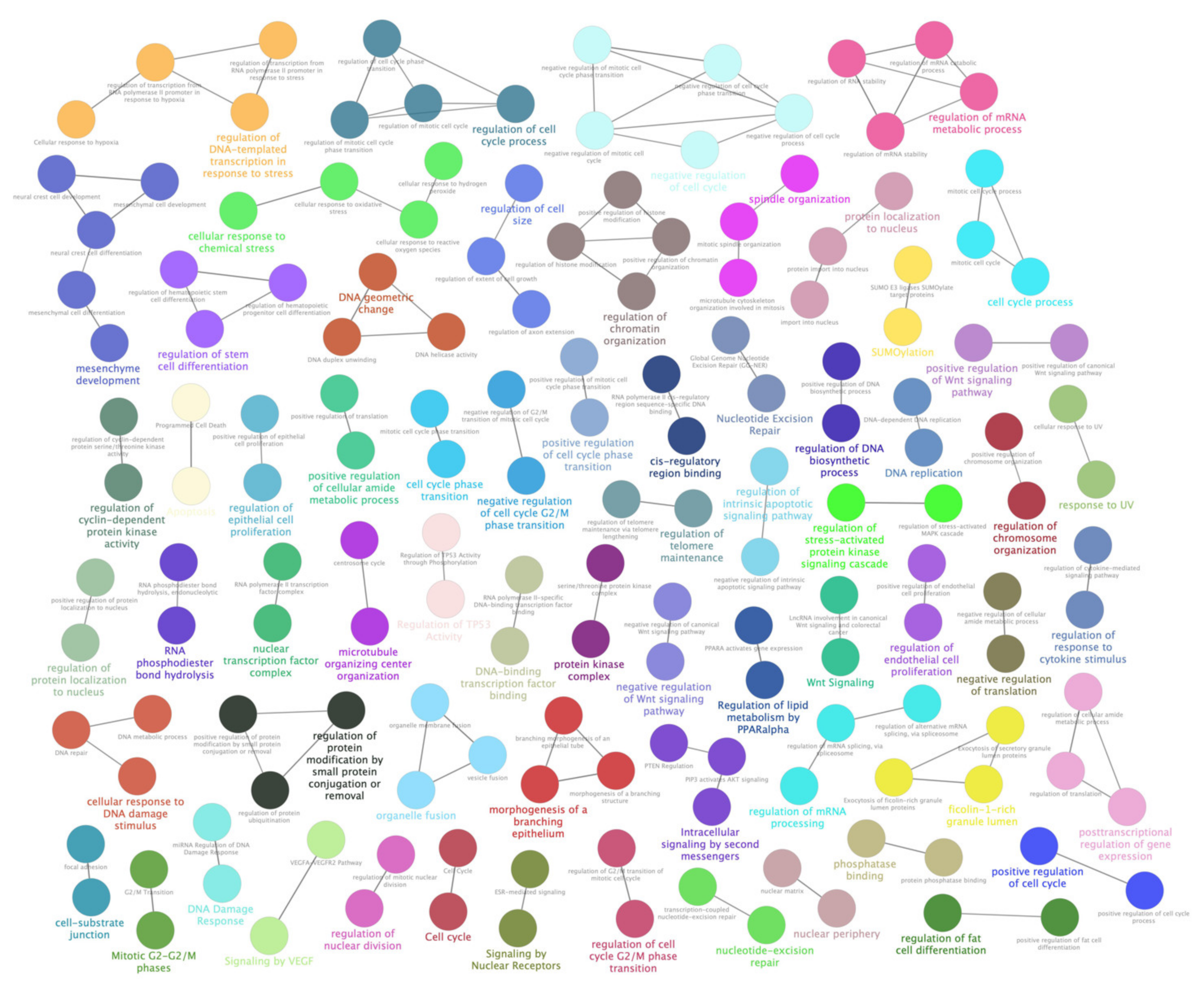
3.5. LncRNA-miRNA Predicted Interactions Enforced the Hypothesis of RPE Cellular Metabolism Damages Induced by Oxidative Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Deposition
References
- Hombach, S.; Kretz, M. Non-coding RNAs: Classification, Biology and Functioning. Adv. Exp. Med. Biol. 2016, 937, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Tsagakis, I.; Douka, K.; Birds, I.; Aspden, J.L. Long non-coding RNAs in development and disease: Conservation to mechanisms. J. Pathol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gaiti, F.; Calcino, A.D.; Tanurdzic, M.; Degnan, B.M. Origin and evolution of the metazoan non-coding regulatory genome. Dev. Biol. 2017, 427, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Jarroux, J.; Morillon, A.; Pinskaya, M. History, Discovery, and Classification of lncRNAs. Adv. Exp. Med. Biol. 2017, 1008, 1–46. [Google Scholar] [CrossRef]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef]
- Ransohoff, J.D.; Wei, Y.; Khavari, P.A. The functions and unique features of long intergenic non-coding RNA. Nat. Rev. Mol. Cell Biol. 2018, 19, 143–157. [Google Scholar] [CrossRef]
- Belousova, E.A.; Filipenko, M.L.; Kushlinskii, N.E. Circular RNA: New Regulatory Molecules. Bull. Exp. Biol. Med. 2018, 164, 803–815. [Google Scholar] [CrossRef]
- Hamazaki, N.; Nakashima, K.; Hayashi, K.; Imamura, T. Detection of Bidirectional Promoter-Derived lncRNAs from Small-Scale Samples Using Pre-Amplification-Free Directional RNA-seq Method. Methods Mol. Biol. 2017, 1605, 83–103. [Google Scholar] [CrossRef]
- Dykes, I.M.; Emanueli, C. Transcriptional and Post-transcriptional Gene Regulation by Long Non-coding RNA. Genom. Proteom. Bioinform. 2017, 15, 177–186. [Google Scholar] [CrossRef]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef]
- Dahariya, S.; Paddibhatla, I.; Kumar, S.; Raghuwanshi, S.; Pallepati, A.; Gutti, R.K. Long non-coding RNA: Classification, biogenesis and functions in blood cells. Mol. Immunol. 2019, 112, 82–92. [Google Scholar] [CrossRef]
- Jain, S.; Thakkar, N.; Chhatai, J.; Pal Bhadra, M.; Bhadra, U. Long non-coding RNA: Functional agent for disease traits. RNA Biol. 2017, 14, 522–535. [Google Scholar] [CrossRef] [PubMed]
- Wawrzyniak, O.; Zarebska, Z.; Rolle, K.; Gotz-Wieckowska, A. Circular and long non-coding RNAs and their role in ophthalmologic diseases. Acta Biochim. Pol. 2018, 65, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.; Scimone, C.; Rinaldi, C.; D’Angelo, R.; Sidoti, A. Non-coding RNAome of RPE cells under oxidative stress suggests unknown regulative aspects of Retinitis pigmentosa etiopathogenesis. Sci. Rep. 2018, 8, 16638. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Jiang, C.; Qin, B.; Liu, G.; Ji, J.; Sun, X.; Xu, M.; Ding, S.; Zhu, M.; Huang, G.; et al. LncRNA ZNF503-AS1 promotes RPE differentiation by downregulating ZNF503 expression. Cell Death Dis. 2017, 8, e3046. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dong, Y.; Wang, Y.; Gao, J.; Lv, J.; Sun, J.; Li, M.; Wang, M.; Zhao, Z.; Wang, J.; et al. Long non-coding RNAs in ocular diseases: New and potential therapeutic targets. FEBS J. 2019, 286, 2261–2272. [Google Scholar] [CrossRef]
- Rochet, E.; Appukuttan, B.; Ma, Y.; Ashander, L.M.; Smith, J.R. Expression of Long Non-Coding RNAs by Human Retinal Muller Glial Cells Infected with Clonal and Exotic Virulent Toxoplasma gondii. Noncod. RNA 2019, 5. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Yao, J.; Liu, C.; Hu, H.T.; Li, X.M.; Ge, H.M.; Zhou, Y.F.; Shan, K.; Jiang, Q.; Yan, B. Long non-coding RNA MEG3 silencing protects against light-induced retinal degeneration. Biochem. Biophys. Res. Commun. 2018, 496, 1236–1242. [Google Scholar] [CrossRef]
- Yang, M.; Wei, W. Long non-coding RNAs in retinoblastoma. Pathol. Res. Pract. 2019, 215, 152435. [Google Scholar] [CrossRef]
- Cisse, Y.; Bai, L.; Chen, M.T. LncRNAs in ocular neovascularizations. Int. J. Ophthalmol. 2019, 12, 1959–1965. [Google Scholar] [CrossRef]
- Matsui, M.; Corey, D.R. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 2017, 16, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, P.A.; Strauss, R.W.; Lu, L.; Hafiz, G.; Wolfson, Y.; Shah, S.M.; Sophie, R.; Mir, T.A.; Scholl, H.P. Is There Excess Oxidative Stress and Damage in Eyes of Patients with Retinitis Pigmentosa? Antioxid. Redox Signal. 2015, 23, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.R.; Fishkin, N.; Zhou, J.; Cai, B.; Jang, Y.P.; Krane, S.; Itagaki, Y.; Nakanishi, K. A2E, a byproduct of the visual cycle. Vision Res. 2003, 43, 2983–2990. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 21 March 2020).
- Okonechnikov, K.; Conesa, A.; Garcia-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- CLC Genomics Workbench 20.0. Available online: https://digitalinsights.qiagen.com (accessed on 21 March 2020).
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2010, 26, 493–500. [Google Scholar] [CrossRef]
- Pereira, M.B.; Wallroth, M.; Jonsson, V.; Kristiansson, E. Comparison of normalization methods for the analysis of metagenomic gene abundance data. BMC Genom. 2018, 19, 274. [Google Scholar] [CrossRef]
- Casper, J.; Zweig, A.S.; Villarreal, C.; Tyner, C.; Speir, M.L.; Rosenbloom, K.R.; Raney, B.J.; Lee, C.M.; Lee, B.T.; Karolchik, D.; et al. The UCSC Genome Browser database: 2018 update. Nucleic Acids Res. 2018, 46, D762–D769. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Giron, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef]
- Volders, P.J.; Verheggen, K.; Menschaert, G.; Vandepoele, K.; Martens, L.; Vandesompele, J.; Mestdagh, P. An update on LNCipedia: A database for annotated human lncRNA sequences. Nucleic Acids Res. 2015, 43, D174–D180. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Liu, T.; Cui, T.; Wang, Z.; Zhang, Y.; Tan, P.; Huang, Y.; Yu, J.; Wang, D. RNAInter in 2020: RNA interactome repository with increased coverage and annotation. Nucleic Acids Res. 2020, 48, D189–D197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Tan, P.; Wang, L.; Jin, N.; Li, Y.; Zhang, L.; Yang, H.; Hu, Z.; Zhang, L.; Hu, C.; et al. RNALocate: A resource for RNA subcellular localizations. Nucleic Acids Res. 2017, 45, D135–D138. [Google Scholar] [CrossRef]
- Cui, T.; Zhang, L.; Huang, Y.; Yi, Y.; Tan, P.; Zhao, Y.; Hu, Y.; Xu, L.; Li, E.; Wang, D. MNDR v2.0: An updated resource of ncRNA-disease associations in mammals. Nucleic Acids Res. 2018, 46, D371–D374. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Huang, Y.; Kang, J.; Li, K.; Bi, X.; Zhang, T.; Jin, N.; Hu, Y.; Tan, P.; Zhang, L.; et al. ncRDeathDB: A comprehensive bioinformatics resource for deciphering network organization of the ncRNA-mediated cell death system. Autophagy 2015, 11, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Rappaport, N.; Fishilevich, S.; Nudel, R.; Twik, M.; Belinky, F.; Plaschkes, I.; Stein, T.I.; Cohen, D.; Oz-Levi, D.; Safran, M.; et al. Rational confederation of genes and diseases: NGS interpretation via GeneCards, MalaCards and VarElect. Biomed. Eng. Online 2017, 16, 72. [Google Scholar] [CrossRef]
- Braschi, B.; Denny, P.; Gray, K.; Jones, T.; Seal, R.; Tweedie, S.; Yates, B.; Bruford, E. Genenames.org: The HGNC and VGNC resources in 2019. Nucleic Acids Res. 2019, 47, D786–D792. [Google Scholar] [CrossRef]
- Paraskevopoulou, M.D.; Vlachos, I.S.; Karagkouni, D.; Georgakilas, G.; Kanellos, I.; Vergoulis, T.; Zagganas, K.; Tsanakas, P.; Floros, E.; Dalamagas, T.; et al. DIANA-LncBase v2: Indexing microRNA targets on non-coding transcripts. Nucleic Acids Res. 2016, 44, D231–D238. [Google Scholar] [CrossRef]
- Bao, Z.; Yang, Z.; Huang, Z.; Zhou, Y.; Cui, Q.; Dong, D. LncRNADisease 2.0: An updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2019, 47, D1034–D1037. [Google Scholar] [CrossRef]
- Cheng, L.; Wang, P.; Tian, R.; Wang, S.; Guo, Q.; Luo, M.; Zhou, W.; Liu, G.; Jiang, H.; Jiang, Q. LncRNA2Target v2.0: A comprehensive database for target genes of lncRNAs in human and mouse. Nucleic Acids Res. 2019, 47, D140–D144. [Google Scholar] [CrossRef]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Song, X.; Wang, K. lncScore: Alignment-free identification of long noncoding RNA from assembled novel transcripts. Sci. Rep. 2016, 6, 34838. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [PubMed]
- Wucher, V.; Legeai, F.; Hedan, B.; Rizk, G.; Lagoutte, L.; Leeb, T.; Jagannathan, V.; Cadieu, E.; David, A.; Lohi, H.; et al. FEELnc: A tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 2017, 45, e57. [Google Scholar] [CrossRef] [PubMed]
- Szabo, L.; Morey, R.; Palpant, N.J.; Wang, P.L.; Afari, N.; Jiang, C.; Parast, M.M.; Murry, C.E.; Laurent, L.C.; Salzman, J. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 2015, 16, 126. [Google Scholar] [CrossRef]
- Zhang, X.O.; Dong, R.; Zhang, Y.; Zhang, J.L.; Luo, Z.; Zhang, J.; Chen, L.L.; Yang, L. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 2016, 26, 1277–1287. [Google Scholar] [CrossRef]
- Song, X.; Zhang, N.; Han, P.; Moon, B.S.; Lai, R.K.; Wang, K.; Lu, W. Circular RNA profile in gliomas revealed by identification tool UROBORUS. Nucleic Acids Res. 2016, 44, e87. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Guo, W.T.N.; Stephen, G.; Milne, I.; Calixto, C.; Waugh, R.; Brown, J.W.; Zhang, R. 3D RNA-seq—A powerful and flexible tool for rapid and accurate differential expression and alternative splicing analysis of RNA-seq data for biologists. bioRxiv 2019, 656686. [Google Scholar] [CrossRef]
- Ge, Y.; Sealfon, S.C.; Speed, T.P. Some Step-down Procedures Controlling the False Discovery Rate under Dependence. Stat. Sin. 2008, 18, 881–904. [Google Scholar] [PubMed]
- Silver, N.; Best, S.; Jiang, J.; Thein, S.L. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol. 2006, 7, 33. [Google Scholar] [CrossRef]
- Andersen, C.L.; Jensen, J.L.; Orntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper--Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.ibm.com/analytics/spss-statistics-software (accessed on 21 March 2012).
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Bindea, G.; Galon, J.; Mlecnik, B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Hon, C.C.; Ramilowski, J.A.; Harshbarger, J.; Bertin, N.; Rackham, O.J.; Gough, J.; Denisenko, E.; Schmeier, S.; Poulsen, T.M.; Severin, J.; et al. An atlas of human long non-coding RNAs with accurate 5’ ends. Nature 2017, 543, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Sanchez Calle, A.; Kawamura, Y.; Yamamoto, Y.; Takeshita, F.; Ochiya, T. Emerging roles of long non-coding RNA in cancer. Cancer Sci. 2018, 109, 2093–2100. [Google Scholar] [CrossRef] [PubMed]
- Ruhle, F.; Stoll, M. Long non-coding RNA Databases in Cardiovascular Research. Genom. Proteom. Bioinform. 2016, 14, 191–199. [Google Scholar] [CrossRef]
- Li, L.; Zhuang, Y.; Zhao, X.; Li, X. Long Non-coding RNA in Neuronal Development and Neurological Disorders. Front. Genet. 2018, 9, 744. [Google Scholar] [CrossRef]
- Vencken, S.F.; Greene, C.M.; McKiernan, P.J. Non-coding RNA as lung disease biomarkers. Thorax 2015, 70, 501–503. [Google Scholar] [CrossRef]
- Riva, P.; Ratti, A.; Venturin, M. The Long Non-Coding RNAs in Neurodegenerative Diseases: Novel Mechanisms of Pathogenesis. Curr. Alzheimer Res. 2016, 13, 1219–1231. [Google Scholar] [CrossRef]
- Birtel, J.; Gliem, M.; Oishi, A.; Muller, P.L.; Herrmann, P.; Holz, F.G.; Mangold, E.; Knapp, M.; Bolz, H.J.; Charbel Issa, P. Genetic testing in patients with retinitis pigmentosa: Features of unsolved cases. Clin. Exp. Ophthalmol. 2019, 47, 779–786. [Google Scholar] [CrossRef]
- Karali, M.; Banfi, S. Non-coding RNAs in retinal development and function. Hum. Genet. 2019, 138, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Strettoi, E.; Gargini, C.; Novelli, E.; Sala, G.; Piano, I.; Gasco, P.; Ghidoni, R. Inhibition of ceramide biosynthesis preserves photoreceptor structure and function in a mouse model of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2010, 107, 18706–18711. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.; Scimone, C.; Nicocia, G.; D’Angelo, R.; Sidoti, A. Role of oxidative stress in Retinitis pigmentosa: New involved pathways by an RNA-Seq analysis. Cell Cycle 2019, 18, 84–104. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.; Scimone, C.; Nicocia, G.; Denaro, L.; Robledo, R.; Sidoti, A.; D’Angelo, R. GLO1 gene polymorphisms and their association with retinitis pigmentosa: A case-control study in a Sicilian population. Mol. Biol. Rep. 2018, 45, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Shinjo, K.; Katsushima, K. Long non-coding RNAs as an epigenetic regulator in human cancers. Cancer Sci. 2017, 108, 1927–1933. [Google Scholar] [CrossRef] [PubMed]
- Manelyte, L.; Strohner, R.; Gross, T.; Langst, G. Chromatin targeting signals, nucleosome positioning mechanism and non-coding RNA-mediated regulation of the chromatin remodeling complex NoRC. PLoS Genet. 2014, 10, e1004157. [Google Scholar] [CrossRef]
- An, H.; Williams, N.G.; Shelkovnikova, T.A. NEAT1 and paraspeckles in neurodegenerative diseases: A missing lnc found? Noncod. RNA Res. 2018, 3, 243–252. [Google Scholar] [CrossRef]
- Zhang, X.; Hamblin, M.H.; Yin, K.J. The long noncoding RNA Malat1: Its physiological and pathophysiological functions. RNA Biol. 2017, 14, 1705–1714. [Google Scholar] [CrossRef]
- Yao, J.; Wang, X.Q.; Li, Y.J.; Shan, K.; Yang, H.; Wang, Y.N.; Yao, M.D.; Liu, C.; Li, X.M.; Shen, Y.; et al. Long non-coding RNA MALAT1 regulates retinal neurodegeneration through CREB signaling. EMBO Mol. Med. 2016, 8, 346–362. [Google Scholar] [CrossRef]
- Li, X.J. Long non-coding RNA nuclear paraspeckle assembly transcript 1 inhibits the apoptosis of retina Muller cells after diabetic retinopathy through regulating miR-497/brain-derived neurotrophic factor axis. Diab. Vasc. Dis. Res. 2018, 15, 204–213. [Google Scholar] [CrossRef]
- Thapar, R. Regulation of DNA Double-Strand Break Repair by Non-Coding RNAs. Molecules 2018, 23, 2789. [Google Scholar] [CrossRef]
- Dhanoa, J.K.; Sethi, R.S.; Verma, R.; Arora, J.S.; Mukhopadhyay, C.S. Long non-coding RNA: Its evolutionary relics and biological implications in mammals: A review. J. Anim. Sci. Technol. 2018, 60, 25. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, Y.; Sun, Y.; Qin, L.; Yang, Y. LncRNA TUG1 affects cell viability by regulating glycolysis in osteosarcoma cells. Gene 2018, 674, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, M.; Yang, H.; Mao, L.; He, Q.; Jin, H.; Ye, Z.M.; Luo, X.Y.; Xia, Y.P.; Hu, B. LncRNA TUG1 sponges microRNA-9 to promote neurons apoptosis by up-regulated Bcl2l11 under ischemia. Biochem. Biophys. Res. Commun. 2017, 485, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, F.; Xiao, H.; Han, F. Long noncoding RNA BDNF-AS inversely regulated BDNF and modulated high-glucose induced apoptosis in human retinal pigment epithelial cells. J. Cell Biochem. 2018, 119, 817–823. [Google Scholar] [CrossRef]
- Millar, C.A.; Powell, K.A.; Hickson, G.R.; Bader, M.F.; Gould, G.W. Evidence for a role for ADP-ribosylation factor 6 in insulin-stimulated glucose transporter-4 (GLUT4) trafficking in 3T3-L1 adipocytes. J. Biol. Chem. 1999, 274, 17619–17625. [Google Scholar] [CrossRef]
- Ellis, B.C.; Graham, L.D.; Molloy, P.L. CRNDE, a long non-coding RNA responsive to insulin/IGF signaling, regulates genes involved in central metabolism. Biochim. Biophys. Acta 2014, 1843, 372–386. [Google Scholar] [CrossRef]
- Zhang, Y.; Xi, X.; Mei, Y.; Zhao, X.; Zhou, L.; Ma, M.; Liu, S.; Zha, X.; Yang, Y. High-glucose induces retinal pigment epithelium mitochondrial pathways of apoptosis and inhibits mitophagy by regulating ROS/PINK1/Parkin signal pathway. Biomed. Pharmacother. 2019, 111, 1315–1325. [Google Scholar] [CrossRef]
- Tarchick, M.J.; Cutler, A.H.; Trobenter, T.D.; Kozlowski, M.R.; Makowski, E.R.; Holoman, N.; Shao, J.; Shen, B.; Anand-Apte, B.; Samuels, I.S. Endogenous insulin signaling in the RPE contributes to the maintenance of rod photoreceptor function in diabetes. Exp. Eye Res. 2019, 180, 63–74. [Google Scholar] [CrossRef]
- Kang, M.K.; Lee, E.J.; Kim, Y.H.; Kim, D.Y.; Oh, H.; Kim, S.I.; Kang, Y.H. Chrysin Ameliorates Malfunction of Retinoid Visual Cycle through Blocking Activation of AGE-RAGE-ER Stress in Glucose-Stimulated Retinal Pigment Epithelial Cells and Diabetic Eyes. Nutrients 2018, 10, 1046. [Google Scholar] [CrossRef]
- Kuan, C.T.; Chang, J.; Mansson, J.E.; Li, J.; Pegram, C.; Fredman, P.; McLendon, R.E.; Bigner, D.D. Multiple phenotypic changes in mice after knockout of the B3gnt5 gene, encoding Lc3 synthase--a key enzyme in lacto-neolacto ganglioside synthesis. BMC Dev. Biol. 2010, 10, 114. [Google Scholar] [CrossRef]
- Jun, S.; Datta, S.; Wang, L.; Pegany, R.; Cano, M.; Handa, J.T. The impact of lipids, lipid oxidation, and inflammation on AMD, and the potential role of miRNAs on lipid metabolism in the RPE. Exp. Eye Res. 2019, 181, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, F.; Mao, Y.; Finnemann, S.C. Advanced Analysis of Photoreceptor Outer Segment Phagocytosis by RPE Cells in Culture. Methods Mol. Biol. 2019, 1834, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.D.; Ng, S.Y.; Lloyd, M.; Eddington, S.; Sun, H.; Nathans, J.; Bok, D.; Radu, R.A.; Travis, G.H. Peropsin modulates transit of vitamin A from retina to retinal pigment epithelium. J. Biol. Chem. 2017, 292, 21407–21416. [Google Scholar] [CrossRef] [PubMed]
- Tachikawa, M.; Akanuma, S.I.; Imai, T.; Okayasu, S.; Tomohiro, T.; Hatanaka, Y.; Hosoya, K.I. Multiple Cellular Transport and Binding Processes of Unesterified Docosahexaenoic Acid in Outer Blood-Retinal Barrier Retinal Pigment Epithelial Cells. Biol. Pharm. Bull. 2018, 41, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Storti, F.; Raphael, G.; Griesser, V.; Klee, K.; Drawnel, F.; Willburger, C.; Scholz, R.; Langmann, T.; Von Eckardstein, A.; Fingerle, J.; et al. Regulated efflux of photoreceptor outer segment-derived cholesterol by human RPE cells. Exp. Eye Res. 2017, 165, 65–77. [Google Scholar] [CrossRef]
- Biswas, L.; Zhou, X.; Dhillon, B.; Graham, A.; Shu, X. Retinal pigment epithelium cholesterol efflux mediated by the 18 kDa translocator protein, TSPO, a potential target for treating age-related macular degeneration. Hum. Mol. Genet. 2017, 26, 4327–4339. [Google Scholar] [CrossRef]
- Yanagi, Y. Role of Peoxisome Proliferator Activator Receptor gamma on Blood Retinal Barrier Breakdown. PPAR Res. 2008, 2008, 679237. [Google Scholar] [CrossRef]
- Ershov, A.V.; Bazan, N.G. Photoreceptor phagocytosis selectively activates PPARgamma expression in retinal pigment epithelial cells. J. Neurosci. Res. 2000, 60, 328–337. [Google Scholar] [CrossRef]
- Lin, J.H.; Lavail, M.M. Misfolded proteins and retinal dystrophies. Adv. Exp. Med. Biol. 2010, 664, 115–121. [Google Scholar] [CrossRef]
- Sokolov, M.; Yadav, R.P.; Brooks, C.; Artemyev, N.O. Chaperones and retinal disorders. Adv. Protein Chem. Struct. Biol. 2019, 114, 85–117. [Google Scholar] [CrossRef]
- Lundkvist, A.; Reichenbach, A.; Betsholtz, C.; Carmeliet, P.; Wolburg, H.; Pekny, M. Under stress, the absence of intermediate filaments from Muller cells in the retina has structural and functional consequences. J. Cell Sci. 2004, 117, 3481–3488. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, R.; Ranchon-Cole, I.; Paris, A.; Herzine, A.; Perche, A.; Laurenceau, D.; Bertrand, P.; Cercy, C.; Pichon, J.; Mortaud, S.; et al. Visual sensorial impairments in neurodevelopmental disorders: Evidence for a retinal phenotype in Fragile X Syndrome. PLoS ONE 2014, 9, e105996. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Saxena, S.; Ruia, S.; Prasad, S.; Singh, V.; Khanna, V.; Staffa, R.; Gaspar, L.; Kruzliak, P. Increased levels of N(epsilon)- Carboxy methyl lysine (N(epsilon)-CML) are associated with topographic alterations in retinal pigment epithelium: A preliminary study. J. Diabetes Complicat. 2016, 30, 868–872. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Gene ID (ENSEMBL) | Primer Forward (5′ → 3′) | Primer Reverse (5′ → 3′) | Length (bp) | TM (°C) |
|---|---|---|---|---|---|
| AC105052.4 | ENSG00000279168 | GTGTGATAAGATACTGCACTTGG | GGATTTCGCCACGTTGCC | 131 | 61 |
| LINC00968 | ENSG00000246430 | CCACCATCCCATTGAGAACC | TTAGCTGGGAAGGATGAATGC | 108 | 60 |
| AL645940.1 | ENSG00000272217 | TAGGCTTAGGGTGGGTCAGG | TTGTCTGGTGGCAAGATCCC | 132 | 62 |
| PSMG3-AS1 | ENSG00000230487 | GGAAATGTGGGAGGGATGGC | GGGCTCCGACATTGAAGATGG | 137 | 63 |
| RDH10-AS1 | ENSG00000250295 | TGACTACAGCGAGCAACAGC | TCCACTGAGACGGAAACTGC | 138 | 62.5 |
| SAMD12-AS1 | ENSG00000281641 | CAAGGGAGGCAGGACTTTACG | AGTGTCCCTGATGCGAAACG | 125 | 63 |
| GABPB1-AS1 | ENSG00000244879 | TGTCTCATCTCAGTTTCCACAGG | GCAGCACTCTAATCCATCAGC | 120 | 62 |
| NEAT1 | ENSG00000245532 | TCATGAGCGAAGTGAAATTGC | AATAGACGCAGCTCAGAACC | 110 | 60 |
| AC068580.3 | ENSG00000235027 | CGCGCTAGGACAATCAGG | GGAAGCCCAAGACTCACAGG | 107 | 63 |
| AC013451.2 | ENSG00000258976 | CCAACTCAAACCAAATGAAGGG | CCGAGGTGCCTGTAACATCC | 126 | 62 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donato, L.; Scimone, C.; Alibrandi, S.; Rinaldi, C.; Sidoti, A.; D’Angelo, R. Transcriptome Analyses of lncRNAs in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa. Antioxidants 2020, 9, 318. https://doi.org/10.3390/antiox9040318
Donato L, Scimone C, Alibrandi S, Rinaldi C, Sidoti A, D’Angelo R. Transcriptome Analyses of lncRNAs in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa. Antioxidants. 2020; 9(4):318. https://doi.org/10.3390/antiox9040318
Chicago/Turabian StyleDonato, Luigi, Concetta Scimone, Simona Alibrandi, Carmela Rinaldi, Antonina Sidoti, and Rosalia D’Angelo. 2020. "Transcriptome Analyses of lncRNAs in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa" Antioxidants 9, no. 4: 318. https://doi.org/10.3390/antiox9040318
APA StyleDonato, L., Scimone, C., Alibrandi, S., Rinaldi, C., Sidoti, A., & D’Angelo, R. (2020). Transcriptome Analyses of lncRNAs in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa. Antioxidants, 9(4), 318. https://doi.org/10.3390/antiox9040318