
Antioxidant Systems as Modulators of Ferroptosis: Focus on Transcription Factors


 , and
, and 
Abstract
1. Introduction
2. Ferroptosis as a Biological Program: Features and General Mechanisms
2.1. Role of Iron in Ferroptosis
2.2. Iron Accumulation and Lipid Peroxidation
2.3. Lipid Peroxidation by Non-Enzymatic and Enzymatic Reactions
2.4. Ferroptosis and Physio-Pathological Processes
2.4.1. Ferroptosis in Aging
2.4.2. Ferroptosis in Cancer Cells
2.4.3. Ferroptosis in Leukemia
2.4.4. Ferroptosis in Heart Failure
2.4.5. Ferroptosis in Neurodegenerative Diseases
3. Redox Homeostasis and Antioxidant Systems: Links to Ferroptosis
3.1. ROS/RNS and Redox Homeostasis in Ferroptosis
3.1.1. ROS/RNS Sources in Ferroptosis
3.1.2. Process of Lipid Peroxidation
- (1)
- The radical-chain process (or radical chain reaction) consists of three sequential non-enzymatic events: initiation, propagation, and termination (Figure 2). Free reactive oxygen-centered radicals, in particular HO• and HOO•, mainly originate from Fenton reactions and can initiate the lipid peroxidation process of different types of PL-PUFAs, frequently those present in biological membranes. In fact, by abstracting a hydrogen atom from a methylene carbon inside the acyl chain double bounds, HO• and HOO• leave an unpaired electron on the carbon, generating a reactive carbon-centered lipid radical (PL-PUFA•); this last can also originate in a spontaneous manner inside the acyl chain harboring the “C=C” double bond (autooxidation/autocatalytic) [130]. Usually, this lipid radical undergoes molecular rearrangement to form an internal conjugated diene, which by reacting with O2 rapidly produces a lipid peroxyl radical (PL-PUFA-OO•). These molecules can subsequently remove hydrogen atoms from adjacent PL-PUFA chains or can combine with each other in several ways (propagation event), producing lipid hydroperoxides (PL-PUFA-OOH), especially if they encounter metals like iron or copper, that by Fenton reactions can also push lipid autoxidation [131]. Thus, if not interrupted by chain-breaking antioxidants (a termination step), an internal radical propagation and peroxidation of lipid radical species can result in a large amount of lipid peroxides [132,133]. The comparatively low dissociation energy of O-O bonds causes PL-PUFA-OOH cleavage, which produces a variety of secondary oxidation products, many of which have an oxygen-containing functionality and a shortened carbon chain with strong electrophilic tendencies. The most common electrophiles are aldehydic, oxo-, and epoxy groups, which can be found at various points along the hydrocarbon chain. Either the truncated phospholipid or the remaining shorter PUFA fragment can form an electrophilic group. Furthermore, persistent exposure to iron and/or copper leads to the decomposition of lipid hydroperoxides with the production of harmful carbonyl compounds like unsaturated 4-hydroxynonenal (4-HNE), malondialdehyde (MDA), and acrolein or end products [134] that can further foster destabilization of cell membranes, finally evoking the breakdown of membrane integrity and consequently ferroptosis [135,136].
- (2)
- The second mechanism involves the non-radical singlet oxygen (1O2), originating from O2 by light energy transfer and/or from endogenous enzymatic reactions involving COX, LOX, and myeloperoxidase enzymes, or from photosensitizer endogenous agents like bilirubin, porphyrins, flavins, pterins, melanin/melanin precursors, vitamin K, and B6 vitamers that could absorb light and transmit energy to O2 producing 1O2 (Figure 2) [137,138,139,140]. By representing an excited state of molecular oxygen, singlet oxygen is highly electrophilic and can rapidly react with various molecules, including unsaturated lipids, especially those present in cell membranes. The mechanism differs from lipid free radical autoxidation in that the singlet oxygen directly reacts with the double bond producing lipid peroxyl radicals, more similar to the reaction performed by the HOO• radical.
- (3)
- Lipid peroxidation by direct enzymatic mechanisms principally involves members of the lipoxygenase (LOX) family coupled to ACSL4/LPCAT3 activity and the oxidoreductase NADPH-cytochrome P450 reductase (POR) isoforms as well as fatty acyl-CoA reductase1 (FAR1), as discussed in the next section.
3.1.3. Enzyme-Mediated Lipid Peroxidation (Figure 2 and Figure 4)
LOX Enzymes
ACSL4 and LPCAT3 Enzymes
POR and CYB5R1 Enzymes
FAR1 Enzymes
3.2. Antioxidant Defenses and Lipid Peroxidation
3.2.1. Mechanisms That Prevent or Intercept ROS/RNS and Lipid Peroxides
3.2.2. Enzymatic Mechanisms That Protect against Lipid Peroxidation
GPX4/Glutathione/Cysteine
Peroxiredoxin 6 (H2O2 and Lipid Peroxides Decomposition and Repair of Membrane)
HMGCR/Mevalonate Pathway (Production of IPP and CoQ10)
FSP1/Coenzyme Q10 (GSH-Independent)
Mitochondrial DHODH/GPD2/CoQ10 (GSH-Independent)
GCH1/Tetrahydrobiopterin (BH4)
Microsomal Glutathione Transferase 1 (MGST1) (Inactivation of ALOX5 and/or Autophagy)
AKR1C Family Members (Scavenging of HNE)
3.3. Other Protective Mechanisms (Composition of Phospholipid Membranes)
3.3.1. Stearoyl-CoA Desaturase 1 (SCD1)
3.3.2. Acyl-CoA Synthetase Long Chain Family Member 3 (ACSL3)
3.3.3. MBOAT1 and MBOAT2/LPCAT4
4. Antioxidant Systems and Transcription Factors
4.1. NRF2 Pathway
4.2. ATF Signaling
4.3. TFAP2 Pathway
4.4. JAK-STAT Pathway
4.5. NF-κB Pathway
4.6. p53 Pathway
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, S.; Shen, J.; Jiang, J.; Wang, F.; Min, J. Targeting Ferroptosis Opens New Avenues for the Development of Novel Therapeutics. Signal Transduct. Target. Ther. 2023, 8, 372. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Y.; Min, J.; Wang, F. Zooming in and out of Ferroptosis in Human Disease. Front. Med. 2023, 17, 173–206. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat. Rev. Mol. Cell Biol 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.; Zhuang, L.; Gan, B. Targeting Ferroptosis as a Vulnerability in Cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, W.; Abdul Razak, S.R.; Han, T.; Ahmad, N.H.; Li, X. Ferroptosis as a Potential Target for Cancer Therapy. Cell Death Dis. 2023, 14, 460. [Google Scholar] [CrossRef]
- Xu, Y.; Zhao, J.; Zhao, Y.; Zhou, L.; Qiao, H.; Xu, Q.; Liu, Y. The Role of Ferroptosis in Neurodegenerative Diseases. Mol. Biol. Rep. 2023, 50, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Coradduzza, D.; Congiargiu, A.; Chen, Z.; Zinellu, A.; Carru, C.; Medici, S. Ferroptosis and Senescence: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 3658. [Google Scholar] [CrossRef]
- Chen, Z.; Jiang, J.; Fu, N.; Chen, L. Targetting Ferroptosis for Blood Cell-Related Diseases. J. Drug Target. 2022, 30, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, Y.; Zhou, S.; Feng, Q.; Lu, Y.; Liu, D.; Liu, Z. Novel Insight into Ferroptosis in Kidney Diseases. Am. J. Nephrol. 2023, 54, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Ardehali, H.; Min, J.; Wang, F. The Molecular and Metabolic Landscape of Iron and Ferroptosis in Cardiovascular Disease. Nat. Rev. Cardiol. 2023, 20, 7–23. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Pope, L.E.; Dixon, S.J. Regulation of Ferroptosis by Lipid Metabolism. Trends Cell Biol. 2023, 33, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R. Ferroptosis Turns 10: Emerging Mechanisms, Physiological Functions, and Therapeutic Applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Chen, X.; Li, J.; Comish, P.; Kang, R.; Tang, D. Transcription Factors in Ferroptotic Cell Death. Cancer Gene Ther. 2020, 27, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Lin, B.; Jin, W.; Tang, L.; Hu, S.; Cai, R. NRF2, a Superstar of Ferroptosis. Antioxidants 2023, 12, 1739. [Google Scholar] [CrossRef] [PubMed]
- Kist, M.; Vucic, D. Cell Death Pathways: Intricate Connections and Disease Implications. EMBO J. 2021, 40, e106700. [Google Scholar] [CrossRef]
- Land, W.G. Regulated Cell Death. In Damage-Associated Molecular Patterns in Human Diseases; Springer International Publishing: Cham, Switzerland, 2018; pp. 427–466. ISBN 978-3-319-78654-4. [Google Scholar]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxidative Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Metodiewa, D.; Kośka, C. Reactive Oxygen Species and Reactive Nitrogen Species: Relevance to Cyto(Neuro)Toxic Events and Neurologic Disorders. An Overview. Neurotox. Res. 2000, 1, 197–233. [Google Scholar] [CrossRef]
- Bayır, H.; Anthonymuthu, T.S.; Tyurina, Y.Y.; Patel, S.J.; Amoscato, A.A.; Lamade, A.M.; Yang, Q.; Vladimirov, G.K.; Philpott, C.C.; Kagan, V.E. Achieving Life through Death: Redox Biology of Lipid Peroxidation in Ferroptosis. Cell Chem. Biol. 2020, 27, 387–408. [Google Scholar] [CrossRef]
- Torii, S.; Shintoku, R.; Kubota, C.; Yaegashi, M.; Torii, R.; Sasaki, M.; Suzuki, T.; Mori, M.; Yoshimoto, Y.; Takeuchi, T.; et al. An Essential Role for Functional Lysosomes in Ferroptosis of Cancer Cells. Biochem. J. 2016, 473, 769–777. [Google Scholar] [CrossRef]
- Armenta, D.A.; Laqtom, N.N.; Alchemy, G.; Dong, W.; Morrow, D.; Poltorack, C.D.; Nathanson, D.A.; Abu-Remalieh, M.; Dixon, S.J. Ferroptosis Inhibition by Lysosome-Dependent Catabolism of Extracellular Protein. Cell Chem. Biol. 2022, 29, 1588–1600.e7. [Google Scholar] [CrossRef]
- Sousa, L.; Oliveira, M.M.; Pessôa, M.T.C.; Barbosa, L.A. Iron Overload: Effects on Cellular Biochemistry. Clin. Chim. Acta 2020, 504, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Yiannikourides, A.; Latunde-Dada, G.O. A Short Review of Iron Metabolism and Pathophysiology of Iron Disorders. Medicines 2019, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Lanceta, L.; Li, C.; Choi, A.M.; Eaton, J.W. Haem Oxygenase-1 Overexpression Alters Intracellular Iron Distribution. Biochem. J. 2013, 449, 189–194. [Google Scholar] [CrossRef]
- Kwon, M.-Y.; Park, E.; Lee, S.-J.; Chung, S.W. Heme Oxygenase-1 Accelerates Erastin-Induced Ferroptotic Cell Death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef]
- Chang, L.-C.; Chiang, S.-K.; Chen, S.-E.; Yu, Y.-L.; Chou, R.-H.; Chang, W.-C. Heme Oxygenase-1 Mediates BAY 11-7085 Induced Ferroptosis. Cancer Lett. 2018, 416, 124–137. [Google Scholar] [CrossRef]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayır, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-Targeted Induction of Dual Ferroptotic Mechanisms Eradicates High-Risk Neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, J.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Tang, D. Ferroptosis Is a Type of Autophagy-Dependent Cell Death. Semin. Cancer Biol. 2020, 66, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular Mechanisms and Health Implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron Homeostasis and Oxidative Stress: An Intimate Relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef] [PubMed]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 Oxidoreductase Contributes to Phospholipid Peroxidation in Ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef]
- Carlsen, C.U.; Møller, J.K.S.; Skibsted, L.H. Heme-Iron in Lipid Oxidation. Coord. Chem. Rev. 2005, 249, 485–498. [Google Scholar] [CrossRef]
- Hajeyah, A.A.; Griffiths, W.J.; Wang, Y.; Finch, A.J.; O’Donnell, V.B. The Biosynthesis of Enzymatically Oxidized Lipids. Front. Endocrinol. 2020, 11, 591819. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Kim, W.K.; Bae, K.-H.; Lee, S.C.; Lee, E.-W. Lipid Metabolism and Ferroptosis. Biology 2021, 10, 184. [Google Scholar] [CrossRef]
- Wu, P.; Zhang, X.; Duan, D.; Zhao, L. Organelle-Specific Mechanisms in Crosstalk between Apoptosis and Ferroptosis. Oxidative Med. Cell. Longev. 2023, 2023, 3400147. [Google Scholar] [CrossRef]
- Mortensen, M.S.; Ruiz, J.; Watts, J.L. Polyunsaturated Fatty Acids Drive Lipid Peroxidation during Ferroptosis. Cells 2023, 12, 804. [Google Scholar] [CrossRef]
- Zhang, X.; Ning, X.; He, X.; Sun, X.; Yu, X.; Cheng, Y.; Yu, R.-Q.; Wu, Y. Fatty Acid Composition Analyses of Commercially Important Fish Species from the Pearl River Estuary, China. PLoS ONE 2020, 15, e0228276. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Lee, J.-Y.; Oh, M.; Lee, E.-W. An Integrated View of Lipid Metabolism in Ferroptosis Revisited via Lipidomic Analysis. Exp. Mol. Med. 2023, 55, 1620–1631. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Conrad, M. Iron and Ferroptosis: A Still Ill-defined Liaison. IUBMB Life 2017, 69, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lu, S.; Wu, L.-L.; Yang, L.; Yang, L.; Wang, J. The Diversified Role of Mitochondria in Ferroptosis in Cancer. Cell Death Dis. 2023, 14, 519. [Google Scholar] [CrossRef] [PubMed]
- Gan, B. Mitochondrial Regulation of Ferroptosis. J. Cell Biol. 2021, 220, e202105043. [Google Scholar] [CrossRef]
- Kagan, V.E.; Tyurina, Y.Y.; Sun, W.Y.; Vlasova, I.I.; Dar, H.; Tyurin, V.A.; Amoscato, A.A.; Mallampalli, R.; Van Der Wel, P.C.A.; He, R.R.; et al. Redox Phospholipidomics of Enzymatically Generated Oxygenated Phospholipids as Specific Signals of Programmed Cell Death. Free Radic. Biol. Med. 2020, 147, 231–241. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological Inhibition of Cystine-Glutamate Exchange Induces Endoplasmic Reticulum Stress and Ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Hu, F.; Feng, H.; Linkermann, A.; Min, W.; Stockwell, B.R. Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. ACS Chem. Biol. 2018, 13, 1013–1020. [Google Scholar] [CrossRef]
- von Krusenstiern, A.N.; Robson, R.N.; Qian, N.; Qiu, B.; Hu, F.; Reznik, E.; Smith, N.; Zandkarimi, F.; Estes, V.M.; Dupont, M.; et al. Identification of Essential Sites of Lipid Peroxidation in Ferroptosis. Nat. Chem. Biol. 2023, 19, 719–730. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Lettieri-Barbato, D.; Aquilano, K.; Punziano, C.; Minopoli, G.; Faraonio, R. MicroRNAs, Long Non-Coding RNAs, and Circular RNAs in the Redox Control of Cell Senescence. Antioxidants 2022, 11, 480. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, E.; Morales-Pison, S.; Urbina, F.; Solari, A. Aging Hallmarks and the Role of Oxidative Stress. Antioxidants 2023, 12, 651. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Z.; Ren, Y.; Wang, Y.; Fang, J.; Yue, H.; Ma, S.; Guan, F. Aging and Age-related Diseases: From Mechanisms to Therapeutic Strategies. Biogerontology 2021, 22, 165–187. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, R.S.; Han, S.M.; Leeuwenburgh, C.; Xiao, R. Iron Homeostasis and Organismal Aging. Ageing Res. Rev. 2021, 72, 101510. [Google Scholar] [CrossRef]
- Mazhar, M.; Din, A.U.; Ali, H.; Yang, G.; Ren, W.; Wang, L.; Fan, X.; Yang, S. Implication of Ferroptosis in Aging. Cell Death Discov. 2021, 7, 149. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Wang, H.; Tian, W.; Zeng, J.; Huang, Y.; Luo, H. Targeting Ferroptosis for Cancer Therapy: Iron Metabolism and Anticancer Immunity. Am. J. Cancer Res. 2021, 11, 5508–5525. [Google Scholar]
- Zhang, R.; Chen, J.; Wang, S.; Zhang, W.; Zheng, Q.; Cai, R. Ferroptosis in Cancer Progression. Cells 2023, 12, 1820. [Google Scholar] [CrossRef]
- Feng, S.; Tang, D.; Wang, Y.; Li, X.; Bao, H.; Tang, C.; Dong, X.; Li, X.; Yang, Q.; Yan, Y.; et al. The Mechanism of Ferroptosis and Its Related Diseases. Mol. Biomed. 2023, 4, 33. [Google Scholar] [CrossRef]
- Chen, H.; Wang, C.; Liu, Z.; He, X.; Tang, W.; He, L.; Feng, Y.; Liu, D.; Yin, Y.; Li, T. Ferroptosis and Its Multifaceted Role in Cancer: Mechanisms and Therapeutic Approach. Antioxidants 2022, 11, 1504. [Google Scholar] [CrossRef]
- Trombetti, S.; Cesaro, E.; Catapano, R.; Sessa, R.; Lo Bianco, A.; Izzo, P.; Grosso, M. Oxidative Stress and ROS-Mediated Signaling in Leukemia: Novel Promising Perspectives to Eradicate Chemoresistant Cells in Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 2470. [Google Scholar] [CrossRef]
- Trombetti, S.; Iaccarino, N.; Riccio, P.; Sessa, R.; Catapano, R.; Salvatore, M.; Luka, S.; de Nicola, S.; Izzo, P.; Roperto, S.; et al. Over-Expressed GATA-1S, the Short Isoform of the Hematopoietic Transcriptional Factor GATA-1, Inhibits Ferroptosis in K562 Myeloid Leukemia Cells by Preventing Lipid Peroxidation. Antioxidants 2023, 12, 537. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Fan, X.; Zhang, X.; Ju, S. Ferroptosis in Tumors and Its Relationship to Other Programmed Cell Death: Role of Non-Coding RNAs. J. Transl. Med. 2023, 21, 514. [Google Scholar] [CrossRef]
- Zhang, L.; Hou, N.; Chen, B.; Kan, C.; Han, F.; Zhang, J.; Sun, X. Post-Translational Modifications of P53 in Ferroptosis: Novel Pharmacological Targets for Cancer Therapy. Front. Pharmacol. 2022, 13, 908772. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gu, W. P53 in Ferroptosis Regulation: The New Weapon for the Old Guardian. Cell Death Differ. 2022, 29, 895–910. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wang, W.; Zhang, W. Ferroptosis and the Bidirectional Regulatory Factor P53. Cell Death Discov. 2023, 9, 197. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Cai, H.; Ren, Y.; Chen, S.; Wang, Y.; Chu, L. Ferroptosis and Tumor Immunotherapy: A Promising Combination Therapy for Tumors. Front. Oncol. 2023, 13, 1119369. [Google Scholar] [CrossRef]
- Wang, S.; He, X.; Wu, Q.; Jiang, L.; Chen, L.; Yu, Y.; Zhang, P.; Huang, X.; Wang, J.; Ju, Z.; et al. Transferrin Receptor 1-Mediated Iron Uptake Plays an Essential Role in Hematopoiesis. Haematologica 2020, 105, 2071–2082. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, C.; Sun, Q.; Li, Y.; Zhou, C.; Sun, C. Susceptibility of Acute Myeloid Leukemia Cells to Ferroptosis and Evasion Strategies. Front. Mol. Biosci. 2023, 10, 1275774. [Google Scholar] [CrossRef]
- Hole, P.S.; Darley, R.L.; Tonks, A. Do Reactive Oxygen Species Play a Role in Myeloid Leukemias? Blood 2011, 117, 5816–5826. [Google Scholar] [CrossRef]
- Udensi, U.K.; Tchounwou, P.B. Dual Effect of Oxidative Stress on Leukemia Cancer Induction and Treatment. J. Exp. Clin. Cancer Res. 2014, 33, 106. [Google Scholar] [CrossRef] [PubMed]
- Khoshtabiat, L.; Mahdavi, M.; Dehghan, G.; Rashidi, M.R. Oxidative Stress-Induced Apoptosis in Chronic Myelogenous Leukemia K562 Cells by an Active Compound from the Dithio- Carbamate Family. Asian Pac. J. Cancer Prev. 2016, 17, 4267–4273. [Google Scholar] [PubMed]
- Kaweme, N.M.; Zhou, S.; Changwe, G.J.; Zhou, F. The Significant Role of Redox System in Myeloid Leukemia: From Pathogenesis to Therapeutic Applications. Biomark. Res. 2020, 8, 63. [Google Scholar] [CrossRef]
- Weber, S.; Parmon, A.; Kurrle, N.; Schnütgen, F.; Serve, H. The Clinical Significance of Iron Overload and Iron Metabolism in Myelodysplastic Syndrome and Acute Myeloid Leukemia. Front. Immunol. 2020, 11, 627662. [Google Scholar] [CrossRef]
- Ma, W.; Wei, S.; Zhang, B.; Li, W. Molecular Mechanisms of Cardiomyocyte Death in Drug-Induced Cardiotoxicity. Front. Cell Dev. Biol. 2020, 8, 434. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Zhang, X.; Huang, B.; Wang, S.; Zhou, W.; Li, C.; Li, X.; Wang, J.; Yang, N. Disulfiram, a Ferroptosis Inducer, Triggers Lysosomal Membrane Permeabilization by Up-Regulating ROS in Glioblastoma. Onco Targets Ther. 2020, 13, 10631–10640. [Google Scholar] [CrossRef] [PubMed]
- Shirazi, L.F.; Bissett, J.; Romeo, F.; Mehta, J.L. Role of Inflammation in Heart Failure. Curr. Atheroscler. Rep. 2017, 19, 27. [Google Scholar] [CrossRef]
- Huang, S.; Frangogiannis, N.G. Anti-Inflammatory Therapies in Myocardial Infarction: Failures, Hopes and Challenges. Br. J. Pharmacol. 2018, 175, 1377–1400. [Google Scholar] [CrossRef]
- Luo, M.-Y.; Su, J.-H.; Gong, S.-X.; Liang, N.; Huang, W.-Q.; Chen, W.; Wang, A.-P.; Tian, Y. Ferroptosis: New Dawn for Overcoming the Cardio-Cerebrovascular Diseases. Front. Cell Dev. Biol. 2021, 9, 733908. [Google Scholar] [CrossRef]
- Liu, B.; Zhao, C.; Li, H.; Chen, X.; Ding, Y.; Xu, S. Puerarin Protects against Heart Failure Induced by Pressure Overload through Mitigation of Ferroptosis. Biochem. Biophys. Res. Commun. 2018, 497, 233–240. [Google Scholar] [CrossRef]
- Qin, Y.; Qiao, Y.; Wang, D.; Tang, C.; Yan, G. Ferritinophagy and Ferroptosis in Cardiovascular Disease: Mechanisms and Potential Applications. Biomed. Pharmacother. 2021, 141, 111872. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, J. Ferroptosis: A New Strategy for Cardiovascular Disease. Front. Cardiovasc. Med. 2023, 10, 1241282. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Zhu, W.; Wei, L.; Zhao, J. Iron Homeostasis Imbalance and Ferroptosis in Brain Diseases. MedComm 2023, 4, e298. [Google Scholar] [CrossRef]
- Lin, K.-J.; Chen, S.-D.; Lin, K.-L.; Liou, C.-W.; Lan, M.-Y.; Chuang, Y.-C.; Wang, P.-W.; Lee, J.-J.; Wang, F.-S.; Lin, H.-Y.; et al. Iron Brain Menace: The Involvement of Ferroptosis in Parkinson Disease. Cells 2022, 11, 3829. [Google Scholar] [CrossRef]
- Ndayisaba, A.; Kaindlstorfer, C.; Wenning, G.K. Iron in Neurodegeneration-Cause or Consequence? Front. Neurosci. 2019, 13, 180. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Zheng, K.; Li, S.; Ren, C.; Shen, Y.; Tian, L.; Zhu, H.; Zhou, Z.; Jiang, Y. Insight into the Potential Role of Ferroptosis in Neurodegenerative Diseases. Front. Cell. Neurosci. 2022, 16, 1005182. [Google Scholar] [CrossRef]
- Reichert, C.O.; de Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; de Lima Barros, P.; Levy, D.; Bydlowski, S.P. Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8765. [Google Scholar] [CrossRef]
- Patanè, G.T.; Putaggio, S.; Tellone, E.; Barreca, D.; Ficarra, S.; Maffei, C.; Calderaro, A.; Laganà, G. Ferroptosis: Emerging Role in Diseases and Potential Implication of Bioactive Compounds. Int. J. Mol. Sci. 2023, 24, 17279. [Google Scholar] [CrossRef]
- Ding, X.; Gao, L.; Han, Z.; Eleuteri, S.; Shi, W.; Shen, Y.; Song, Z.; Su, M.; Yang, Q.; Qu, Y.; et al. Ferroptosis in Parkinson’s Disease: Molecular Mechanisms and Therapeutic Potential. Ageing Res. Rev. 2023, 91, 102077. [Google Scholar] [CrossRef]
- Zhou, Y.; Lin, W.; Rao, T.; Zheng, J.; Zhang, T.; Zhang, M.; Lin, Z. Ferroptosis and Its Potential Role in the Nervous System Diseases. JIR 2022, 15, 1555–1574. [Google Scholar] [CrossRef]
- Yu, Y.; Yan, Y.; Niu, F.; Wang, Y.; Chen, X.; Su, G.; Liu, Y.; Zhao, X.; Qian, L.; Liu, P.; et al. Ferroptosis: A Cell Death Connecting Oxidative Stress, Inflammation and Cardiovascular Diseases. Cell Death Discov. 2021, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Kong, X.-Y.; Yao, Y.; Wang, X.-A.; Yang, W.; Wu, H.; Li, S.; Ding, J.-W.; Yang, J. The Critical Role and Molecular Mechanisms of Ferroptosis in Antioxidant Systems: A Narrative Review. Ann. Transl. Med. 2022, 10, 368. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen Peroxide as a Central Redox Signaling Molecule in Physiological Oxidative Stress: Oxidative Eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Tretter, V.; Hochreiter, B.; Zach, M.L.; Krenn, K.; Klein, K.U. Understanding Cellular Redox Homeostasis: A Challenge for Precision Medicine. Int. J. Mol. Sci. 2021, 23, 106. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Cueto, R.; Effi, C.; Zhang, Y.; Tan, H.; Qin, X.; Ji, Y.; Yang, X.; Wang, H. Biochemical Basis and Metabolic Interplay of Redox Regulation. Redox Biol. 2019, 26, 101284. [Google Scholar] [CrossRef]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Kodama, R.; Kato, M.; Furuta, S.; Ueno, S.; Zhang, Y.; Matsuno, K.; Yabe-Nishimura, C.; Tanaka, E.; Kamata, T. ROS-Generating Oxidases Nox1 and Nox4 Contribute to Oncogenic Ras-Induced Premature Senescence. Genes Cells 2013, 18, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Carroll, K.S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Skonieczna, M.; Hejmo, T.; Poterala-Hejmo, A.; Cieslar-Pobuda, A.; Buldak, R.J. NADPH Oxidases: Insights into Selected Functions and Mechanisms of Action in Cancer and Stem Cells. Oxidative Med. Cell. Longev. 2017, 2017, 9420539. [Google Scholar] [CrossRef] [PubMed]
- Park, M.W.; Cha, H.W.; Kim, J.; Kim, J.H.; Yang, H.; Yoon, S.; Boonpraman, N.; Yi, S.S.; Yoo, I.D.; Moon, J.-S. NOX4 Promotes Ferroptosis of Astrocytes by Oxidative Stress-Induced Lipid Peroxidation via the Impairment of Mitochondrial Metabolism in Alzheimer’s Diseases. Redox Biol. 2021, 41, 101947. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor P53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef]
- Yang, W.-H.; Ding, C.-K.C.; Sun, T.; Rupprecht, G.; Lin, C.-C.; Hsu, D.; Chi, J.-T. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep. 2019, 28, 2501–2508.e4. [Google Scholar] [CrossRef]
- Yang, W.-H.; Huang, Z.; Wu, J.; Ding, C.-K.C.; Murphy, S.K.; Chi, J.-T. A TAZ-ANGPTL4-NOX2 Axis Regulates Ferroptotic Cell Death and Chemoresistance in Epithelial Ovarian Cancer. Mol. Cancer Res. 2020, 18, 79–90. [Google Scholar] [CrossRef]
- Ju, H.-Q.; Lin, J.-F.; Tian, T.; Xie, D.; Xu, R.-H. NADPH Homeostasis in Cancer: Functions, Mechanisms and Therapeutic Implications. Signal Transduct. Target. Ther. 2020, 5, 231. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef]
- Suzuki, S.; Venkatesh, D.; Kanda, H.; Nakayama, A.; Hosokawa, H.; Lee, E.; Miki, T.; Stockwell, B.R.; Yokote, K.; Tanaka, T.; et al. GLS2 Is a Tumor Suppressor and a Regulator of Ferroptosis in Hepatocellular Carcinoma. Cancer Res. 2022, 82, 3209–3222. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Lee, J.; You, J.H.; Kim, D.; Roh, J.-L. Dihydrolipoamide Dehydrogenase Regulates Cystine Deprivation-Induced Ferroptosis in Head and Neck Cancer. Redox Biol. 2020, 30, 101418. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Goncalves, R.L.S.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-Oxoacid Dehydrogenase Complexes in Mitochondria Can Produce Superoxide/Hydrogen Peroxide at Much Higher Rates than Complex I. J. Biol. Chem. 2014, 289, 8312–8325. [Google Scholar] [CrossRef]
- Song, X.; Liu, J.; Kuang, F.; Chen, X.; Zeh, H.J.; Kang, R.; Kroemer, G.; Xie, Y.; Tang, D. PDK4 Dictates Metabolic Resistance to Ferroptosis by Suppressing Pyruvate Oxidation and Fatty Acid Synthesis. Cell Rep. 2021, 34, 108767. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial Membrane Potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, A.; Winge, D.R. Metal Acquisition and Availability in the Mitochondria. Chem. Rev. 2009, 109, 4708–4721. [Google Scholar] [CrossRef] [PubMed]
- Lipper, C.H.; Stofleth, J.T.; Bai, F.; Sohn, Y.-S.; Roy, S.; Mittler, R.; Nechushtai, R.; Onuchic, J.N.; Jennings, P.A. Redox-Dependent Gating of VDAC by mitoNEET. Proc. Natl. Acad. Sci. USA 2019, 116, 19924–19929. [Google Scholar] [CrossRef] [PubMed]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-Dependent Oxidative Cell Death Involving Voltage-Dependent Anion Channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-Mediated Ferroptosis Defence Is a Targetable Vulnerability in Cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Mao, C.; Kondiparthi, L.; Poyurovsky, M.V.; Olszewski, K.; Gan, B. A Ferroptosis Defense Mechanism Mediated by Glycerol-3-Phosphate Dehydrogenase 2 in Mitochondria. Proc. Natl. Acad. Sci. USA 2022, 119, e2121987119. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R.; Darash-Yahana, M.; Sohn, Y.S.; Bai, F.; Song, L.; Cabantchik, I.Z.; Jennings, P.A.; Onuchic, J.N.; Nechushtai, R. NEET Proteins: A New Link Between Iron Metabolism, Reactive Oxygen Species, and Cancer. Antioxid. Redox Signal. 2019, 30, 1083–1095. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. CISD1 Inhibits Ferroptosis by Protection against Mitochondrial Lipid Peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844. [Google Scholar] [CrossRef]
- Bagheri, M.; Nair, R.R.; Singh, K.K.; Saini, D.K. ATM-ROS-iNOS Axis Regulates Nitric Oxide Mediated Cellular Senescence. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 177–190. [Google Scholar] [CrossRef]
- Pourbagher-Shahri, A.M.; Farkhondeh, T.; Talebi, M.; Kopustinskiene, D.M.; Samarghandian, S.; Bernatoniene, J. An Overview of NO Signaling Pathways in Aging. Molecules 2021, 26, 4533. [Google Scholar] [CrossRef] [PubMed]
- Wink, D.A.; Miranda, K.M.; Espey, M.G.; Pluta, R.M.; Hewett, S.J.; Colton, C.; Vitek, M.; Feelisch, M.; Grisham, M.B. Mechanisms of the Antioxidant Effects of Nitric Oxide. Antioxid. Redox Signal. 2001, 3, 203–213. [Google Scholar] [CrossRef]
- Radi, R. Peroxynitrite, a Stealthy Biological Oxidant. J. Biol. Chem. 2013, 288, 26464–26472. [Google Scholar] [CrossRef] [PubMed]
- White, C.R.; Brock, T.A.; Chang, L.Y.; Crapo, J.; Briscoe, P.; Ku, D.; Bradley, W.A.; Gianturco, S.H.; Gore, J.; Freeman, B.A. Superoxide and Peroxynitrite in Atherosclerosis. Proc. Natl. Acad. Sci. USA 1994, 91, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Mastrogiovanni, M.; Trostchansky, A.; Rubbo, H. Fatty Acid Nitration in Human Low-Density Lipoprotein. Arch. Biochem. Biophys. 2020, 679, 108190. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Pratt, D.A. The Chemical Basis of Ferroptosis. Nat. Chem. Biol. 2019, 15, 1137–1147. [Google Scholar] [CrossRef]
- Do, Q.; Zhang, R.; Hooper, G.; Xu, L. Differential Contributions of Distinct Free Radical Peroxidation Mechanisms to the Induction of Ferroptosis. JACS Au 2023, 3, 1100–1117. [Google Scholar] [CrossRef]
- Yin, H.; Xu, L.; Porter, N.A. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid Peroxidation and Ferroptosis: The Role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Porter, N.A.; Schneider, C.; Brash, A.R.; Yin, H. Formation of 4-Hydroxynonenal from Cardiolipin Oxidation: Intramolecular Peroxyl Radical Addition and Decomposition. Free Radic. Biol. Med. 2011, 50, 166–178. [Google Scholar] [CrossRef]
- Riegman, M.; Sagie, L.; Galed, C.; Levin, T.; Steinberg, N.; Dixon, S.J.; Wiesner, U.; Bradbury, M.S.; Niethammer, P.; Zaritsky, A.; et al. Ferroptosis Occurs through an Osmotic Mechanism and Propagates Independently of Cell Rupture. Nat. Cell Biol. 2020, 22, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Fujii, J.; Soma, Y.; Matsuda, Y. Biological Action of Singlet Molecular Oxygen from the Standpoint of Cell Signaling, Injury and Death. Molecules 2023, 28, 4085. [Google Scholar] [CrossRef]
- Onyango, A.N. Endogenous Generation of Singlet Oxygen and Ozone in Human and Animal Tissues: Mechanisms, Biological Significance, and Influence of Dietary Components. Oxidative Med. Cell. Longev. 2016, 2016, 2398573. [Google Scholar] [CrossRef]
- Murotomi, K.; Umeno, A.; Shichiri, M.; Tanito, M.; Yoshida, Y. Significance of Singlet Oxygen Molecule in Pathologies. Int. J. Mol. Sci. 2023, 24, 2739. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.J.; Zhao, Y.; Yu, T.C.; Maytin, E.V.; Anand, S.; Hasan, T.; Pogue, B.W. Singlet Molecular Oxygen: From COIL Lasers to Photodynamic Cancer Therapy. J. Phys. Chem. B 2023, 127, 2289–2301. [Google Scholar] [CrossRef]
- Kuhn, H.; Banthiya, S.; van Leyen, K. Mammalian Lipoxygenases and Their Biological Relevance. Biochim. Biophys. Acta 2015, 1851, 308–330. [Google Scholar] [CrossRef]
- Schneider, C.; Pratt, D.A.; Porter, N.A.; Brash, A.R. Control of Oxygenation in Lipoxygenase and Cyclooxygenase Catalysis. Chem. Biol. 2007, 14, 473–488. [Google Scholar] [CrossRef]
- Pratt, D.A. Targeting Lipoxygenases to Suppress Ferroptotic Cell Death. Proc. Natl. Acad. Sci. USA 2023, 120, e2309317120. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Mikulska-Ruminska, K.; Anthonymuthu, T.S.; Levkina, A.; Shrivastava, I.H.; Kapralov, A.A.; Bayır, H.; Kagan, V.E.; Bahar, I. NO● Represses the Oxygenation of Arachidonoyl PE by 15LOX/PEBP1: Mechanism and Role in Ferroptosis. Int. J. Mol. Sci. 2021, 22, 5253. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of Polyunsaturated Fatty Acids by Lipoxygenases Drives Ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed]
- Magtanong, L.; Ko, P.-J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef] [PubMed]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-Mediated Generation of Lipid Peroxides Enhances Ferroptosis Induced by Erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 Is Required for P53-Mediated Tumour Suppression through a Distinct Ferroptosis Pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St Croix, C.M.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.e26. [Google Scholar] [CrossRef] [PubMed]
- Dar, H.H.; Mikulska-Ruminska, K.; Tyurina, Y.Y.; Luci, D.K.; Yasgar, A.; Samovich, S.N.; Kapralov, A.A.; Souryavong, A.B.; Tyurin, V.A.; Amoscato, A.A.; et al. Discovering Selective Antiferroptotic Inhibitors of the 15LOX/PEBP1 Complex Noninterfering with Biosynthesis of Lipid Mediators. Proc. Natl. Acad. Sci. USA 2023, 120, e2218896120. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Li, J.; Song, Y.; Luo, C. ACSL4-Mediated Ferroptosis and Its Potential Role in Central Nervous System Diseases and Injuries. Int. J. Mol. Sci. 2023, 24, 10021. [Google Scholar] [CrossRef]
- Lagrost, L.; Masson, D. The Expanding Role of Lyso-Phosphatidylcholine Acyltransferase-3 (LPCAT3), a Phospholipid Remodeling Enzyme, in Health and Disease. Curr. Opin. Lipidol. 2022, 33, 193–198. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 Dictates Ferroptosis Sensitivity by Shaping Cellular Lipid Composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Ai, Y.; Sun, Q.; Ma, Y.; Cao, Y.; Wang, J.; Zhang, Z.; Wang, X. Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol. Cell 2021, 81, 355–369.e10. [Google Scholar] [CrossRef]
- Reed, A.; Ichu, T.-A.; Milosevich, N.; Melillo, B.; Schafroth, M.A.; Otsuka, Y.; Scampavia, L.; Spicer, T.P.; Cravatt, B.F. LPCAT3 Inhibitors Remodel the Polyunsaturated Phospholipid Content of Human Cells and Protect from Ferroptosis. ACS Chem. Biol. 2022, 17, 1607–1618. [Google Scholar] [CrossRef]
- Cui, J.; Wang, Y.; Tian, X.; Miao, Y.; Ma, L.; Zhang, C.; Xu, X.; Wang, J.; Fang, W.; Zhang, X. LPCAT3 Is Transcriptionally Regulated by YAP/ZEB/EP300 and Collaborates with ACSL4 and YAP to Determine Ferroptosis Sensitivity. Antioxid. Redox Signal. 2023, 39, 491–511. [Google Scholar] [CrossRef]
- Yang, Y.; Zhu, T.; Wang, X.; Xiong, F.; Hu, Z.; Qiao, X.; Yuan, X.; Wang, D. ACSL3 and ACSL4, Distinct Roles in Ferroptosis and Cancers. Cancers 2022, 14, 5896. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Feng, Y.; Zandkarimi, F.; Wang, H.; Zhang, Z.; Kim, J.; Cai, Y.; Gu, W.; Stockwell, B.R.; Jiang, X. Ferroptosis Surveillance Independent of GPX4 and Differentially Regulated by Sex Hormones. Cell 2023, 186, 2748–2764.e22. [Google Scholar] [CrossRef]
- Pandey, A.V.; Flück, C.E. NADPH P450 Oxidoreductase: Structure, Function, and Pathology of Diseases. Pharmacol. Ther. 2013, 138, 229–254. [Google Scholar] [CrossRef]
- Dean, J.M.; Lodhi, I.J. Structural and Functional Roles of Ether Lipids. Protein Cell 2018, 9, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kroemer, G. Peroxisome: The New Player in Ferroptosis. Signal Transduct. Target. Ther. 2020, 5, 273. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zhuang, L.; Gan, B. Ether Phospholipids Govern Ferroptosis. J. Genet. Genom. 2021, 48, 517–519. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Henry, W.S.; Ricq, E.L.; Graham, E.T.; Phadnis, V.V.; Maretich, P.; Paradkar, S.; Boehnke, N.; Deik, A.A.; Reinhardt, F.; et al. Plasticity of Ether Lipids Promotes Ferroptosis Susceptibility and Evasion. Nature 2020, 585, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Liu, D.; Gu, W.; Chu, B. Peroxisome-Driven Ether-Linked Phospholipids Biosynthesis Is Essential for Ferroptosis. Cell Death Differ. 2021, 28, 2536–2551. [Google Scholar] [CrossRef] [PubMed]
- Marsh, K.G.; Arrieta, A.; Thuerauf, D.J.; Blackwood, E.A.; MacDonnell, L.; Glembotski, C.C. The Peroxisomal Enzyme, FAR1, Is Induced during ER Stress in an ATF6-Dependent Manner in Cardiac Myocytes. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1813–H1821. [Google Scholar] [CrossRef]
- Ahmadinejad, F.; Geir Møller, S.; Hashemzadeh-Chaleshtori, M.; Bidkhori, G.; Jami, M.-S. Molecular Mechanisms behind Free Radical Scavengers Function against Oxidative Stress. Antioxidants 2017, 6, 51. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide Dismutases: Dual Roles in Controlling ROS Damage and Regulating ROS Signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Amos, A.; Jiang, N.; Zong, D.; Gu, J.; Zhou, J.; Yin, L.; He, X.; Xu, Y.; Wu, L. Depletion of SOD2 Enhances Nasopharyngeal Carcinoma Cell Radiosensitivity via Ferroptosis Induction Modulated by DHODH Inhibition. BMC Cancer 2023, 23, 117. [Google Scholar] [CrossRef]
- Boukalova, S.; Hubackova, S.; Milosevic, M.; Ezrova, Z.; Neuzil, J.; Rohlena, J. Dihydroorotate Dehydrogenase in Oxidative Phosphorylation and Cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165759. [Google Scholar] [CrossRef]
- Riccio, P.; Sessa, R.; de Nicola, S.; Petruzziello, F.; Trombetti, S.; Menna, G.; Pepe, G.; Maddalena, P.; Izzo, P.; Grosso, M. GATA-1 Isoforms Differently Contribute to the Production and Compartmentation of Reactive Oxygen Species in the Myeloid Leukemia Cell Line K562. J. Cell. Physiol. 2019, 234, 20829–20846. [Google Scholar] [CrossRef] [PubMed]
- Trombetti, S.; Sessa, R.; Catapano, R.; Rinaldi, L.; Lo Bianco, A.; Feliciello, A.; Izzo, P.; Grosso, M. Exploring the Leukemogenic Potential of GATA-1S, the Shorter Isoform of GATA-1: Novel Insights into Mechanisms Hampering Respiratory Chain Complex II Activity and Limiting Oxidative Phosphorylation Efficiency. Antioxidants 2021, 10, 1603. [Google Scholar] [CrossRef] [PubMed]
- Zeida, A.; Trujillo, M.; Ferrer-Sueta, G.; Denicola, A.; Estrin, D.A.; Radi, R. Catalysis of Peroxide Reduction by Fast Reacting Protein Thiols. Chem. Rev. 2019, 119, 10829–10855. [Google Scholar] [CrossRef] [PubMed]
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef]
- Hwang, J.S.; Kim, E.; Lee, H.G.; Lee, W.J.; Won, J.P.; Hur, J.; Fujii, J.; Seo, H.G. Peroxisome Proliferator-Activated Receptor δ Rescues xCT-Deficient Cells from Ferroptosis by Targeting Peroxisomes. Biomed. Pharmacother. 2021, 143, 112223. [Google Scholar] [CrossRef]
- Kanner, J.; Harel, S.; Granit, R. Nitric Oxide, an Inhibitor of Lipid Oxidation by Lipoxygenase, Cyclooxygenase and Hemoglobin. Lipids 1992, 27, 46–49. [Google Scholar] [CrossRef]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox Lipid Reprogramming Commands Susceptibility of Macrophages and Microglia to Ferroptotic Death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef]
- Homma, T.; Kobayashi, S.; Conrad, M.; Konno, H.; Yokoyama, C.; Fujii, J. Nitric Oxide Protects against Ferroptosis by Aborting the Lipid Peroxidation Chain Reaction. Nitric Oxide 2021, 115, 34–43. [Google Scholar] [CrossRef]
- Astudillo, A.M.; Balboa, M.A.; Balsinde, J. Compartmentalized Regulation of Lipid Signaling in Oxidative Stress and Inflammation: Plasmalogens, Oxidized Lipids and Ferroptosis as New Paradigms of Bioactive Lipid Research. Prog. Lipid Res. 2023, 89, 101207. [Google Scholar] [CrossRef]
- Morgan, N.V.; Westaway, S.K.; Morton, J.E.V.; Gregory, A.; Gissen, P.; Sonek, S.; Cangul, H.; Coryell, J.; Canham, N.; Nardocci, N.; et al. PLA2G6, Encoding a Phospholipase A2, Is Mutated in Neurodegenerative Disorders with High Brain Iron. Nat. Genet. 2006, 38, 752–754. [Google Scholar] [CrossRef]
- Paisan-Ruiz, C.; Bhatia, K.P.; Li, A.; Hernandez, D.; Davis, M.; Wood, N.W.; Hardy, J.; Houlden, H.; Singleton, A.; Schneider, S.A. Characterization of PLA2G6 as a Locus for Dystonia-Parkinsonism. Ann. Neurol. 2009, 65, 19–23. [Google Scholar] [CrossRef]
- Astudillo, A.M.; Balboa, M.A.; Balsinde, J. Selectivity of Phospholipid Hydrolysis by Phospholipase A2 Enzymes in Activated Cells Leading to Polyunsaturated Fatty Acid Mobilization. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Chu, B.; Yang, X.; Liu, Z.; Jin, Y.; Kon, N.; Rabadan, R.; Jiang, X.; Stockwell, B.R.; Gu, W. iPLA2β-Mediated Lipid Detoxification Controls P53-Driven Ferroptosis Independent of GPX4. Nat. Commun. 2021, 12, 3644. [Google Scholar] [CrossRef]
- Beharier, O.; Tyurin, V.A.; Goff, J.P.; Guerrero-Santoro, J.; Kajiwara, K.; Chu, T.; Tyurina, Y.Y.; St Croix, C.M.; Wallace, C.T.; Parry, S.; et al. PLA2G6 Guards Placental Trophoblasts against Ferroptotic Injury. Proc. Natl. Acad. Sci. USA 2020, 117, 27319–27328. [Google Scholar] [CrossRef]
- Sun, W.-Y.; Tyurin, V.A.; Mikulska-Ruminska, K.; Shrivastava, I.H.; Anthonymuthu, T.S.; Zhai, Y.-J.; Pan, M.-H.; Gong, H.-B.; Lu, D.-H.; Sun, J.; et al. Phospholipase iPLA2β Averts Ferroptosis by Eliminating a Redox Lipid Death Signal. Nat. Chem. Biol. 2021, 17, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Toppo, S.; Flohé, L.; Ursini, F.; Vanin, S.; Maiorino, M. Catalytic Mechanisms and Specificities of Glutathione Peroxidases: Variations of a Basic Scheme. Biochim. Biophys. Acta 2009, 1790, 1486–1500. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Flohé, L. Regulatory Phenomena in the Glutathione Peroxidase Superfamily. Antioxid. Redox Signal. 2020, 33, 498–516. [Google Scholar] [CrossRef]
- Xie, Y.; Kang, R.; Klionsky, D.J.; Tang, D. GPX4 in Cell Death, Autophagy, and Disease. Autophagy 2023, 19, 2621–2638. [Google Scholar] [CrossRef] [PubMed]
- Maiorino, M.; Conrad, M.; Ursini, F. GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid. Redox Signal. 2018, 29, 61–74. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef]
- Schnurr, K.; Belkner, J.; Ursini, F.; Schewe, T.; Kühn, H. The Selenoenzyme Phospholipid Hydroperoxide Glutathione Peroxidase Controls the Activity of the 15-Lipoxygenase with Complex Substrates and Preserves the Specificity of the Oxygenation Products. J. Biol. Chem. 1996, 271, 4653–4658. [Google Scholar] [CrossRef]
- Imai, H.; Hirao, F.; Sakamoto, T.; Sekine, K.; Mizukura, Y.; Saito, M.; Kitamoto, T.; Hayasaka, M.; Hanaoka, K.; Nakagawa, Y. Early Embryonic Lethality Caused by Targeted Disruption of the Mouse PHGPx Gene. Biochem. Biophys. Res. Commun. 2003, 305, 278–286. [Google Scholar] [CrossRef]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The Selenoprotein GPX4 Is Essential for Mouse Development and Protects from Radiation and Oxidative Damage Insults. Free Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef]
- Garry, M.R.; Kavanagh, T.J.; Faustman, E.M.; Sidhu, J.S.; Liao, R.; Ware, C.; Vliet, P.A.; Deeb, S.S. Sensitivity of Mouse Lung Fibroblasts Heterozygous for GPx4 to Oxidative Stress. Free Radic. Biol. Med. 2008, 44, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- de Haan, J.B.; Bladier, C.; Lotfi-Miri, M.; Taylor, J.; Hutchinson, P.; Crack, P.J.; Hertzog, P.; Kola, I. Fibroblasts Derived from Gpx1 Knockout Mice Display Senescent-like Features and Are Susceptible to H2O2-Mediated Cell Death. Free Radic. Biol. Med. 2004, 36, 53–64. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in Mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Zuo, S.; Yu, J.; Pan, H.; Lu, L. Novel Insights on Targeting Ferroptosis in Cancer Therapy. Biomark. Res. 2020, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Takada, K. Reactive Oxygen Species in Cancer: Current Findings and Future Directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of Oxidative Stress as an Anticancer Strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Combs, J.A.; DeNicola, G.M. The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival. Cancers 2019, 11, 678. [Google Scholar] [CrossRef]
- Bonifácio, V.D.B.; Pereira, S.A.; Serpa, J.; Vicente, J.B. Cysteine Metabolic Circuitries: Druggable Targets in Cancer. Br. J. Cancer 2021, 124, 862–879. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; DeNicola, G.M. The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol. 2020, 30, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Bencze, K.Z.; Stemmler, T.L.; Philpott, C.C. A Cytosolic Iron Chaperone That Delivers Iron to Ferritin. Science 2008, 320, 1207–1210. [Google Scholar] [CrossRef]
- Patel, S.J.; Protchenko, O.; Shakoury-Elizeh, M.; Baratz, E.; Jadhav, S.; Philpott, C.C. The Iron Chaperone and Nucleic Acid-Binding Activities of Poly(rC)-Binding Protein 1 Are Separable and Independently Essential. Proc. Natl. Acad. Sci. USA 2021, 118, e2104666118. [Google Scholar] [CrossRef] [PubMed]
- Soula, M.; Weber, R.A.; Zilka, O.; Alwaseem, H.; La, K.; Yen, F.; Molina, H.; Garcia-Bermudez, J.; Pratt, D.A.; Birsoy, K. Metabolic Determinants of Cancer Cell Sensitivity to Canonical Ferroptosis Inducers. Nat. Chem. Biol. 2020, 16, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.H.; Miyamoto, M.; Sastre, A.; Schnaar, R.L.; Coyle, J.T. Glutamate Toxicity in a Neuronal Cell Line Involves Inhibition of Cystine Transport Leading to Oxidative Stress. Neuron 1989, 2, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and Expression of a Plasma Membrane Cystine/Glutamate Exchange Transporter Composed of Two Distinct Proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef]
- Pader, I.; Sengupta, R.; Cebula, M.; Xu, J.; Lundberg, J.O.; Holmgren, A.; Johansson, K.; Arnér, E.S.J. Thioredoxin-Related Protein of 14 kDa Is an Efficient L-Cystine Reductase and S-Denitrosylase. Proc. Natl. Acad. Sci. USA 2014, 111, 6964–6969. [Google Scholar] [CrossRef]
- Poltorack, C.D.; Dixon, S.J. Understanding the Role of Cysteine in Ferroptosis: Progress & Paradoxes. FEBS J. 2022, 289, 374–385. [Google Scholar] [CrossRef]
- Cramer, S.L.; Saha, A.; Liu, J.; Tadi, S.; Tiziani, S.; Yan, W.; Triplett, K.; Lamb, C.; Alters, S.E.; Rowlinson, S.; et al. Systemic Depletion of L-Cyst(e)Ine with Cyst(e)Inase Increases Reactive Oxygen Species and Suppresses Tumor Growth. Nat. Med. 2017, 23, 120–127. [Google Scholar] [CrossRef]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino Acid Transporter SLC7A11/xCT at the Crossroads of Regulating Redox Homeostasis and Nutrient Dependency of Cancer. Cancer Commun. 2018, 38, 12. [Google Scholar] [CrossRef]
- Liu, M.-R.; Zhu, W.-T.; Pei, D.-S. System Xc-: A Key Regulatory Target of Ferroptosis in Cancer. Investig. New Drugs 2021, 39, 1123–1131. [Google Scholar] [CrossRef]
- Yan, Y.; Teng, H.; Hang, Q.; Kondiparthi, L.; Lei, G.; Horbath, A.; Liu, X.; Mao, C.; Wu, S.; Zhuang, L.; et al. SLC7A11 Expression Level Dictates Differential Responses to Oxidative Stress in Cancer Cells. Nat. Commun. 2023, 14, 3673. [Google Scholar] [CrossRef]
- Griffith, O.W.; Bridges, R.J.; Meister, A. Evidence That the Gamma-Glutamyl Cycle Functions in Vivo Using Intracellular Glutathione: Effects of Amino Acids and Selective Inhibition of Enzymes. Proc. Natl. Acad. Sci. USA 1978, 75, 5405–5408. [Google Scholar] [CrossRef]
- Hayashima, K.; Katoh, H. Expression of Gamma-Glutamyltransferase 1 in Glioblastoma Cells Confers Resistance to Cystine Deprivation-Induced Ferroptosis. J. Biol. Chem. 2022, 298, 101703. [Google Scholar] [CrossRef]
- Xu, G.; Wang, J.; Zhang, Y.; Chen, Z.; Deng, R. GGT1 Suppresses the Development of Ferroptosis and Autophagy in Mouse Retinal Ganglion Cell Through Targeting GCLC. Eye Brain 2023, 15, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.A.; Pérez-Sala, D. Mammalian Sulfur Amino Acid Metabolism: A Nexus Between Redox Regulation, Nutrition, Epigenetics, and Detoxification. Antioxid. Redox Signal. 2018, 29, 408–452. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, W.; Chen, Q.; Zheng, Z.; Jiang, X.; Xue, Y.; Lin, D. TXNRD1: A Key Regulator Involved in the Ferroptosis of CML Cells Induced by Cysteine Depletion In Vitro. Oxidative Med. Cell. Longev. 2021, 2021, 7674565. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Xu, H.; Cao, C.; Cao, J.; Zhang, Y.; Zhang, C.; Qiao, R.; Ming, W.; Li, Y.; Ren, H.; et al. Neutral Amino Acid Transporter SLC38A2 Protects Renal Medulla from Hyperosmolarity-Induced Ferroptosis. eLife 2023, 12, e80647. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-F.; Klein Geltink, R.I.; Parker, S.J.; Sorensen, P.H. Transsulfuration, Minor Player or Crucial for Cysteine Homeostasis in Cancer. Trends Cell Biol. 2022, 32, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Regulators of the Transsulfuration Pathway. Br. J. Pharmacol. 2019, 176, 583–593. [Google Scholar] [CrossRef]
- Hayano, M.; Yang, W.S.; Corn, C.K.; Pagano, N.C.; Stockwell, B.R. Loss of Cysteinyl-tRNA Synthetase (CARS) Induces the Transsulfuration Pathway and Inhibits Ferroptosis Induced by Cystine Deprivation. Cell Death Differ. 2016, 23, 270–278. [Google Scholar] [CrossRef]
- Wang, L.; Cai, H.; Hu, Y.; Liu, F.; Huang, S.; Zhou, Y.; Yu, J.; Xu, J.; Wu, F. A Pharmacological Probe Identifies Cystathionine β-Synthase as a New Negative Regulator for Ferroptosis. Cell Death Dis. 2018, 9, 1005. [Google Scholar] [CrossRef]
- Cao, J.; Chen, X.; Jiang, L.; Lu, B.; Yuan, M.; Zhu, D.; Zhu, H.; He, Q.; Yang, B.; Ying, M. DJ-1 Suppresses Ferroptosis through Preserving the Activity of S-Adenosyl Homocysteine Hydrolase. Nat. Commun. 2020, 11, 1251. [Google Scholar] [CrossRef]
- Liao, T.; Xu, X.; Ye, X.; Yan, J. DJ-1 Upregulates the Nrf2/GPX4 Signal Pathway to Inhibit Trophoblast Ferroptosis in the Pathogenesis of Preeclampsia. Sci. Rep. 2022, 12, 2934. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Sato, M.; Kasakoshi, T.; Tsutsui, T.; Sugimoto, M.; Osaki, M.; Okada, F.; Igarashi, K.; Hiratake, J.; Homma, T.; et al. Cystathionine Is a Novel Substrate of Cystine/Glutamate Transporter: Implications for Immune Function. J. Biol. Chem. 2015, 290, 8778–8788. [Google Scholar] [CrossRef]
- Peng, H.; Zhu, M.; Kong, W.; Tang, C.; Du, J.; Huang, Y.; Jin, H. L-Cystathionine Protects against Oxidative Stress and DNA Damage Induced by Oxidized Low-Density Lipoprotein in THP-1-Derived Macrophages. Front. Pharmacol. 2023, 14, 1161542. [Google Scholar] [CrossRef]
- Fisher, A.B.; Vasquez-Medina, J.P.; Dodia, C.; Sorokina, E.M.; Tao, J.-Q.; Feinstein, S.I. Peroxiredoxin 6 Phospholipid Hydroperoxidase Activity in the Repair of Peroxidized Cell Membranes. Redox Biol. 2018, 14, 41–46. [Google Scholar] [CrossRef]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against Oxidative Stress and Modulators of Peroxide Signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B.; Dodia, C.; Sorokina, E.M.; Li, H.; Zhou, S.; Raabe, T.; Feinstein, S.I. A Novel Lysophosphatidylcholine Acyl Transferase Activity Is Expressed by Peroxiredoxin 6. J. Lipid Res. 2016, 57, 587–596. [Google Scholar] [CrossRef]
- Fisher, A.B. Peroxiredoxin 6 in the Repair of Peroxidized Cell Membranes and Cell Signaling. Arch. Biochem. Biophys. 2017, 617, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Arevalo, J.A.; Vázquez-Medina, J.P. The Role of Peroxiredoxin 6 in Cell Signaling. Antioxidants 2018, 7, 172. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Chen, X.-B.; Hong, Y.-C.; Zhu, H.; He, Q.-J.; Yang, B.; Ying, M.-D.; Cao, J. Identification of PRDX6 as a Regulator of Ferroptosis. Acta Pharmacol. Sin. 2019, 40, 1334–1342. [Google Scholar] [CrossRef]
- Liao, J.; Xie, S.-S.; Deng, Y.; Wu, D.-D.; Meng, H.; Lan, W.-F.; Dai, P. PRDX6-Mediated Pulmonary Artery Endothelial Cell Ferroptosis Contributes to Monocrotaline-Induced Pulmonary Hypertension. Microvasc. Res. 2023, 146, 104471. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front. Immunol. 2020, 11, 484. [Google Scholar] [CrossRef]
- Zhang, Q.; Hu, Y.; Hu, J.-E.; Ding, Y.; Shen, Y.; Xu, H.; Chen, H.; Wu, N. Sp1-Mediated Upregulation of Prdx6 Expression Prevents Podocyte Injury in Diabetic Nephropathy via Mitigation of Oxidative Stress and Ferroptosis. Life Sci. 2021, 278, 119529. [Google Scholar] [CrossRef]
- Liu, J.; Sun, L.; Chen, D.; Huo, X.; Tian, X.; Li, J.; Liu, M.; Yu, Z.; Zhang, B.; Yang, Y.; et al. Prdx6-Induced Inhibition of Ferroptosis in Epithelial Cells Contributes to Liquiritin-Exerted Alleviation of Colitis. Food Funct. 2022, 13, 9470–9480. [Google Scholar] [CrossRef]
- Yang, L.; Fan, X.; Zhou, C.; Wang, Z.; Cui, Z.; Wu, X.; Xu, Z.; Yang, J.; Zhang, X. Construction and Validation of a Novel Ferroptosis-Related Prognostic Signature for Lung Adenocarcinoma. Transl. Lung Cancer Res. 2023, 12, 1766–1781. [Google Scholar] [CrossRef]
- Li, Z.; Xiao, J.; Liu, M.; Cui, J.; Lian, B.; Sun, Y.; Li, C. Notch3 Regulates Ferroptosis via ROS-Induced Lipid Peroxidation in NSCLC Cells. FEBS Open Bio 2022, 12, 1197–1205. [Google Scholar] [CrossRef]
- Holstein, S.A.; Hohl, R.J. Isoprenoids: Remarkable Diversity of Form and Function. Lipids 2004, 39, 293–309. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a Therapy-Resistant State of Cancer Cells on a Lipid Peroxidase Pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Conrad, M. Selenium and GPX4, a Vital Symbiosis. Free Radic. Biol. Med. 2018, 127, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Warner, G.J.; Berry, M.J.; Moustafa, M.E.; Carlson, B.A.; Hatfield, D.L.; Faust, J.R. Inhibition of Selenoprotein Synthesis by Selenocysteine tRNA[Ser]Sec Lacking Isopentenyladenosine. J. Biol. Chem. 2000, 275, 28110–28119. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global Survey of Cell Death Mechanisms Reveals Metabolic Regulation of Ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Pallotti, F.; Bergamini, C.; Lamperti, C.; Fato, R. The Roles of Coenzyme Q in Disease: Direct and Indirect Involvement in Cellular Functions. Int. J. Mol. Sci. 2021, 23, 128. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 Is a Glutathione-Independent Ferroptosis Suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Zeng, F.; Chen, X.; Deng, G. The Anti-Ferroptotic Role of FSP1: Current Molecular Mechanism and Therapeutic Approach. Mol. Biomed. 2022, 3, 37. [Google Scholar] [CrossRef]
- Li, W.; Liang, L.; Liu, S.; Yi, H.; Zhou, Y. FSP1: A Key Regulator of Ferroptosis. Trends Mol. Med. 2023, 29, 753–764. [Google Scholar] [CrossRef]
- Mishima, E.; Ito, J.; Wu, Z.; Nakamura, T.; Wahida, A.; Doll, S.; Tonnus, W.; Nepachalovich, P.; Eggenhofer, E.; Aldrovandi, M.; et al. A Non-Canonical Vitamin K Cycle Is a Potent Ferroptosis Suppressor. Nature 2022, 608, 778–783. [Google Scholar] [CrossRef]
- Lv, Y.; Liang, C.; Sun, Q.; Zhu, J.; Xu, H.; Li, X.; Li, Y.-Y.; Wang, Q.; Yuan, H.; Chu, B.; et al. Structural Insights into FSP1 Catalysis and Ferroptosis Inhibition. Nat. Commun. 2023, 14, 5933. [Google Scholar] [CrossRef] [PubMed]
- Dai, E.; Zhang, W.; Cong, D.; Kang, R.; Wang, J.; Tang, D. AIFM2 Blocks Ferroptosis Independent of Ubiquinol Metabolism. Biochem. Biophys. Res. Commun. 2020, 523, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Dai, E.; Meng, L.; Kang, R.; Wang, X.; Tang, D. ESCRT-III-Dependent Membrane Repair Blocks Ferroptosis. Biochem. Biophys. Res. Commun. 2020, 522, 415–421. [Google Scholar] [CrossRef]
- Liu, J.; Kang, R.; Tang, D. ESCRT-III-Mediated Membrane Repair in Cell Death and Tumor Resistance. Cancer Gene Ther. 2021, 28, 1–4. [Google Scholar] [CrossRef]
- Yoshioka, H.; Kawamura, T.; Muroi, M.; Kondoh, Y.; Honda, K.; Kawatani, M.; Aono, H.; Waldmann, H.; Watanabe, N.; Osada, H. Identification of a Small Molecule That Enhances Ferroptosis via Inhibition of Ferroptosis Suppressor Protein 1 (FSP1). ACS Chem. Biol. 2022, 17, 483–491. [Google Scholar] [CrossRef]
- Zhang, S.; Gou, S.; Zhang, Q.; Yong, X.; Gan, B.; Jia, D. FSP1 Oxidizes NADPH to Suppress Ferroptosis. Cell Res. 2023, 33, 967–970. [Google Scholar] [CrossRef]
- Koppula, P.; Lei, G.; Zhang, Y.; Yan, Y.; Mao, C.; Kondiparthi, L.; Shi, J.; Liu, X.; Horbath, A.; Das, M.; et al. A Targetable CoQ-FSP1 Axis Drives Ferroptosis- and Radiation-Resistance in KEAP1 Inactive Lung Cancers. Nat. Commun. 2022, 13, 2206. [Google Scholar] [CrossRef]
- Lee, J.; Roh, J.-L. Unleashing Ferroptosis in Human Cancers: Targeting Ferroptosis Suppressor Protein 1 for Overcoming Therapy Resistance. Antioxidants 2023, 12, 1218. [Google Scholar] [CrossRef] [PubMed]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Mráček, T.; Drahota, Z.; Houštěk, J. The Function and the Role of the Mitochondrial Glycerol-3-Phosphate Dehydrogenase in Mammalian Tissues. Biochim. Biophys. Acta 2013, 1827, 401–410. [Google Scholar] [CrossRef]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef]
- Hu, Q.; Wei, W.; Wu, D.; Huang, F.; Li, M.; Li, W.; Yin, J.; Peng, Y.; Lu, Y.; Zhao, Q.; et al. Blockade of GCH1/BH4 Axis Activates Ferritinophagy to Mitigate the Resistance of Colorectal Cancer to Erastin-Induced Ferroptosis. Front. Cell Dev. Biol. 2022, 10, 810327. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liang, W.; Huo, D.; Wang, H.; Wang, Y.; Cong, C.; Zhang, C.; Yan, S.; Gao, M.; Su, X.; et al. SPY1 Inhibits Neuronal Ferroptosis in Amyotrophic Lateral Sclerosis by Reducing Lipid Peroxidation through Regulation of GCH1 and TFR1. Cell Death Differ. 2023, 30, 369–382. [Google Scholar] [CrossRef]
- Kuang, F.; Liu, J.; Xie, Y.; Tang, D.; Kang, R. MGST1 Is a Redox-Sensitive Repressor of Ferroptosis in Pancreatic Cancer Cells. Cell Chem. Biol. 2021, 28, 765–775.e5. [Google Scholar] [CrossRef] [PubMed]
- Johansson, K.; Järvliden, J.; Gogvadze, V.; Morgenstern, R. Multiple Roles of Microsomal Glutathione Transferase 1 in Cellular Protection: A Mechanistic Study. Free Radic. Biol. Med. 2010, 49, 1638–1645. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, R.; Zhang, J.; Johansson, K. Microsomal Glutathione Transferase 1: Mechanism and Functional Roles. Drug Metab. Rev. 2011, 43, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Peng, N. Microsomal Glutathione S-Transferase 1 Targets the Autophagy Signaling Pathway to Suppress Ferroptosis in Gastric Carcinoma Cells. Hum. Exp. Toxicol. 2023, 42, 9603271231172915. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Cai, X.; Kang, R.; Liu, J.; Tang, D. Autophagy-Dependent Ferroptosis in Cancer. Antioxid. Redox Signal. 2023, 39, 79–101. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Chen, P.; Liu, J.; Zhu, S.; Kroemer, G.; Klionsky, D.J.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Clockophagy Is a Novel Selective Autophagy Process Favoring Ferroptosis. Sci. Adv. 2019, 5, eaaw2238. [Google Scholar] [CrossRef]
- Rižner, T.L.; Penning, T.M. Role of Aldo-Keto Reductase Family 1 (AKR1) Enzymes in Human Steroid Metabolism. Steroids 2014, 79, 49–63. [Google Scholar] [CrossRef]
- Burczynski, M.E.; Sridhar, G.R.; Palackal, N.T.; Penning, T.M. The Reactive Oxygen Species--and Michael Acceptor-Inducible Human Aldo-Keto Reductase AKR1C1 Reduces the Alpha,Beta-Unsaturated Aldehyde 4-Hydroxy-2-Nonenal to 1,4-Dihydroxy-2-Nonene. J. Biol. Chem. 2001, 276, 2890–2897. [Google Scholar] [CrossRef]
- Gagliardi, M.; Cotella, D.; Santoro, C.; Corà, D.; Barlev, N.A.; Piacentini, M.; Corazzari, M. Aldo-Keto Reductases Protect Metastatic Melanoma from ER Stress-Independent Ferroptosis. Cell Death Dis. 2019, 10, 902. [Google Scholar] [CrossRef]
- Zuo, X.; Zeng, H.; Wang, B.; Yang, X.; He, D.; Wang, L.; Ouyang, H.; Yuan, J. AKR1C1 Protects Corneal Epithelial Cells Against Oxidative Stress-Mediated Ferroptosis in Dry Eye. Investig. Ophthalmol. Vis. Sci. 2022, 63, 3. [Google Scholar] [CrossRef]
- Tesfay, L.; Paul, B.T.; Konstorum, A.; Deng, Z.; Cox, A.O.; Lee, J.; Furdui, C.M.; Hegde, P.; Torti, F.M.; Torti, S.V. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res. 2019, 79, 5355–5366. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Wang, X.; Song, S.; Wang, Y.; Dan, Q.; Ge, H. Targeting Stearoyl-Coa Desaturase Enhances Radiation Induced Ferroptosis and Immunogenic Cell Death in Esophageal Squamous Cell Carcinoma. OncoImmunology 2022, 11, 2101769. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Zhu, J.; Wu, J.; Thompson, C.B.; Jiang, X. Oncogenic Activation of PI3K-AKT-mTOR Signaling Suppresses Ferroptosis via SREBP-Mediated Lipogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 31189–31197. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhou, J.; Hooi, S.C.; Jiang, Y.-M.; Lu, G.-D. Fatty Acid Activation in Carcinogenesis and Cancer Development: Essential Roles of Long-Chain Acyl-CoA Synthetases. Oncol. Lett. 2018, 16, 1390–1396. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Gui, G.; Zhang, L.; Qin, A.; Zhou, C.; Zha, X. Overview of Methionine Adenosyltransferase 2A (MAT2A) as an Anticancer Target: Structure, Function, and Inhibitors. J. Med. Chem. 2022, 65, 9531–9547. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Kong, P.; Huang, Y.; Wang, J.; Liu, X.; Hu, Y.; Chen, X.; Du, C.; Yang, H. Activation of MAT2A-ACSL3 Pathway Protects Cells from Ferroptosis in Gastric Cancer. Free Radic. Biol. Med. 2022, 181, 288–299. [Google Scholar] [CrossRef]
- Shakya, A.; McKee, N.W.; Dodson, M.; Chapman, E.; Zhang, D.D. Anti-Ferroptotic Effects of Nrf2: Beyond the Antioxidant Response. Mol. Cells 2023, 46, 165–175. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The Selective Autophagy Substrate P62 Activates the Stress Responsive Transcription Factor Nrf2 through Inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Cao, J.Y.; Poddar, A.; Magtanong, L.; Lumb, J.H.; Mileur, T.R.; Reid, M.A.; Dovey, C.M.; Wang, J.; Locasale, J.W.; Stone, E.; et al. A Genome-Wide Haploid Genetic Screen Identifies Regulators of Glutathione Abundance and Ferroptosis Sensitivity. Cell Rep. 2019, 26, 1544–1556.e8. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, T.; Gouge, J. Nrf2 in Cancer, Detoxifying Enzymes and Cell Death Programs. Antioxidants 2021, 10, 1030. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 Enhances Resistance of Cancer Cells to Chemotherapeutic Drugs, the Dark Side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Pouremamali, F.; Pouremamali, A.; Dadashpour, M.; Soozangar, N.; Jeddi, F. An Update of Nrf2 Activators and Inhibitors in Cancer Prevention/Promotion. Cell Commun. Signal. 2022, 20, 100. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 Pathway Protects against Ferroptosis in Hepatocellular Carcinoma Cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kim, M.-J.; Han, T.-H.; Lee, J.-Y.; Kim, S.; Kim, H.; Oh, K.-J.; Kim, W.K.; Han, B.-S.; Bae, K.-H.; et al. FSP1 Confers Ferroptosis Resistance in KEAP1 Mutant Non-Small Cell Lung Carcinoma in NRF2-Dependent and -Independent Manner. Cell Death Dis. 2023, 14, 567. [Google Scholar] [CrossRef] [PubMed]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef]
- Anandhan, A.; Dodson, M.; Shakya, A.; Chen, J.; Liu, P.; Wei, Y.; Tan, H.; Wang, Q.; Jiang, Z.; Yang, K.; et al. NRF2 Controls Iron Homeostasis and Ferroptosis through HERC2 and VAMP8. Sci. Adv. 2023, 9, eade9585. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Rojo, A.I.; Velasco, D.; De Sagarra, R.M.; Cuadrado, A. Glycogen Synthase Kinase-3β Inhibits the Xenobiotic and Antioxidant Cell Response by Direct Phosphorylation and Nuclear Exclusion of the Transcription Factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef]
- Takahashi, N.; Cho, P.; Selfors, L.M.; Kuiken, H.J.; Kaul, R.; Fujiwara, T.; Harris, I.S.; Zhang, T.; Gygi, S.P.; Brugge, J.S. 3D Culture Models with CRISPR Screens Reveal Hyperactive NRF2 as a Prerequisite for Spheroid Formation via Regulation of Proliferation and Ferroptosis. Mol. Cell 2020, 80, 828–844.e6. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, R.; Jaiswal, A.K. Nrf2 and Nrf1 in Association with Jun Proteins Regulate Antioxidant Response Element-Mediated Expression and Coordinated Induction of Genes Encoding Detoxifying Enzymes. Oncogene 1998, 17, 3145–3156. [Google Scholar] [CrossRef]
- He, C.H.; Gong, P.; Hu, B.; Stewart, D.; Choi, M.E.; Choi, A.M.K.; Alam, J. Identification of Activating Transcription Factor 4 (ATF4) as an Nrf2-Interacting Protein. J. Biol. Chem. 2001, 276, 20858–20865. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; Zhang, H.; Hu, S.-J.; Liao, S.-Y.; Tao, D.-C.; Tan, X.-L.; Yi, M.; Leng, X.-Y.; Wang, Z.-K.; Shi, J.-Y.; et al. NR0B1 Suppresses Ferroptosis through Upregulation of NRF2/c-JUN-CBS Signaling Pathway in Lung Cancer Cells. Am. J. Cancer Res. 2023, 13, 5174–5196. [Google Scholar]
- Anandhan, A.; Dodson, M.; Schmidlin, C.J.; Liu, P.; Zhang, D.D. Breakdown of an Ironclad Defense System: The Critical Role of NRF2 in Mediating Ferroptosis. Cell Chem. Biol. 2020, 27, 436–447. [Google Scholar] [CrossRef]
- Chen, D.; Fan, Z.; Rauh, M.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N. ATF4 Promotes Angiogenesis and Neuronal Cell Death and Confers Ferroptosis in a xCT-Dependent Manner. Oncogene 2017, 36, 5593–5608. [Google Scholar] [CrossRef] [PubMed]
- Bhoumik, A.; Lopez-Bergami, P.; Ronai, Z. ATF2 on the Double-Activating Transcription Factor and DNA Damage Response Protein. Pigment Cell Res. 2007, 20, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Li, Y.; Wu, Y.; Wang, M.; Lu, Y.; Fang, Z.; Wang, H.; Li, Y. Increased ATF2 Expression Predicts Poor Prognosis and Inhibits Sorafenib-Induced Ferroptosis in Gastric Cancer. Redox Biol. 2023, 59, 102564. [Google Scholar] [CrossRef]
- Wang, L.; Chen, Y.; Mi, Y.; Qiao, J.; Jin, H.; Li, J.; Lu, Z.; Wang, Q.; Zou, Z. ATF2 Inhibits Ani-Tumor Effects of BET Inhibitor in a Negative Feedback Manner by Attenuating Ferroptosis. Biochem. Biophys. Res. Commun. 2021, 558, 216–223. [Google Scholar] [CrossRef]
- Ju, H.; Yun, H.; Kim, Y.; Nam, Y.J.; Lee, S.; Lee, J.; Jeong, S.M.; Heo, J.; Kwon, H.; Cho, Y.S.; et al. Activating Transcription Factor-2 Supports the Antioxidant Capacity and Ability of Human Mesenchymal Stem Cells to Prevent Asthmatic Airway Inflammation. Exp. Mol. Med. 2023, 55, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Huang, Y.; Zhou, X.; Xiang, Z.; Yang, Z.; Meng, D.; Wu, D.; Zhang, J.; Yang, J. ATF3 and Its Emerging Role in Atherosclerosis: A Narrative Review. Cardiovasc. Diagn. Ther. 2022, 12, 926–942. [Google Scholar] [CrossRef] [PubMed]
- Ku, H.-C.; Cheng, C.-F. Master Regulator Activating Transcription Factor 3 (ATF3) in Metabolic Homeostasis and Cancer. Front. Endocrinol. 2020, 11, 556. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Du, T.; Yang, H.; Lei, L.; Guo, M.; Ding, H.-F.; Zhang, J.; Wang, H.; Chen, X.; et al. ATF3 Promotes Erastin-Induced Ferroptosis by Suppressing System Xc−. Cell Death Differ. 2020, 27, 662–675. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yan, J.; Zhao, Q.; Zhang, Y.; Zhang, Y. ATF3 Promotes Ferroptosis in Sorafenib-Induced Cardiotoxicity by Suppressing Slc7a11 Expression. Front. Pharmacol. 2022, 13, 904314. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, Z.; Li, C.; Zhang, Z.; Lu, S.; Wang, X.; Liang, Q.; Zhu, X.; Pan, C.; Wang, Q.; et al. SIRT1 Activated by AROS Sensitizes Glioma Cells to Ferroptosis via Induction of NAD+ Depletion-Dependent Activation of ATF3. Redox Biol. 2024, 69, 103030. [Google Scholar] [CrossRef]
- Ameri, K.; Harris, A.L. Activating Transcription Factor 4. Int. J. Biochem. Cell Biol. 2008, 40, 14–21. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, Q.; Sun, X.; Zeh, H.J.; Lotze, M.T.; Kang, R.; Tang, D. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017, 77, 2064–2077. [Google Scholar] [CrossRef]
- Meinert, M.; Jessen, C.; Hufnagel, A.; Kreß, J.K.C.; Burnworth, M.; Däubler, T.; Gallasch, T.; Xavier Da Silva, T.N.; Dos Santos, A.F.; Ade, C.P.; et al. Thiol Starvation Triggers Melanoma State Switching in an ATF4 and NRF2-Dependent Manner. Redox Biol. 2024, 70, 103011. [Google Scholar] [CrossRef]
- Gao, R.; Kalathur, R.K.R.; Coto-Llerena, M.; Ercan, C.; Buechel, D.; Shuang, S.; Piscuoglio, S.; Dill, M.T.; Camargo, F.D.; Christofori, G.; et al. YAP/TAZ and ATF4 Drive Resistance to Sorafenib in Hepatocellular Carcinoma by Preventing Ferroptosis. EMBO Mol. Med. 2021, 13, e14351. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Zhang, P.; Liu, J.; Wang, R.; Kaufman, R.J.; Yaden, B.C.; Karin, M. ATF4 Suppresses Hepatocarcinogenesis by Inducing SLC7A11 (xCT) to Block Stress-Related Ferroptosis. J. Hepatol. 2023, 79, 362–377. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Luo, Y.; Liang, Z.; Li, X.; Kołat, D.; Zhao, L.; Xiong, W. Crucial Role of the Transcription Factors Family Activator Protein 2 in Cancer: Current Clue and Views. J. Transl. Med. 2023, 21, 371. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-X.; Yang, G.; Yang, Y.; Yan, J.; Tang, X.-Y.; Pan, Q. TFAP2A Is a Novel Regulator That Modulates Ferroptosis in Gallbladder Carcinoma Cells via the Nrf2 Signalling Axis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4745–4755. [Google Scholar] [CrossRef] [PubMed]
- Alim, I.; Caulfield, J.T.; Chen, Y.; Swarup, V.; Geschwind, D.H.; Ivanova, E.; Seravalli, J.; Ai, Y.; Sansing, L.H.; Ste.Marie, E.J.; et al. Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell 2019, 177, 1262–1279.e25. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Guo, S.; Xu, M.; Ma, B.; Liu, R.; Xu, Y.; Zhang, Y. TFAP2C-Mediated lncRNA PCAT1 Inhibits Ferroptosis in Docetaxel-Resistant Prostate Cancer Through c-Myc/miR-25-3p/SLC7A11 Signaling. Front. Oncol. 2022, 12, 862015. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fang, Z.-M.; Yi, X.; Wei, X.; Jiang, D.-S. The Interaction between Ferroptosis and Inflammatory Signaling Pathways. Cell Death Dis. 2023, 14, 205. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT Signaling Pathway: From Bench to Clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Hu, Q.; Bian, Q.; Rong, D.; Wang, L.; Song, J.; Huang, H.-S.; Zeng, J.; Mei, J.; Wang, P.-Y. JAK/STAT Pathway: Extracellular Signals, Diseases, Immunity, and Therapeutic Regimens. Front. Bioeng. Biotechnol. 2023, 11, 1110765. [Google Scholar] [CrossRef]
- Harrison, D.A. The JAK/STAT Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011205. [Google Scholar] [CrossRef]
- Tolomeo, M.; Cascio, A. The Multifaced Role of STAT3 in Cancer and Its Implication for Anticancer Therapy. Int. J. Mol. Sci. 2021, 22, 603. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-Stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904.e5. [Google Scholar] [CrossRef]
- Wang, W.; Green, M.; Choi, J.E.; Gijón, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8+ T Cells Regulate Tumour Ferroptosis during Cancer Immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhu, D.; Luo, B.; Kou, W.; Cheng, Y.; Zhu, Y. IFNγ Enhances Ferroptosis by Increasing JAK-STAT Pathway Activation to Suppress SLCA711 Expression in Adrenocortical Carcinoma. Oncol. Rep. 2022, 47, 97. [Google Scholar] [CrossRef]
- Kong, R.; Wang, N.; Han, W.; Bao, W.; Lu, J. IFNγ-Mediated Repression of System Xc− Drives Vulnerability to Induced Ferroptosis in Hepatocellular Carcinoma Cells. J. Leukoc. Biol. 2021, 110, 301–314. [Google Scholar] [CrossRef]
- Wei, T.; Zhang, M.; Zheng, X.; Xie, T.; Wang, W.; Zou, J.; Li, Y.; Li, H.; Cai, J.; Wang, X.; et al. Interferon-γ Induces Retinal Pigment Epithelial Cell Ferroptosis by a JAK1-2/STAT1/SLC7A11 Signaling Pathway in Age-related Macular Degeneration. FEBS J. 2022, 289, 1968–1983. [Google Scholar] [CrossRef]
- Gao, H.; Bai, Y.; Jia, Y.; Zhao, Y.; Kang, R.; Tang, D.; Dai, E. Ferroptosis Is a Lysosomal Cell Death Process. Biochem. Biophys. Res. Commun. 2018, 503, 1550–1556. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, S.; Li, H.; Lou, L.; Huang, Q.; Zhang, Z.; Mo, J.; Li, M.; Lu, J.; Zhu, K.; Chu, Y.; et al. Inhibition of STAT3-Ferroptosis Negative Regulatory Axis Suppresses Tumor Growth and Alleviates Chemoresistance in Gastric Cancer. Redox Biol. 2022, 52, 102317. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, K. The Induction of Ferroptosis by Impairing STAT3/Nrf2/GPx4 Signaling Enhances the Sensitivity of Osteosarcoma Cells to Cisplatin. Cell Biol. Int. 2019, 43, 1245–1256. [Google Scholar] [CrossRef]
- Luo, Y.; Gao, X.; Zou, L.; Lei, M.; Feng, J.; Hu, Z. Bavachin Induces Ferroptosis through the STAT3/P53/SLC7A11 Axis in Osteosarcoma Cells. Oxidative Med. Cell. Longev. 2021, 2021, 1783485. [Google Scholar] [CrossRef]
- Linher-Melville, K.; Haftchenary, S.; Gunning, P.; Singh, G. Signal Transducer and Activator of Transcription 3 and 5 Regulate System Xc- and Redox Balance in Human Breast Cancer Cells. Mol. Cell Biochem. 2015, 405, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Sung, B.; Aggarwal, B.B. Nuclear Factor-kappaB Activation: From Bench to Bedside. Exp. Biol. Med. 2008, 233, 21–31. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in NF-κB Signaling Pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B.; Kaltschmidt, C. NF-kappaB in the Nervous System. Cold Spring Harb. Perspect. Biol. 2009, 1, a001271. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Tan, S.; Zhou, Y.; Lin, J.; Wang, H.; Oyang, L.; Tian, Y.; Liu, L.; Su, M.; Wang, H.; et al. Role of the NFκB-Signaling Pathway in Cancer. OTT 2018, 11, 2063–2073. [Google Scholar] [CrossRef]
- Zinatizadeh, M.R.; Schock, B.; Chalbatani, G.M.; Zarandi, P.K.; Jalali, S.A.; Miri, S.R. The Nuclear Factor Kappa B (NF-κB) Signaling in Cancer Development and Immune Diseases. Genes Dis. 2021, 8, 287–297. [Google Scholar] [CrossRef]
- Liu, G.-H.; Qu, J.; Shen, X. NF-κB/P65 Antagonizes Nrf2-ARE Pathway by Depriving CBP from Nrf2 and Facilitating Recruitment of HDAC3 to MafK. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2008, 1783, 713–727. [Google Scholar] [CrossRef]
- Xu, M.; Tao, J.; Yang, Y.; Tan, S.; Liu, H.; Jiang, J.; Zheng, F.; Wu, B. Ferroptosis Involves in Intestinal Epithelial Cell Death in Ulcerative Colitis. Cell Death Dis. 2020, 11, 86. [Google Scholar] [CrossRef]
- Yan, N.; Xu, Z.; Qu, C.; Zhang, J. Dimethyl Fumarate Improves Cognitive Deficits in Chronic Cerebral Hypoperfusion Rats by Alleviating Inflammation, Oxidative Stress, and Ferroptosis via NRF2/ARE/NF-κB Signal Pathway. Int. Immunopharmacol. 2021, 98, 107844. [Google Scholar] [CrossRef]
- Li, S.; He, Y.; Chen, K.; Sun, J.; Zhang, L.; He, Y.; Yu, H.; Li, Q. RSL3 Drives Ferroptosis through NF-κB Pathway Activation and GPX4 Depletion in Glioblastoma. Oxidative Med. Cell. Longev. 2021, 2021, 2915019. [Google Scholar] [CrossRef]
- Schmitt, A.; Xu, W.; Bucher, P.; Grimm, M.; Konantz, M.; Horn, H.; Zapukhlyak, M.; Berning, P.; Brändle, M.; Jarboui, M.-A.; et al. Dimethyl Fumarate Induces Ferroptosis and Impairs NF-κB/STAT3 Signaling in DLBCL. Blood 2021, 138, 871–884. [Google Scholar] [CrossRef]
- Wang, Y.; Feng, J.; Zhao, L.; Zhao, M.; Wei, X.; Geng, Y.; Yuan, H.; Hou, C.; Zhang, H.; Wang, G.; et al. Aspirin Triggers Ferroptosis in Hepatocellular Carcinoma Cells through Restricting NF-κB P65-Activated SLC7A11 Transcription. Acta Pharmacol. Sin. 2023, 44, 1712–1724. [Google Scholar] [CrossRef]
- Jin, H.; Zhu, Z.; Yu, P.; Wang, G.; Zhang, J.; Li, J.; Ai, R.; Li, Z.; Tian, Y.; Xu, W.; et al. Myrislignan Attenuates Lipopolysaccharide-induced Inflammation Reaction in Murine Macrophage Cells Through Inhibition of NF-κB Signalling Pathway Activation. Phytother. Res. 2012, 26, 1320–1326. [Google Scholar] [CrossRef]
- Zhou, Y.; Qian, W.; Li, X.; Wei, W. NF-κB Inhibitor Myrislignan Induces Ferroptosis of Glioblastoma Cells via Regulating Epithelial-Mesenchymal Transformation in a Slug-Dependent Manner. Oxidative Med. Cell. Longev. 2023, 2023, 7098313. [Google Scholar] [CrossRef]
- Xiao, X.; Yeoh, B.S.; Vijay-Kumar, M. Lipocalin 2: An Emerging Player in Iron Homeostasis and Inflammation. Annu. Rev. Nutr. 2017, 37, 103–130. [Google Scholar] [CrossRef]
- Yao, F.; Deng, Y.; Zhao, Y.; Mei, Y.; Zhang, Y.; Liu, X.; Martinez, C.; Su, X.; Rosato, R.R.; Teng, H.; et al. A Targetable LIFR−NF-κB−LCN2 Axis Controls Liver Tumorigenesis and Vulnerability to Ferroptosis. Nat. Commun. 2021, 12, 7333. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.L.; Freund, S.M.V.; Veprintsev, D.B.; Bycroft, M.; Fersht, A.R. Regulation of DNA Binding of P53 by Its C-Terminal Domain. J. Mol. Biol. 2004, 342, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.R. P300/CBP/P53 Interaction and Regulation of the P53 Response. Eur. J. Biochem. 2001, 268, 2773–2778. [Google Scholar] [CrossRef] [PubMed]
- Pflaum, J.; Schlosser, S.; Müller, M. P53 Family and Cellular Stress Responses in Cancer. Front. Oncol. 2014, 4, 285. [Google Scholar] [CrossRef]
- Mallette, F.A.; Calabrese, V.; Ilangumaran, S.; Ferbeyre, G. SOCS1, a Novel Interaction Partner of P53 Controlling Oncogene-Induced Senescence. Aging 2010, 2, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Saint-Germain, E.; Mignacca, L.; Vernier, M.; Bobbala, D.; Ilangumaran, S.; Ferbeyre, G. SOCS1 Regulates Senescence and Ferroptosis by Modulating the Expression of P53 Target Genes. Aging 2017, 9, 2137–2162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zheng, Q.; Yue, X.; Yuan, Z.; Ling, J.; Yuan, Y.; Liang, Y.; Sun, A.; Liu, Y.; Li, H.; et al. ZNF498 Promotes Hepatocellular Carcinogenesis by Suppressing P53-Mediated Apoptosis and Ferroptosis via the Attenuation of P53 Ser46 Phosphorylation. J. Exp. Clin. Cancer Res. 2022, 41, 79. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a P53-Mediated Activity during Tumour Suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, Y.; Zhang, J.; Yan, W.; Jung, Y.-S.; Chen, M.; Huang, E.; Lloyd, K.; Duan, Y.; Wang, J.; et al. Ferredoxin Reductase Is Critical for P53-Dependent Tumor Suppression via Iron Regulatory Protein 2. Genes Dev. 2017, 31, 1243–1256. [Google Scholar] [CrossRef]
- Zhang, Z.; Guo, M.; Shen, M.; Kong, D.; Zhang, F.; Shao, J.; Tan, S.; Wang, S.; Chen, A.; Cao, P.; et al. The BRD7-P53-SLC25A28 Axis Regulates Ferroptosis in Hepatic Stellate Cells. Redox Biol. 2020, 36, 101619. [Google Scholar] [CrossRef]
- Ou, Y.; Wang, S.-J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 Engages Polyamine Metabolism with P53-Mediated Ferroptotic Responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef]
- Ma, X.-H.; Liu, J.-H.-Z.; Liu, C.-Y.; Sun, W.-Y.; Duan, W.-J.; Wang, G.; Kurihara, H.; He, R.-R.; Li, Y.-F.; Chen, Y.; et al. ALOX15-Launched PUFA-Phospholipids Peroxidation Increases the Susceptibility of Ferroptosis in Ischemia-Induced Myocardial Damage. Signal Transduct. Target. Ther. 2022, 7, 288. [Google Scholar] [CrossRef]
- Fedorka, S.R.; So, K.; Al-Hamashi, A.A.; Gad, I.; Shah, R.; Kholodovych, V.; Alqahtani, H.D.; Taylor, W.R.; Tillekeratne, L.M.V. Small-Molecule Anticancer Agents Kill Cancer Cells by Harnessing Reactive Oxygen Species in an Iron-Dependent Manner. Org. Biomol. Chem. 2018, 16, 1465–1479. [Google Scholar] [CrossRef] [PubMed]
- Kuganesan, N.; Dlamini, S.; Tillekeratne, L.M.V.; Taylor, W.R. Tumor Suppressor P53 Promotes Ferroptosis in Oxidative Stress Conditions Independent of Modulation of Ferroptosis by P21, CDKs, RB, and E2F. J. Biol. Chem. 2021, 297, 101365. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, D.; Stockwell, B.R.; Prives, C. P21 Can Be a Barrier to Ferroptosis Independent of P53. Aging 2020, 12, 17800–17814. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Shang, X.; Sun, K.; Hou, Y.; Zhang, X.; Xu, J.; Liu, H.; Ruan, Z.; Hou, L.; Guo, Z.; et al. P21 Resists Ferroptosis in Osteoarthritic Chondrocytes by Regulating GPX4 Protein Stability. Free. Radic. Biol. Med. 2024, 212, 336–348. [Google Scholar] [CrossRef]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. P53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef]
- Johnson, T.M.; Hammond, E.M.; Giaccia, A.; Attardi, L.D. The p53QS Transactivation-Deficient Mutant Shows Stress-Specific Apoptotic Activity and Induces Embryonic Lethality. Nat. Genet. 2005, 37, 145–152. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Z.; Wang, X.-J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct Interaction between Nrf2 and p21Cip1/WAF1 Upregulates the Nrf2-Mediated Antioxidant Response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef]
- Tarangelo, A.; Dixon, S. The P53-P21 Pathway Inhibits Ferroptosis during Metabolic Stress. Oncotarget 2018, 9, 24572–24573. [Google Scholar] [CrossRef] [PubMed]
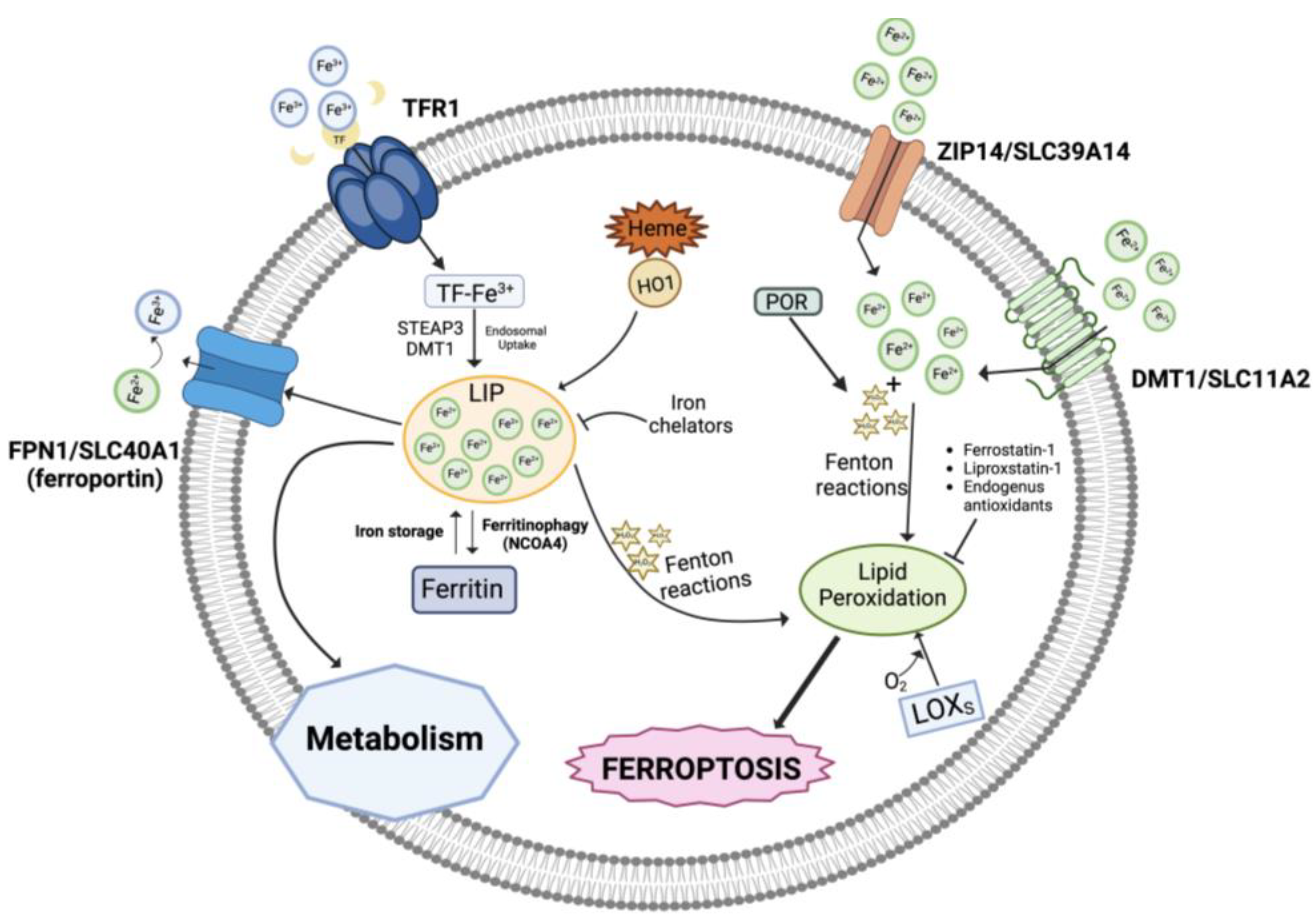
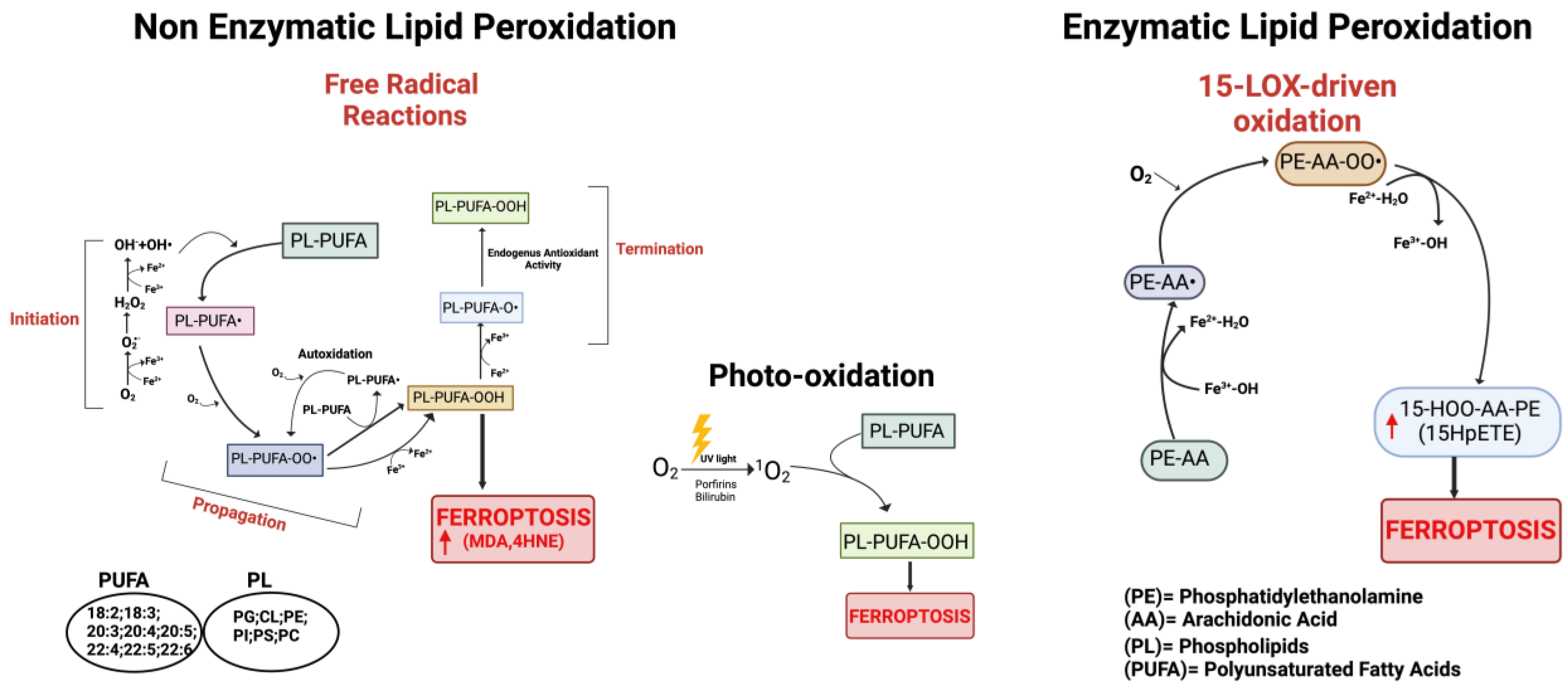
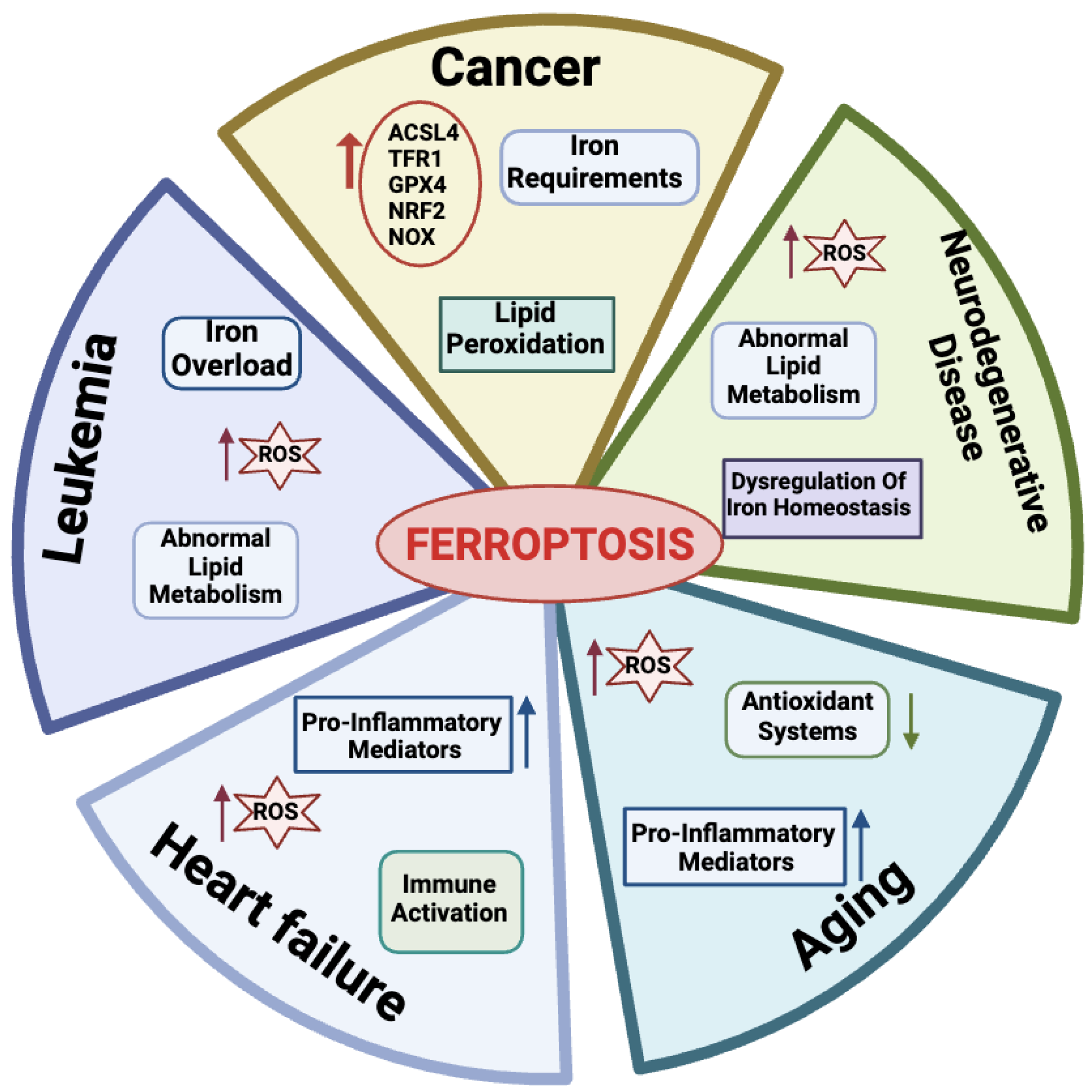
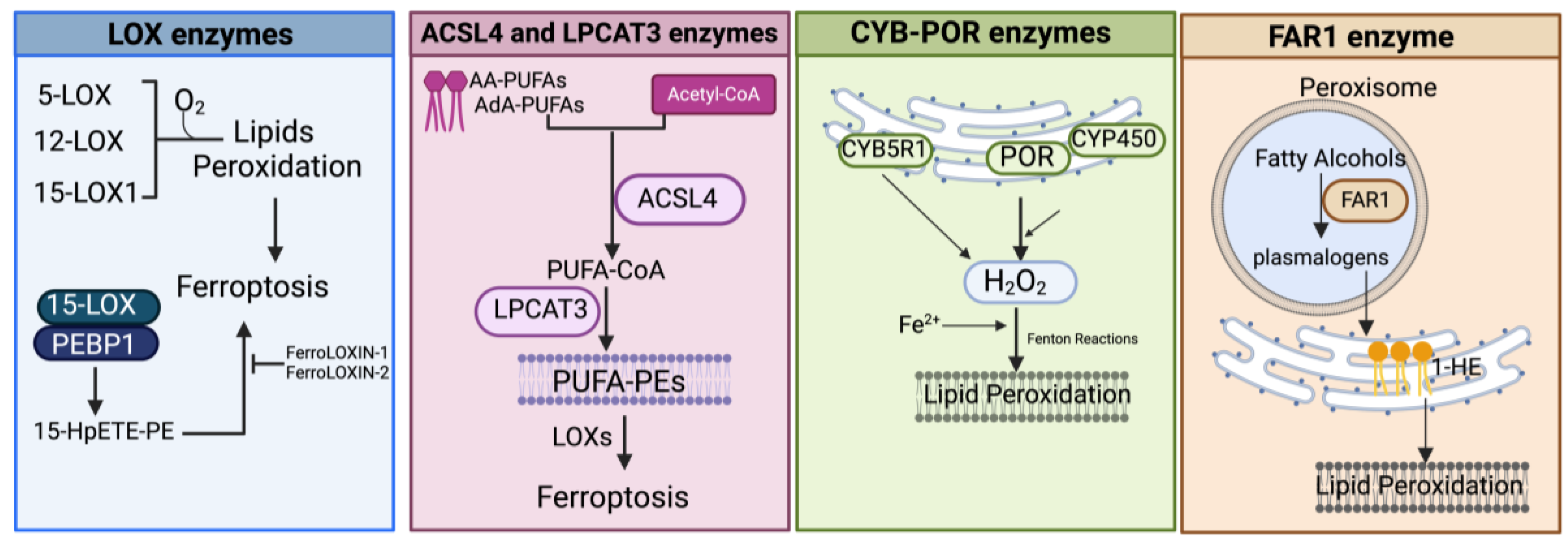
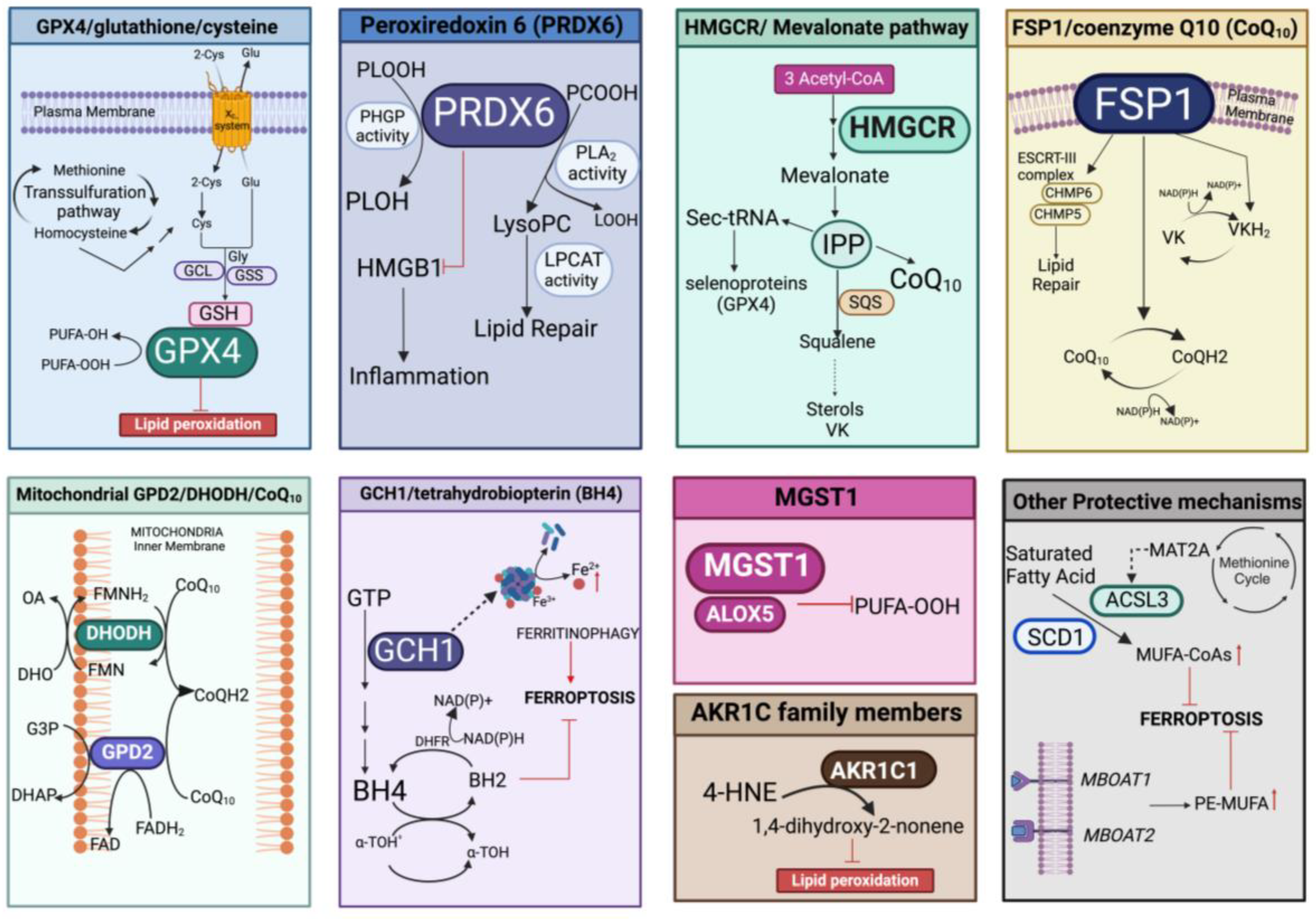


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Direct Target Genes | Transcription Factors and Effects on Target Genes | |
|---|---|---|
| FTH | NRF2 | ⭡ |
| TFAP2 | ⭣ | |
| STAT3 | ⭡ | |
| GPX4 | NRF2 | ⭡⭣ |
| ATF3 | ⭣ | |
| STAT1 | ⭣ | |
| STAT3 | ⭡ | |
| NF-kB | ⭡ | |
| HO1 | NRF2 | ⭡ |
| TFAP2 | ⭣ | |
| NF-kB | ⭡ | |
| NQO1 | NRF2 | ⭡ |
| TFAP2 | ⭡ | |
| NF-kB | ⭡ | |
| SCL7A11 | NRF2 | ⭡ |
| ATF4 | ⭡ | |
| ATF3 | ⭣ | |
| STAT1 | ⭡⭣ | |
| STAT3 | ⭡ | |
| NF-kB | ⭡⭣ | |
| P53 | ⭣ | |
| TXNDR1 | NRF2 | ⭡ |
| TFAP2 | ⭡ | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Punziano, C.; Trombetti, S.; Cesaro, E.; Grosso, M.; Faraonio, R. Antioxidant Systems as Modulators of Ferroptosis: Focus on Transcription Factors. Antioxidants 2024, 13, 298. https://doi.org/10.3390/antiox13030298
Punziano C, Trombetti S, Cesaro E, Grosso M, Faraonio R. Antioxidant Systems as Modulators of Ferroptosis: Focus on Transcription Factors. Antioxidants. 2024; 13(3):298. https://doi.org/10.3390/antiox13030298
Chicago/Turabian StylePunziano, Carolina, Silvia Trombetti, Elena Cesaro, Michela Grosso, and Raffaella Faraonio. 2024. "Antioxidant Systems as Modulators of Ferroptosis: Focus on Transcription Factors" Antioxidants 13, no. 3: 298. https://doi.org/10.3390/antiox13030298
APA StylePunziano, C., Trombetti, S., Cesaro, E., Grosso, M., & Faraonio, R. (2024). Antioxidant Systems as Modulators of Ferroptosis: Focus on Transcription Factors. Antioxidants, 13(3), 298. https://doi.org/10.3390/antiox13030298