

The Roles of Neutrophil-Derived Myeloperoxidase (MPO) in Diseases: The New Progress


Abstract
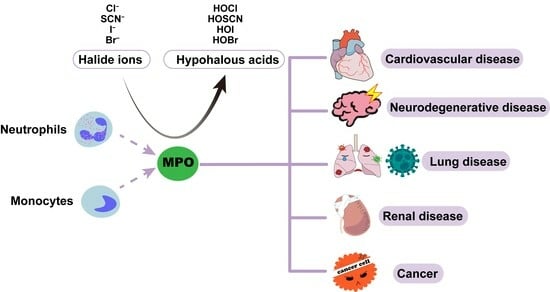
1. Introduction
2. Generation and Structure of MPO
3. MPO-Derived Oxidants
3.1. HOCl
3.2. HOSCN
3.3. ONOO−
4. Role of MPO in Innate Immunity
5. Role of MPO in Diseases
5.1. Cardiovascular Diseases
5.1.1. LDL
5.1.2. HDL
5.1.3. Endothelial Dysfunction
5.2. Neurodegenerative Diseases
5.2.1. Alzheimer’s Disease
5.2.2. Parkinson’s Disease
5.2.3. Multiple Sclerosis
5.3. Cancers
5.4. Renal Diseases
5.5. Lung Diseases and COVID-19
6. MPO Inhibitors in Clinical Trials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Pharmacophore | Inhibitor | Structure | IC50 | Pharmacological Effects | References |
|---|---|---|---|---|---|---|
| Irreversible inhibitors | Hydrazide | 4-ABAH |  | 0.3 μM | Improves neurogenesis after ischemic stroke (mice model); improves endothelial function and reduces atherosclerotic plaque development (mice model) | [158,229,242] |
| Xanthine | AZD4831 |  | 1.5 nM | Downregulates biomarkers associated with HFpEF (clinical trial) | [234,235,243,244] | |
| AZD3241 |  | 630 nM | Attenuates ALI (mice model); improves PD (clinical trial); and enhances immune checkpoint therapy for melanoma | [231,232,233] | ||
| AZD5904 |  | 140 nM | Alleviates the relaxation defect in hypertrophic human cardiomyocytes; enhances human sperm function in vitro | [245,246] | ||
| Thiouracil | PF-06282999 |  | 1.9 μM | Promotes atherosclerotic lesion stabilization and prevents atherosclerotic plaque rupture (mice model) | [236,247] | |
| Guanidine | MPO-IN-28 |  | 44 nM | Protects against endothelial glycocalyx degradation in primary human aortic endothelial cells cultured with plasma of COVID-19 patients | [237,248] | |
| Reversible inhibitors | Hydroxamic acid | SHA |  | 25 μM | No evidence of pharmacological effects; can be used to validate MPO inhibitors in silico | [238] |
| Tyrosine | KYC |  | 7 μM | Reduces bronchopulmonary dysplasia in hyperoxic neonatal rat pups; reduces oxidative injury and preserves neuronal function in MS (mice model); increases vasodilatation in sickle cell disease mice; promotes brain recovery from injury after stroke (mice model) | [240,249,250,251,252] | |
| Hydroxamate | HX1 |  | 5 nM | No evidence of pharmacological effects | [239,253] |
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ortiz-Cerda, T.; Xie, K.; Mojadadi, A.; Witting, P.K. Myeloperoxidase in Health and Disease. Int. J. Mol. Sci. 2023, 24, 7725. [Google Scholar] [CrossRef] [PubMed]
- Bos, A.; Wever, R.; Roos, D. Characterization and quantification of the peroxidase in human monocytes. Biochim. Biophys. Acta 1978, 525, 37–44. [Google Scholar] [CrossRef]
- Siraki, A.G. The many roles of myeloperoxidase: From inflammation and immunity to biomarkers, drug metabolism and drug discovery. Redox Biol. 2021, 46, 102109. [Google Scholar] [CrossRef] [PubMed]
- Agner, K. Verdoperoxidase; a ferment isolated from leucocytes. Acta Physiol. Scand. 1941, 2, 1–64. [Google Scholar]
- Agner, K. Detoxicating effect of verdoperoxidase on toxins. Nature 1947, 159, 271. [Google Scholar] [CrossRef] [PubMed]
- Marzouki, S.M.; Limam, F.; Smaali, M.I.; Ulber, R.; Marzouki, M.N. A new thermostable peroxidase from garlic Allium sativum: Purification, biochemical properties, immobilization, and use in H2O2 detection in milk. Appl. Biochem. Biotechnol. 2005, 127, 201–214. [Google Scholar] [CrossRef]
- Maehly, A.C. [142] Myeloperoxidase. Methods Enzymol. 1955, 2, 794–801. [Google Scholar]
- Klebanoff, S.J. Myeloperoxidase: Friend and foe. J. Leukoc. Biol. 2005, 77, 598–625. [Google Scholar] [CrossRef]
- Klebanoff, S.J. Myeloperoxidase-halide-hydrogen peroxide antibacterial system. J. Bacteriol. 1968, 95, 2131–2138. [Google Scholar] [CrossRef]
- Van Dalen, C.J.; Whitehouse, M.W.; Winterbourn, C.C.; Kettle, A.J. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem. J. 1997, 327 Pt 2, 487–492. [Google Scholar] [CrossRef]
- Prokopowicz, Z.; Marcinkiewicz, J.; Katz, D.R.; Chain, B.M. Neutrophil myeloperoxidase: Soldier and statesman. Arch. Immunol. Ther. Exp. 2012, 60, 43–54. [Google Scholar] [CrossRef]
- Odell, E.W.; Segal, A.W. The bactericidal effects of the respiratory burst and the myeloperoxidase system isolated in neutrophil cytoplasts. Biochim. Biophys. Acta 1988, 971, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Prütz, W.A. Hypochlorous acid interactions with thiols, nucleotides, DNA, and other biological substrates. Arch. Biochem. Biophys. 1996, 332, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; van den Berg, J.J.; Roitman, E.; Kuypers, F.A. Chlorohydrin formation from unsaturated fatty acids reacted with hypochlorous acid. Arch. Biochem. Biophys. 1992, 296, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Karakas, M.; Koenig, W. Myeloperoxidase production by macrophage and risk of atherosclerosis. Curr. Atheroscler. Rep. 2012, 14, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Pattison, D.I.; Davies, M.J. Kinetic analysis of the reactions of hypobromous acid with protein components: Implications for cellular damage and use of 3-bromotyrosine as a marker of oxidative stress. Biochemistry 2004, 43, 4799–4809. [Google Scholar] [CrossRef]
- Brennan, M.L.; Penn, M.S.; Van Lente, F.; Nambi, V.; Shishehbor, M.H.; Aviles, R.J.; Goormastic, M.; Pepoy, M.L.; McErlean, E.S.; Topol, E.J.; et al. Prognostic value of myeloperoxidase in patients with chest pain. N. Engl. J. Med. 2003, 349, 1595–1604. [Google Scholar] [CrossRef]
- Baldus, S.; Heeschen, C.; Meinertz, T.; Zeiher, A.M.; Eiserich, J.P.; Münzel, T.; Simoons, M.L.; Hamm, C.W. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation 2003, 108, 1440–1445. [Google Scholar] [CrossRef]
- Kothari, N.; Keshari, R.S.; Bogra, J.; Kohli, M.; Abbas, H.; Malik, A.; Dikshit, M.; Barthwal, M.K. Increased myeloperoxidase enzyme activity in plasma is an indicator of inflammation and onset of sepsis. J. Crit. Care 2011, 26, 435.e1–435.e7. [Google Scholar] [CrossRef]
- Zhu, A.; Ge, D.; Zhang, J.; Teng, Y.; Yuan, C.; Huang, M.; Adcock, I.M.; Barnes, P.J.; Yao, X. Sputum myeloperoxidase in chronic obstructive pulmonary disease. Eur. J. Med. Res. 2014, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Chelluboina, S.; Jedge, P.; Doke, P.; Palkar, S.; Mishra, A.C.; Arankalle, V.A. Elevated Levels of Neutrophil Activated Proteins, Alpha-Defensins (DEFA1), Calprotectin (S100A8/A9) and Myeloperoxidase (MPO) Are Associated with Disease Severity in COVID-19 Patients. Front. Cell. Infect. Microbiol. 2021, 11, 751232. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Ghanbari, E.P.; Jakobs, K.; Puccini, M.; Reinshagen, L.; Friebel, J.; Haghikia, A.; Kränkel, N.; Landmesser, U.; Rauch-Kröhnert, U. The Role of NETosis and Complement Activation in COVID-19-Associated Coagulopathies. Biomedicines 2023, 11, 1371. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef]
- Odobasic, D.; Kitching, A.R.; Holdsworth, S.R. Neutrophil-Mediated Regulation of Innate and Adaptive Immunity: The Role of Myeloperoxidase. J. Immunol. Res. 2016, 2016, 2349817. [Google Scholar] [CrossRef]
- Noguchi, N.; Nakano, K.; Aratani, Y.; Koyama, H.; Kodama, T.; Niki, E. Role of myeloperoxidase in the neutrophil-induced oxidation of low density lipoprotein as studied by myeloperoxidase-knockout mouse. J. Biochem. 2000, 127, 971–976. [Google Scholar] [CrossRef]
- Van der Veen, B.S.; de Winther, M.P.; Heeringa, P. Myeloperoxidase: Molecular mechanisms of action and their relevance to human health and disease. Antioxid. Redox Signal. 2009, 11, 2899–2937. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M. Posttranslational processing of a human myeloid lysosomal protein, myeloperoxidase. Blood 1987, 70, 1143–1150. [Google Scholar] [CrossRef]
- Nauseef, W.M.; McCormick, S.J.; Clark, R.A. Calreticulin functions as a molecular chaperone in the biosynthesis of myeloperoxidase. J. Biol. Chem. 1995, 270, 4741–4747. [Google Scholar] [CrossRef]
- Olsson, I.; Egesten, A.; Gullberg, U.; Lantz, M.; Strömberg, K.; Winqvist, I. The biosynthesis of neutrophil and eosinophil granule proteins. Folia Histochem. Cytobiol. 1986, 24, 89–97. [Google Scholar]
- Nauseef, W.M. Biosynthesis of human myeloperoxidase. Arch. Biochem. Biophys. 2018, 642, 1–9. [Google Scholar] [CrossRef]
- Ikeda-Saito, M. On the analogy in the structure of the spleen green heme protein and granulocyte myeloperoxidase. FEBS Lett. 1986, 202, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.C.; Krinsky, N.I. The reductive cleavage of myeloperoxidase in half, producing enzymically active hemi-myeloperoxidase. J. Biol. Chem. 1981, 256, 4211–4218. [Google Scholar] [CrossRef] [PubMed]
- Furtmüller, P.G.; Zederbauer, M.; Jantschko, W.; Helm, J.; Bogner, M.; Jakopitsch, C.; Obinger, C. Active site structure and catalytic mechanisms of human peroxidases. Arch. Biochem. Biophys. 2006, 445, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Van Antwerpen, P.; Slomianny, M.C.; Boudjeltia, K.Z.; Delporte, C.; Faid, V.; Calay, D.; Rousseau, A.; Moguilevsky, N.; Raes, M.; Vanhamme, L.; et al. Glycosylation pattern of mature dimeric leukocyte and recombinant monomeric myeloperoxidase: Glycosylation is required for optimal enzymatic activity. J. Biol. Chem. 2010, 285, 16351–16359. [Google Scholar] [CrossRef] [PubMed]
- Kooter, I.M.; Koehler, B.P.; Moguilevsky, N.; Bollen, A.; Wever, R.; Johnson, M.K. The Met243 sulfonium ion linkage is responsible for the anomalous magnetic circular dichroism and optical spectral properties of myeloperoxidase. J. Biol. Inorg. Chem. JBIC Publ. Soc. Biol. Inorg. Chem. 1999, 4, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Kettle, A.J.; Winterbourn, C.C. Myeloperoxidase: A key regulator of neutrophil oxidant production. Redox Rep. Commun. Free Radic. Res. 1997, 3, 3–15. [Google Scholar] [CrossRef]
- Davies, M.J.; Hawkins, C.L. The Role of Myeloperoxidase in Biomolecule Modification, Chronic Inflammation, and Disease. Antioxid. Redox Signal 2020, 32, 957–981. [Google Scholar] [CrossRef]
- Davies, M.J.; Hawkins, C.L.; Pattison, D.I.; Rees, M.D. Mammalian heme peroxidases: From molecular mechanisms to health implications. Antioxid. Redox Signal 2008, 10, 1199–1234. [Google Scholar] [CrossRef]
- Metodiewa, D.; Dunford, H.B. The reactions of horseradish peroxidase, lactoperoxidase, and myeloperoxidase with enzymatically generated superoxide. Arch. Biochem. Biophys. 1989, 272, 245–253. [Google Scholar] [CrossRef]
- Marquez, L.A.; Dunford, H.B. Reaction of compound III of myeloperoxidase with ascorbic acid. J. Biol. Chem. 1990, 265, 6074–6078. [Google Scholar] [CrossRef] [PubMed]
- Delporte, C.; Van Antwerpen, P.; Vanhamme, L.; Roumeguère, T.; Zouaoui Boudjeltia, K. Low-density lipoprotein modified by myeloperoxidase in inflammatory pathways and clinical studies. Mediat. Inflamm. 2013, 2013, 971579. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.L.; Skaff, O.; Senthilmohan, R.; Kettle, A.J.; Davies, M.J. Hypobromous acid and bromamine production by neutrophils and modulation by superoxide. Biochem. J. 2009, 417, 773–781. [Google Scholar] [CrossRef]
- Lymar, S.V.; Hurst, J.K. Role of compartmentation in promoting toxicity of leukocyte-generated strong oxidants. Chem. Res. Toxicol. 1995, 8, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Pattison, D.I.; Davies, M.J. Absolute rate constants for the reaction of hypochlorous acid with protein side chains and peptide bonds. Chem. Res. Toxicol. 2001, 14, 1453–1464. [Google Scholar] [CrossRef]
- Pattison, D.I.; Davies, M.J. Reactions of myeloperoxidase-derived oxidants with biological substrates: Gaining chemical insight into human inflammatory diseases. Curr. Med. Chem. 2006, 13, 3271–3290. [Google Scholar] [CrossRef] [PubMed]
- Ashby, M.T.; Carlson, A.C.; Scott, M.J. Redox buffering of hypochlorous acid by thiocyanate in physiologic fluids. J. Am. Chem. Soc. 2004, 126, 15976–15977. [Google Scholar] [CrossRef]
- Jantschko, W.; Furtmüller, P.G.; Zederbauer, M.; Neugschwandtner, K.; Lehner, I.; Jakopitsch, C.; Arnhold, J.; Obinger, C. Exploitation of the unusual thermodynamic properties of human myeloperoxidase in inhibitor design. Biochem. Pharmacol. 2005, 69, 1149–1157. [Google Scholar] [CrossRef]
- Ashby, M.T. Inorganic chemistry of defensive peroxidases in the human oral cavity. J. Dent. Res. 2008, 87, 900–914. [Google Scholar] [CrossRef]
- Wang, J.; Slungaard, A. Role of eosinophil peroxidase in host defense and disease pathology. Arch. Biochem. Biophys. 2006, 445, 256–260. [Google Scholar] [CrossRef]
- Skaff, O.; Pattison, D.I.; Davies, M.J. Hypothiocyanous acid reactivity with low-molecular-mass and protein thiols: Absolute rate constants and assessment of biological relevance. Biochem. J. 2009, 422, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Hondal, R.J.; Marino, S.M.; Gladyshev, V.N. Selenocysteine in thiol/disulfide-like exchange reactions. Antioxid. Redox Signal 2013, 18, 1675–1689. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.E.; Tan, J.T.; Hawkins, C.L.; Heather, A.K.; Davies, M.J. The myeloperoxidase-derived oxidant HOSCN inhibits protein tyrosine phosphatases and modulates cell signalling via the mitogen-activated protein kinase (MAPK) pathway in macrophages. Biochem. J. 2010, 430, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, M.M.; van Reyk, D.M.; Davies, M.J.; Hawkins, C.L. Hypothiocyanous acid is a more potent inducer of apoptosis and protein thiol depletion in murine macrophage cells than hypochlorous acid or hypobromous acid. Biochem. J. 2008, 414, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Szép, S.; Lu, Z. The antioxidant role of thiocyanate in the pathogenesis of cystic fibrosis and other inflammation-related diseases. Proc. Natl. Acad. Sci. USA 2009, 106, 20515–20519. [Google Scholar] [CrossRef]
- Nunes, P.; Demaurex, N.; Dinauer, M.C. Regulation of the NADPH oxidase and associated ion fluxes during phagocytosis. Traffic 2013, 14, 1118–1131. [Google Scholar] [CrossRef] [PubMed]
- Segal, B.H.; Grimm, M.J.; Khan, A.N.; Han, W.; Blackwell, T.S. Regulation of innate immunity by NADPH oxidase. Free Radic. Biol. Med. 2012, 53, 72–80. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef]
- Hogg, N.; Darley-Usmar, V.M.; Wilson, M.T.; Moncada, S. Production of hydroxyl radicals from the simultaneous generation of superoxide and nitric oxide. Biochem. J. 1992, 281 Pt 2, 419–424. [Google Scholar] [CrossRef]
- Denicola, A.; Rubbo, H.; Rodríguez, D.; Radi, R. Peroxynitrite-mediated cytotoxicity to Trypanosoma cruzi. Arch. Biochem. Biophys. 1993, 304, 279–286. [Google Scholar] [CrossRef]
- Burney, S.; Caulfield, J.L.; Niles, J.C.; Wishnok, J.S.; Tannenbaum, S.R. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat. Res. 1999, 424, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef] [PubMed]
- Van der Loo, B.; Labugger, R.; Skepper, J.N.; Bachschmid, M.; Kilo, J.; Powell, J.M.; Palacios-Callender, M.; Erusalimsky, J.D.; Quaschning, T.; Malinski, T.; et al. Enhanced peroxynitrite formation is associated with vascular aging. J. Exp. Med. 2000, 192, 1731–1744. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.; Denicola, A.; Radi, R. Red blood cells in the metabolism of nitric oxide-derived peroxynitrite. IUBMB Life 2006, 58, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Mihm, M.J.; Wattanapitayakul, S.K.; Piao, S.F.; Hoyt, D.G.; Bauer, J.A. Effects of angiotensin II on vascular endothelial cells: Formation of receptor-mediated reactive nitrogen species. Biochem. Pharmacol. 2003, 65, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.F.; Bruce-Keller, A.J.; Goodman, Y.; Mattson, M.P. Uric acid protects neurons against excitotoxic and metabolic insults in cell culture, and against focal ischemic brain injury in vivo. J. Neurosci. Res. 1998, 53, 613–625. [Google Scholar] [CrossRef]
- Alvarez, S.; Boveris, A. Mitochondrial nitric oxide metabolism in rat muscle during endotoxemia. Free Radic. Biol. Med. 2004, 37, 1472–1478. [Google Scholar] [CrossRef]
- Oh-hashi, K.; Maruyama, W.; Yi, H.; Takahashi, T.; Naoi, M.; Isobe, K. Mitogen-activated protein kinase pathway mediates peroxynitrite-induced apoptosis in human dopaminergic neuroblastoma SH-SY5Y cells. Biochem. Biophys. Res. Commun. 1999, 263, 504–509. [Google Scholar] [CrossRef]
- Cabassi, A.; Dumont, E.C.; Girouard, H.; Bouchard, J.F.; Le Jossec, M.; Lamontagne, D.; Besner, J.G.; de Champlain, J. Effects of chronic N-acetylcysteine treatment on the actions of peroxynitrite on aortic vascular reactivity in hypertensive rats. J. Hypertens. 2001, 19, 1233–1244. [Google Scholar] [CrossRef]
- Ziegler, D.; Nowak, H.; Kempler, P.; Vargha, P.; Low, P.A. Treatment of symptomatic diabetic polyneuropathy with the antioxidant alpha-lipoic acid: A meta-analysis. Diabet. Med. J. Br. Diabet. Assoc. 2004, 21, 114–121. [Google Scholar] [CrossRef]
- Ferrer-Sueta, G.; Radi, R. Chemical biology of peroxynitrite: Kinetics, diffusion, and radicals. ACS Chem. Biol. 2009, 4, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Rizo-Téllez, S.A.; Sekheri, M.; Filep, J.G. Myeloperoxidase: Regulation of Neutrophil Function and Target for Therapy. Antioxidants 2022, 11, 2302. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, S.J. Iodination of bacteria: A bactericidal mechanism. J. Exp. Med. 1967, 126, 1063–1078. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M.; Takeuchi, C.; Ruedi, J.; Gutierrez, A.; Wentworth, P., Jr. Investigating antibody-catalyzed ozone generation by human neutrophils. Proc. Natl. Acad. Sci. USA 2003, 100, 3031–3034. [Google Scholar] [CrossRef] [PubMed]
- Kettle, A.J.; Maroz, A.; Woodroffe, G.; Winterbourn, C.C.; Anderson, R.F. Spectral and kinetic evidence for reaction of superoxide with compound I of myeloperoxidase. Free Radic. Biol. Med. 2011, 51, 2190–2194. [Google Scholar] [CrossRef]
- Marcinkiewicz, J.; Walczewska, M. Neutrophils as Sentinel Cells of the Immune System: A Role of the MPO-halide-system in Innate and Adaptive Immunity. Curr. Med. Chem. 2020, 27, 2840–2851. [Google Scholar] [CrossRef]
- McKenna, S.M.; Davies, K.J. The inhibition of bacterial growth by hypochlorous acid. Possible role in the bactericidal activity of phagocytes. Biochem. J. 1988, 254, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Hirche, T.O.; Gaut, J.P.; Heinecke, J.W.; Belaaouaj, A. Myeloperoxidase plays critical roles in killing Klebsiella pneumoniae and inactivating neutrophil elastase: Effects on host defense. J. Immunol. 2005, 174, 1557–1565. [Google Scholar] [CrossRef]
- Montecucco, F.; Bertolotto, M.; Ottonello, L.; Pende, A.; Dapino, P.; Quercioli, A.; Mach, F.; Dallegri, F. Chlorhexidine prevents hypochlorous acid-induced inactivation of alpha1-antitrypsin. Clin. Exp. Pharmacol. Physiol. 2009, 36, e72–e77. [Google Scholar] [CrossRef]
- Olszowska, E.; Olszowski, S.; Zgliczyński, J.M.; Stelmaszyńska, T. Enhancement of proteinase-mediated degradation of proteins modified by chlorination. Int. J. Biochem. 1989, 21, 799–805. [Google Scholar] [CrossRef]
- Romano, M.; Dri, P.; Da Dalt, L.; Patriarca, P.; Baralle, F.E. Biochemical and molecular characterization of hereditary myeloperoxidase deficiency. Blood 1997, 90, 4126–4134. [Google Scholar] [CrossRef]
- Kutter, D. Prevalence of myeloperoxidase deficiency: Population studies using Bayer-Technicon automated hematology. J. Mol. Med. 1998, 76, 669–675. [Google Scholar] [CrossRef]
- Reynolds, W.F.; Stegeman, C.A.; Tervaert, J.W. -463 G/A myeloperoxidase promoter polymorphism is associated with clinical manifestations and the course of disease in MPO-ANCA-associated vasculitis. Clin. Immunol. 2002, 103, 154–160. [Google Scholar] [CrossRef]
- Weng, K.P.; Hsieh, K.S.; Huang, S.H.; Wu, H.W.; Chien, J.H.; Lin, C.C.; Tang, C.W.; Ou, S.F.; Huang, S.J.; Ger, L.P. Myeloperoxidase genetic polymorphisms and susceptibility to Kawasaki disease in Taiwanese children. J. Microbiol. Immunol. Infect. 2016, 49, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Inside the neutrophil phagosome: Oxidants, myeloperoxidase, and bacterial killing. Blood 1998, 92, 3007–3017. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.A.; Gandhi, A.A.; Meng, H.; Yalavarthi, S.; Vreede, A.P.; Estes, S.K.; Palmer, O.R.; Bockenstedt, P.L.; Pinsky, D.J.; Greve, J.M.; et al. Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat. Commun. 2019, 10, 1916. [Google Scholar] [CrossRef]
- Frantzeskaki, F.; Armaganidis, A.; Orfanos, S.E. Immunothrombosis in Acute Respiratory Distress Syndrome: Cross Talks between Inflammation and Coagulation. Respir. Int. Rev. Thorac. Dis. 2017, 93, 212–225. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef]
- Klinke, A.; Nussbaum, C.; Kubala, L.; Friedrichs, K.; Rudolph, T.K.; Rudolph, V.; Paust, H.J.; Schröder, C.; Benten, D.; Lau, D.; et al. Myeloperoxidase attracts neutrophils by physical forces. Blood 2011, 117, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Parry, M.F.; Root, R.K.; Metcalf, J.A.; Delaney, K.K.; Kaplow, L.S.; Richar, W.J. Myeloperoxidase deficiency: Prevalence and clinical significance. Ann. Intern. Med. 1981, 95, 293–301. [Google Scholar] [CrossRef]
- Vanhamme, L.; Zouaoui Boudjeltia, K.; Van Antwerpen, P.; Delporte, C. The other myeloperoxidase: Emerging functions. Arch. Biochem. Biophys. 2018, 649, 1–14. [Google Scholar] [CrossRef]
- Baldus, S.; Heitzer, T.; Eiserich, J.P.; Lau, D.; Mollnau, H.; Ortak, M.; Petri, S.; Goldmann, B.; Duchstein, H.J.; Berger, J.; et al. Myeloperoxidase enhances nitric oxide catabolism during myocardial ischemia and reperfusion. Free Radic. Biol. Med. 2004, 37, 902–911. [Google Scholar] [CrossRef]
- Trentini, A.; Rosta, V.; Spadaro, S.; Bellini, T.; Rizzo, P.; Vieceli Dalla Sega, F.; Passaro, A.; Zuliani, G.; Gentili, V.; Campo, G.; et al. Development, optimization and validation of an absolute specific assay for active myeloperoxidase (MPO) and its application in a clinical context: Role of MPO specific activity in coronary artery disease. Clin. Chem. Lab. Med. 2020, 58, 1749–1758. [Google Scholar] [CrossRef]
- Matsuo, Y.; Onodera, H.; Shiga, Y.; Nakamura, M.; Ninomiya, M.; Kihara, T.; Kogure, K. Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat. Effects of neutrophil depletion. Stroke 1994, 25, 1469–1475. [Google Scholar] [CrossRef]
- Forghani, R.; Kim, H.J.; Wojtkiewicz, G.R.; Bure, L.; Wu, Y.; Hayase, M.; Wei, Y.; Zheng, Y.; Moskowitz, M.A.; Chen, J.W. Myeloperoxidase propagates damage and is a potential therapeutic target for subacute stroke. J. Cereb. Blood Flow Metab. 2015, 35, 485–493. [Google Scholar] [CrossRef]
- Kim, H.J.; Wei, Y.; Wojtkiewicz, G.R.; Lee, J.Y.; Moskowitz, M.A.; Chen, J.W. Reducing myeloperoxidase activity decreases inflammation and increases cellular protection in ischemic stroke. J. Cereb. Blood Flow Metab. 2019, 39, 1864–1877. [Google Scholar] [CrossRef]
- Li, J.; Cao, T.; Wei, Y.; Zhang, N.; Zhou, Z.; Wang, Z.; Li, J.; Zhang, Y.; Wang, S.; Wang, P.; et al. A Review of Novel Cardiac Biomarkers in Acute or Chronic Cardiovascular Diseases: The Role of Soluble ST2 (sST2), Lipoprotein-Associated Phospholipase A2 (Lp-PLA2), Myeloperoxidase (MPO), and Procalcitonin (PCT). Dis. Markers 2021, 2021, 6258865. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.D.; Gransar, H.; Narula, J.; Shaw, L.; Moon, J.H.; Miranda-Peats, R.; Rozanski, A.; Hayes, S.W.; Thomson, L.E.; Friedman, J.D.; et al. Myeloperoxidase, subclinical atherosclerosis, and cardiovascular disease events. JACC Cardiovasc. Imaging 2009, 2, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Nadel, J.; Tumanov, S.; Kong, S.M.Y.; Chen, W.; Giannotti, N.; Sivasubramaniam, V.; Rashid, I.; Ugander, M.; Jabbour, A.; Stocker, R. Intraplaque Myeloperoxidase Activity as Biomarker of Unstable Atheroma and Adverse Clinical Outcomes in Human Atherosclerosis. JACC Adv. 2023, 2, 100310. [Google Scholar] [CrossRef]
- Rashid, I.; Maghzal, G.J.; Chen, Y.C.; Cheng, D.; Talib, J.; Newington, D.; Ren, M.; Vajandar, S.K.; Searle, A.; Maluenda, A.; et al. Myeloperoxidase is a potential molecular imaging and therapeutic target for the identification and stabilization of high-risk atherosclerotic plaque. Eur. Heart J. 2018, 39, 3301–3310. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, A.; Dunn, J.L.; Rateri, D.L.; Heinecke, J.W. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Investig. 1994, 94, 437–444. [Google Scholar] [CrossRef]
- Chen, W.; Tumanov, S.; Kong, S.M.Y.; Cheng, D.; Michaëlsson, E.; Bongers, A.; Power, C.; Ayer, A.; Stocker, R. Therapeutic inhibition of MPO stabilizes pre-existing high risk atherosclerotic plaque. Redox Biol. 2022, 58, 102532. [Google Scholar] [CrossRef]
- Dhote, V.; Balaraman, R. Anti-oxidant activity mediated neuroprotective potential of trimetazidine on focal cerebral ischaemia-reperfusion injury in rats. Clin. Exp. Pharmacol. Physiol. 2008, 35, 630–637. [Google Scholar] [CrossRef]
- Podrez, E.A.; Schmitt, D.; Hoff, H.F.; Hazen, S.L. Myeloperoxidase-generated reactive nitrogen species convert LDL into an atherogenic form in vitro. J. Clin. Investig. 1999, 103, 1547–1560. [Google Scholar] [CrossRef]
- Abu-Soud, H.M.; Hazen, S.L. Nitric oxide is a physiological substrate for mammalian peroxidases. J. Biol. Chem. 2000, 275, 37524–37532. [Google Scholar] [CrossRef]
- Teng, N.; Maghzal, G.J.; Talib, J.; Rashid, I.; Lau, A.K.; Stocker, R. The roles of myeloperoxidase in coronary artery disease and its potential implication in plaque rupture. Redox Rep. Commun. Free Radic. Res. 2017, 22, 51–73. [Google Scholar] [CrossRef]
- Sugiyama, S.; Kugiyama, K.; Aikawa, M.; Nakamura, S.; Ogawa, H.; Libby, P. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: Involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1309–1314. [Google Scholar] [CrossRef]
- Podrez, E.A.; Febbraio, M.; Sheibani, N.; Schmitt, D.; Silverstein, R.L.; Hajjar, D.P.; Cohen, P.A.; Frazier, W.A.; Hoff, H.F.; Hazen, S.L. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J. Clin. Investig. 2000, 105, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; McCall, M.R.; Frei, B. Oxidation of LDL by myeloperoxidase and reactive nitrogen species: Reaction pathways and antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1716–1723. [Google Scholar] [CrossRef] [PubMed]
- Hazen, S.L.; Heinecke, J.W. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J. Clin. Investig. 1997, 99, 2075–2081. [Google Scholar] [CrossRef]
- Schindhelm, R.K.; van der Zwan, L.P.; Teerlink, T.; Scheffer, P.G. Myeloperoxidase: A useful biomarker for cardiovascular disease risk stratification? Clin. Chem. 2009, 55, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Geovanini, G.R.; Libby, P. Atherosclerosis and inflammation: Overview and updates. Clin. Sci. 2018, 132, 1243–1252. [Google Scholar] [CrossRef]
- Stocker, R.; Keaney, J.F., Jr. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef]
- Prasad, A.; Tsimikas, S. Candidate biomarkers for the detection of coronary plaque destabilization and rupture. Curr. Atheroscler. Rep. 2008, 10, 309–317. [Google Scholar] [CrossRef]
- Zhang, Y.; Zanotti, I.; Reilly, M.P.; Glick, J.M.; Rothblat, G.H.; Rader, D.J. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation 2003, 108, 661–663. [Google Scholar] [CrossRef]
- Pan, B.; Yu, B.; Ren, H.; Willard, B.; Pan, L.; Zu, L.; Shen, X.; Ma, Y.; Li, X.; Niu, C.; et al. High-density lipoprotein nitration and chlorination catalyzed by myeloperoxidase impair its effect of promoting endothelial repair. Free Radic. Biol. Med. 2013, 60, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Hama, S.Y.; Anantharamaiah, G.M.; Hassan, K.; Hough, G.P.; Watson, A.D.; Reddy, S.T.; Sevanian, A.; Fonarow, G.C.; Fogelman, A.M. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: Steps 2 and 3. J. Lipid Res. 2000, 41, 1495–1508. [Google Scholar] [CrossRef]
- Kontush, A.; Chapman, M.J. Functionally defective high-density lipoprotein: A new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol. Rev. 2006, 58, 342–374. [Google Scholar] [CrossRef]
- Davidson, W.S.; Thompson, T.B. The structure of apolipoprotein A-I in high density lipoproteins. J. Biol. Chem. 2007, 282, 22249–22253. [Google Scholar] [CrossRef]
- Ertek, S. High-density Lipoprotein (HDL) Dysfunction and the Future of HDL. Curr. Vasc. Pharmacol. 2018, 16, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Johansen, O.; Abdelnoor, M.; Brekke, M.; Seljeflot, I.; Høstmark, A.T.; Arnesen, H. Predictors of restenosis after coronary angioplasty. A study on demographic and metabolic variables. Scand. Cardiovasc. J. SCJ 2001, 35, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; DiDonato, J.A.; Levison, B.S.; Schmitt, D.; Li, L.; Wu, Y.; Buffa, J.; Kim, T.; Gerstenecker, G.S.; Gu, X.; et al. An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat. Med. 2014, 20, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Nukuna, B.; Brennan, M.L.; Sun, M.; Goormastic, M.; Settle, M.; Schmitt, D.; Fu, X.; Thomson, L.; Fox, P.L.; et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J. Clin. Investig. 2004, 114, 529–541. [Google Scholar] [CrossRef]
- Jin, Z.; Zhou, L.; Tian, R.; Lu, N. Myeloperoxidase Targets Apolipoprotein A-I for Site-Specific Tyrosine Chlorination in Atherosclerotic Lesions and Generates Dysfunctional High-Density Lipoprotein. Chem. Res. Toxicol. 2021, 34, 1672–1680. [Google Scholar] [CrossRef]
- Shao, B.; Tang, C.; Sinha, A.; Mayer, P.S.; Davenport, G.D.; Brot, N.; Oda, M.N.; Zhao, X.Q.; Heinecke, J.W. Humans with atherosclerosis have impaired ABCA1 cholesterol efflux and enhanced high-density lipoprotein oxidation by myeloperoxidase. Circ. Res. 2014, 114, 1733–1742. [Google Scholar] [CrossRef]
- Marsche, G.; Furtmüller, P.G.; Obinger, C.; Sattler, W.; Malle, E. Hypochlorite-modified high-density lipoprotein acts as a sink for myeloperoxidase in vitro. Cardiovasc. Res. 2008, 79, 187–194. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nicholls, S.J.; Hazen, S.L. Myeloperoxidase, modified lipoproteins, and atherogenesis. J. Lipid Res. 2009, 50, S346–S351. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Pennathur, S.; Pagani, I.; Oda, M.N.; Witztum, J.L.; Oram, J.F.; Heinecke, J.W. Modifying apolipoprotein A-I by malondialdehyde, but not by an array of other reactive carbonyls, blocks cholesterol efflux by the ABCA1 pathway. J. Biol. Chem. 2010, 285, 18473–18484. [Google Scholar] [CrossRef] [PubMed]
- May-Zhang, L.S.; Yermalitsky, V.; Huang, J.; Pleasent, T.; Borja, M.S.; Oda, M.N.; Jerome, W.G.; Yancey, P.G.; Linton, M.F.; Davies, S.S. Modification by isolevuglandins, highly reactive γ-ketoaldehydes, deleteriously alters high-density lipoprotein structure and function. J. Biol. Chem. 2018, 293, 9176–9187. [Google Scholar] [CrossRef]
- Kameda, T.; Usami, Y.; Shimada, S.; Haraguchi, G.; Matsuda, K.; Sugano, M.; Kurihara, Y.; Isobe, M.; Tozuka, M. Determination of myeloperoxidase-induced apoAI-apoAII heterodimers in high-density lipoprotein. Ann. Clin. Lab. Sci. 2012, 42, 384–391. [Google Scholar] [PubMed]
- Kameda, T.; Horiuchi, Y.; Shimano, S.; Yano, K.; Lai, S.J.; Ichimura, N.; Tohda, S.; Kurihara, Y.; Tozuka, M.; Ohkawa, R. Effect of myeloperoxidase oxidation and N-homocysteinylation of high-density lipoprotein on endothelial repair function. Biol. Chem. 2022, 403, 265–277. [Google Scholar] [CrossRef]
- Celermajer, D.S.; Sorensen, K.E.; Gooch, V.M.; Spiegelhalter, D.J.; Miller, O.I.; Sullivan, I.D.; Lloyd, J.K.; Deanfield, J.E. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet 1992, 340, 1111–1115. [Google Scholar] [CrossRef]
- Hartman, C.L.; Ford, D.A. MPO (Myeloperoxidase) Caused Endothelial Dysfunction. Arter. Thromb. Vasc. Biol. 2018, 38, 1676–1677. [Google Scholar] [CrossRef]
- Cheng, D.; Talib, J.; Stanley, C.P.; Rashid, I.; Michaëlsson, E.; Lindstedt, E.L.; Croft, K.D.; Kettle, A.J.; Maghzal, G.J.; Stocker, R. Inhibition of MPO (Myeloperoxidase) Attenuates Endothelial Dysfunction in Mouse Models of Vascular Inflammation and Atherosclerosis. Arter. Thromb. Vasc. Biol. 2019, 39, 1448–1457. [Google Scholar] [CrossRef]
- Palmer, R.M.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef]
- Tiruppathi, C.; Naqvi, T.; Wu, Y.; Vogel, S.M.; Minshall, R.D.; Malik, A.B. Albumin mediates the transcytosis of myeloperoxidase by means of caveolae in endothelial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 7699–7704. [Google Scholar] [CrossRef] [PubMed]
- Eiserich, J.P.; Baldus, S.; Brennan, M.L.; Ma, W.; Zhang, C.; Tousson, A.; Castro, L.; Lusis, A.J.; Nauseef, W.M.; White, C.R.; et al. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science 2002, 296, 2391–2394. [Google Scholar] [CrossRef]
- Harrison, D.G. Cellular and molecular mechanisms of endothelial cell dysfunction. J. Clin. Investig. 1997, 100, 2153–2157. [Google Scholar] [CrossRef] [PubMed]
- Etwebi, Z.; Landesberg, G.; Preston, K.; Eguchi, S.; Scalia, R. Mechanistic Role of the Calcium-Dependent Protease Calpain in the Endothelial Dysfunction Induced by MPO (Myeloperoxidase). Hypertension 2018, 71, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Huang, A.; Jeranian, E.; Hou, J.Y.; Wu, T.T.; Thomas, S.R.; Keaney, J.F., Jr. Hypochlorous acid impairs endothelium-derived nitric oxide bioactivity through a superoxide-dependent mechanism. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2028–2033. [Google Scholar] [CrossRef]
- Hazell, L.J.; Arnold, L.; Flowers, D.; Waeg, G.; Malle, E.; Stocker, R. Presence of hypochlorite-modified proteins in human atherosclerotic lesions. J. Clin. Investig. 1996, 97, 1535–1544. [Google Scholar] [CrossRef]
- Luft, J.H. Fine structures of capillary and endocapillary layer as revealed by ruthenium red. Fed. Proc. 1966, 25, 1773–1783. [Google Scholar]
- Constantinescu, A.A.; Vink, H.; Spaan, J.A. Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1541–1547. [Google Scholar] [CrossRef]
- Manchanda, K.; Kolarova, H.; Kerkenpaß, C.; Mollenhauer, M.; Vitecek, J.; Rudolph, V.; Kubala, L.; Baldus, S.; Adam, M.; Klinke, A. MPO (Myeloperoxidase) Reduces Endothelial Glycocalyx Thickness Dependent on Its Cationic Charge. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1859–1867. [Google Scholar] [CrossRef]
- Wagner, B.A.; Buettner, G.R.; Oberley, L.W.; Darby, C.J.; Burns, C.P. Myeloperoxidase is involved in H2O2-induced apoptosis of HL-60 human leukemia cells. J. Biol. Chem. 2000, 275, 22461–22469. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Yoshikawa, N.; Kagota, S.; Nakamura, K.; Haginaka, J.; Kunitomo, M. Elevated circulating levels of markers of oxidative-nitrative stress and inflammation in a genetic rat model of metabolic syndrome. Nitric Oxide Biol. Chem. 2006, 15, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.S.; Katyal, A. Myeloperoxidase: Bridging the gap in neurodegeneration. Neurosci. Biobehav. Rev. 2016, 68, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Simard, A.R.; Rivest, S. Neuroprotective properties of the innate immune system and bone marrow stem cells in Alzheimer’s disease. Mol. Psychiatry 2006, 11, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Maki, R.A.; Tyurin, V.A.; Lyon, R.C.; Hamilton, R.L.; DeKosky, S.T.; Kagan, V.E.; Reynolds, W.F. Aberrant expression of myeloperoxidase in astrocytes promotes phospholipid oxidation and memory deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 2009, 284, 3158–3169. [Google Scholar] [CrossRef] [PubMed]
- Pope, S.K.; Kritchevsky, S.B.; Ambrosone, C.; Yaffe, K.; Tylavsky, F.; Simonsick, E.M.; Rosano, C.; Stewart, S.; Harris, T. Myeloperoxidase polymorphism and cognitive decline in older adults in the Health, Aging, and Body Composition Study. Am. J. Epidemiol. 2006, 163, 1084–1090. [Google Scholar] [CrossRef][Green Version]
- Whiteman, M.; Rose, P.; Siau, J.L.; Cheung, N.S.; Tan, G.S.; Halliwell, B.; Armstrong, J.S. Hypochlorous acid-mediated mitochondrial dysfunction and apoptosis in human hepatoma HepG2 and human fetal liver cells: Role of mitochondrial permeability transition. Free Radic. Biol. Med. 2005, 38, 1571–1584. [Google Scholar] [CrossRef]
- Schraufstätter, I.U.; Browne, K.; Harris, A.; Hyslop, P.A.; Jackson, J.H.; Quehenberger, O.; Cochrane, C.G. Mechanisms of hypochlorite injury of target cells. J. Clin. Investig. 1990, 85, 554–562. [Google Scholar] [CrossRef]
- Kim, H.; Wei, Y.; Lee, J.Y.; Wu, Y.; Zheng, Y.; Moskowitz, M.A.; Chen, J.W. Myeloperoxidase Inhibition Increases Neurogenesis after Ischemic Stroke. J. Pharmacol. Exp. Ther. 2016, 359, 262–272. [Google Scholar] [CrossRef]
- Aratani, Y.; Koyama, H.; Nyui, S.; Suzuki, K.; Kura, F.; Maeda, N. Severe impairment in early host defense against Candida albicans in mice deficient in myeloperoxidase. Infect. Immun. 1999, 67, 1828–1836. [Google Scholar] [CrossRef]
- Serý, O.; Povová, J.; Míšek, I.; Pešák, L.; Janout, V. Molecular mechanisms of neuropathological changes in Alzheimer’s disease: A review. Folia Neuropathol. 2013, 51, 1–9. [Google Scholar] [CrossRef]
- Crawford, F.C.; Freeman, M.J.; Schinka, J.A.; Morris, M.D.; Abdullah, L.I.; Richards, D.; Sevush, S.; Duara, R.; Mullan, M.J. Association between Alzheimer’s disease and a functional polymorphism in the Myeloperoxidase gene. Exp. Neurol. 2001, 167, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Green, P.S.; Mendez, A.J.; Jacob, J.S.; Crowley, J.R.; Growdon, W.; Hyman, B.T.; Heinecke, J.W. Neuronal expression of myeloperoxidase is increased in Alzheimer’s disease. J. Neurochem. 2004, 90, 724–733. [Google Scholar] [CrossRef]
- Wu, C.Y.; Bawa, K.K.; Ouk, M.; Leung, N.; Yu, D.; Lanctôt, K.L.; Herrmann, N.; Pakosh, M.; Swardfager, W. Neutrophil activation in Alzheimer’s disease and mild cognitive impairment: A systematic review and meta-analysis of protein markers in blood and cerebrospinal fluid. Ageing Res. Rev. 2020, 62, 101130. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, W.F.; Rhees, J.; Maciejewski, D.; Paladino, T.; Sieburg, H.; Maki, R.A.; Masliah, E. Myeloperoxidase polymorphism is associated with gender specific risk for Alzheimer’s disease. Exp. Neurol. 1999, 155, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, D.L.; Lefkowitz, S.S. Microglia and myeloperoxidase: A deadly partnership in neurodegenerative disease. Free Radic. Biol. Med. 2008, 45, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Glezer, I.; Simard, A.R.; Rivest, S. Neuroprotective role of the innate immune system by microglia. Neuroscience 2007, 147, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Maki, R.A.; Holzer, M.; Motamedchaboki, K.; Malle, E.; Masliah, E.; Marsche, G.; Reynolds, W.F. Human myeloperoxidase (hMPO) is expressed in neurons in the substantia nigra in Parkinson’s disease and in the hMPO-α-synuclein-A53T mouse model, correlating with increased nitration and aggregation of α-synuclein and exacerbation of motor impairment. Free Radic. Biol. Med. 2019, 141, 115–140. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.K.; Pennathur, S.; Perier, C.; Tieu, K.; Teismann, P.; Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Vonsattel, J.P.; Heinecke, J.W.; et al. Ablation of the inflammatory enzyme myeloperoxidase mitigates features of Parkinson’s disease in mice. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 6594–6600. [Google Scholar] [CrossRef]
- Kim, W.G.; Mohney, R.P.; Wilson, B.; Jeohn, G.H.; Liu, B.; Hong, J.S. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: Role of microglia. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 6309–6316. [Google Scholar] [CrossRef]
- Omiatek, D.M.; Bressler, A.J.; Cans, A.S.; Andrews, A.M.; Heien, M.L.; Ewing, A.G. The real catecholamine content of secretory vesicles in the CNS revealed by electrochemical cytometry. Sci. Rep. 2013, 3, 1447. [Google Scholar] [CrossRef]
- Jeitner, T.M.; Kalogiannis, M.; Patrick, P.A.; Gomolin, I.; Palaia, T.; Ragolia, L.; Brand, D.; Delikatny, E.J. Inflaming the diseased brain: A role for tainted melanins. Biochim. Biophys. Acta 2015, 1852, 937–950. [Google Scholar] [CrossRef]
- Jeitner, T.M.; Kalogiannis, M.; Krasnikov, B.F.; Gomolin, I.; Peltier, M.R.; Moran, G.R. Linking Inflammation and Parkinson Disease: Hypochlorous Acid Generates Parkinsonian Poisons. Toxicol. Sci. Off. J. Soc. Toxicol. 2016, 153, 410. [Google Scholar] [CrossRef]
- Olek, M.J. Multiple Sclerosis. Ann. Intern. Med. 2021, 174, ITC81–ITC96. [Google Scholar] [CrossRef] [PubMed]
- Nagra, R.M.; Becher, B.; Tourtellotte, W.W.; Antel, J.P.; Gold, D.; Paladino, T.; Smith, R.A.; Nelson, J.R.; Reynolds, W.F. Immunohistochemical and genetic evidence of myeloperoxidase involvement in multiple sclerosis. J. Neuroimmunol. 1997, 78, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.; Thomas, T.L.; Betmouni, S.; Scolding, N.; Love, S. Elevated myeloperoxidase activity in white matter in multiple sclerosis. Neurosci. Lett. 2008, 444, 195–198. [Google Scholar] [CrossRef]
- Brennan, M.; Gaur, A.; Pahuja, A.; Lusis, A.J.; Reynolds, W.F. Mice lacking myeloperoxidase are more susceptible to experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2001, 112, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewska-Pniewska, B.; Styczynska, M.; Podlecka, A.; Samocka, R.; Peplonska, B.; Barcikowska, M.; Kwiecinski, H. Association of apolipoprotein E and myeloperoxidase genotypes to clinical course of familial and sporadic multiple sclerosis. Mult. Scler. 2004, 10, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Kantarci, O.H.; Atkinson, E.J.; Hebrink, D.D.; McMurray, C.T.; Weinshenker, B.G. Association of a myeloperoxidase promoter polymorphism with multiple sclerosis. J. Neuroimmunol. 2000, 105, 189–194. [Google Scholar] [CrossRef]
- Dissemond, J.; Weimann, T.K.; Schneider, L.A.; Schneeberger, A.; Scharffetter-Kochanek, K.; Goos, M.; Wagner, S.N. Activated neutrophils exert antitumor activity against human melanoma cells: Reactive oxygen species-induced mechanisms and their modulation by granulocyte-macrophage-colony-stimulating factor. J. Investig. Dermatol. 2003, 121, 936–938. [Google Scholar] [CrossRef]
- Larsson, S.; Nordenson, A.; Glader, P.; Yoshihara, S.; Lindén, A.; Slinde, F. A gender difference in circulating neutrophils in malnourished patients with COPD. Int. J. Chronic Obstr. Pulm. Dis. 2011, 6, 83–88. [Google Scholar] [CrossRef]
- Rainis, T.; Maor, I.; Lanir, A.; Shnizer, S.; Lavy, A. Enhanced oxidative stress and leucocyte activation in neoplastic tissues of the colon. Dig. Dis. Sci. 2007, 52, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.P.; Piedrafita, F.J.; Reynolds, W.F. Peroxisome proliferator-activated receptor gamma ligands regulate myeloperoxidase expression in macrophages by an estrogen-dependent mechanism involving the -463GA promoter polymorphism. J. Biol. Chem. 2004, 279, 8300–8315. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, C.; Casali, B.; Farnetti, E.; Pipitone, N.; Nicoli, D.; Macchioni, P.L.; Cimino, L.; Bajocchi, G.L.; Catanoso, M.G.; Pattacini, L.; et al. -463 G/A myeloperoxidase promoter polymorphism in giant cell arteritis. Ann. Rheum. Dis. 2008, 67, 485–488. [Google Scholar] [CrossRef]
- Reynolds, W.F.; Kumar, A.P.; Piedrafita, F.J. The human myeloperoxidase gene is regulated by LXR and PPARalpha ligands. Biochem. Biophys. Res. Commun. 2006, 349, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Van Schooten, F.J.; Boots, A.W.; Knaapen, A.M.; Godschalk, R.W.; Maas, L.M.; Borm, P.J.; Drent, M.; Jacobs, J.A. Myeloperoxidase (MPO) -463G->A reduces MPO activity and DNA adduct levels in bronchoalveolar lavages of smokers. Cancer Epidemiol. Biomark. Prev. 2004, 13, 828–833. [Google Scholar] [CrossRef]
- Yuzhalin, A.E.; Kutikhin, A.G. Common genetic variants in the myeloperoxidase and paraoxonase genes and the related cancer risk: A review. J. Environ. Sci. Health Part C Environ. Carcinog. Ecotoxicol. Rev. 2012, 30, 287–322. [Google Scholar] [CrossRef]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef]
- Rymaszewski, A.L.; Tate, E.; Yimbesalu, J.P.; Gelman, A.E.; Jarzembowski, J.A.; Zhang, H.; Pritchard, K.A., Jr.; Vikis, H.G. The role of neutrophil myeloperoxidase in models of lung tumor development. Cancers 2014, 6, 1111–1127. [Google Scholar] [CrossRef]
- Saed, G.M.; Ali-Fehmi, R.; Jiang, Z.L.; Fletcher, N.M.; Diamond, M.P.; Abu-Soud, H.M.; Munkarah, A.R. Myeloperoxidase serves as a redox switch that regulates apoptosis in epithelial ovarian cancer. Gynecol. Oncol. 2010, 116, 276–281. [Google Scholar] [CrossRef]
- Mallet, W.G.; Mosebrook, D.R.; Trush, M.A. Activation of (+-)-trans-7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene to diolepoxides by human polymorphonuclear leukocytes or myeloperoxidase. Carcinogenesis 1991, 12, 521–524. [Google Scholar] [CrossRef]
- Kadlubar, F.F.; Butler, M.A.; Kaderlik, K.R.; Chou, H.C.; Lang, N.P. Polymorphisms for aromatic amine metabolism in humans: Relevance for human carcinogenesis. Environ. Health Perspect. 1992, 98, 69–74. [Google Scholar] [CrossRef]
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer 2007, 121, 2381–2386. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Bauer, G. HOCl and the control of oncogenesis. J. Inorg. Biochem. 2018, 179, 10–23. [Google Scholar] [CrossRef]
- An, Z.; Zhao, Z.; Zhao, L.; Yue, Q.; Li, K.; Zhao, B.; Miao, J.; Su, L. The novel HOCl fluorescent probe CAN induced A549 apoptosis by inhibiting chlorination activity of MPO. Bioorganic Med. Chem. Lett. 2020, 30, 127394. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Davies, M.J. Hypochlorite-induced damage to nucleosides: Formation of chloramines and nitrogen-centered radicals. Chem. Res. Toxicol. 2001, 14, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.L.; Davies, M.J. Hypochlorite-induced damage to DNA, RNA, and polynucleotides: Formation of chloramines and nitrogen-centered radicals. Chem. Res. Toxicol. 2002, 15, 83–92. [Google Scholar] [CrossRef]
- Castelão, C.; da Silva, A.P.; Matos, A.; Inácio, Â.; Bicho, M.; Medeiros, R.; Bicho, M.C. Association of myeloperoxidase polymorphism (G463A) with cervix cancer. Mol. Cell. Biochem. 2015, 404, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Noor, S.; Alam, F.; Fatima, F.; Orakzai, S. Myeloperoxidase: Indicator of cardiovascular disease in chronic kidney disease patients of tertiary care hospital of Karachi. JPMA J. Pak. Med. Assoc. 2021, 71, 1128–1132. [Google Scholar] [CrossRef]
- Gröne, H.J.; Gröne, E.F.; Malle, E. Immunohistochemical detection of hypochlorite-modified proteins in glomeruli of human membranous glomerulonephritis. Lab. Investig. J. Tech. Methods Pathol. 2002, 82, 5–14. [Google Scholar] [CrossRef]
- Correa, S.; Pena-Esparragoza, J.K.; Scovner, K.M.; Waikar, S.S.; Mc Causland, F.R. Myeloperoxidase and the Risk of CKD Progression, Cardiovascular Disease, and Death in the Chronic Renal Insufficiency Cohort (CRIC) Study. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2020, 76, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Lehners, A.; Lange, S.; Niemann, G.; Rosendahl, A.; Meyer-Schwesinger, C.; Oh, J.; Stahl, R.; Ehmke, H.; Benndorf, R.; Klinke, A.; et al. Myeloperoxidase deficiency ameliorates progression of chronic kidney disease in mice. Am. J. Physiol. Ren. Physiol. 2014, 307, F407–F417. [Google Scholar] [CrossRef] [PubMed]
- Correa, S.; Curtis, K.A.; Waikar, S.S.; Mc Causland, F.R. Serum Myeloperoxidase, Uric Acid, and the Risk of Atrial Fibrillation in Chronic Kidney Disease. Circ. Arrhythmia Electrophysiol. 2021, 14, e009483. [Google Scholar] [CrossRef]
- Tsai, M.S.; Shaw, H.M.; Li, Y.J.; Lin, M.T.; Lee, W.T.; Chan, K.S. Myeloperoxidase in chronic kidney disease: Role of visceral fat. Nephrology 2014, 19, 136–142. [Google Scholar] [CrossRef]
- Rennke, H.G.; Venkatachalam, M.A. Glomerular permeability: In vivo tracer studies with polyanionic and polycationic ferritins. Kidney Int. 1977, 11, 44–53. [Google Scholar] [CrossRef]
- Johnson, R.J.; Couser, W.G.; Chi, E.Y.; Adler, S.; Klebanoff, S.J. New mechanism for glomerular injury. Myeloperoxidase-hydrogen peroxide-halide system. J. Clin. Investig. 1987, 79, 1379–1387. [Google Scholar] [CrossRef]
- Heinzelmann, M.; Mercer-Jones, M.A.; Passmore, J.C. Neutrophils and renal failure. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 1999, 34, 384–399. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, C.; Newbold, P.; White, P.; Thong, B.; Stone, H.; Stockley, R.A. 3-Chlorotyrosine in sputum of COPD patients: Relationship with airway inflammation. COPD 2010, 7, 411–417. [Google Scholar] [CrossRef]
- Dickerhof, N.; Pearson, J.F.; Hoskin, T.S.; Berry, L.J.; Turner, R.; Sly, P.D.; Kettle, A.J. Oxidative stress in early cystic fibrosis lung disease is exacerbated by airway glutathione deficiency. Free Radic. Biol. Med. 2017, 113, 236–243. [Google Scholar] [CrossRef]
- He, J.; Turino, G.M.; Lin, Y.Y. Characterization of peptide fragments from lung elastin degradation in chronic obstructive pulmonary disease. Exp. Lung Res. 2010, 36, 548–557. [Google Scholar] [CrossRef]
- Magon, N.J.; Turner, R.; Gearry, R.B.; Hampton, M.B.; Sly, P.D.; Kettle, A.J. Oxidation of calprotectin by hypochlorous acid prevents chelation of essential metal ions and allows bacterial growth: Relevance to infections in cystic fibrosis. Free Radic. Biol. Med. 2015, 86, 133–144. [Google Scholar] [CrossRef]
- Zhan, Z.; Lei, Q.; Dai, Y.; Wang, D.; Yu, Q.; Lv, Y.; Li, W. Simultaneous Monitoring of HOCl and Viscosity with Drug-Induced Pyroptosis in Live Cells and Acute Lung Injury. Anal. Chem. 2022, 94, 12144–12151. [Google Scholar] [CrossRef]
- Chniguir, A.; Zioud, F.; Marzaioli, V.; El-Benna, J.; Bachoual, R. Syzygium aromaticum aqueous extract inhibits human neutrophils myeloperoxidase and protects mice from LPS-induced lung inflammation. Pharm. Biol. 2019, 57, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Dickerhof, N.; Huang, J.; Min, E.; Michaëlsson, E.; Lindstedt, E.L.; Pearson, J.F.; Kettle, A.J.; Day, B.J. Myeloperoxidase inhibition decreases morbidity and oxidative stress in mice with cystic fibrosis-like lung inflammation. Free Radic. Biol. Med. 2020, 152, 91–99. [Google Scholar] [CrossRef]
- Haegens, A.; Heeringa, P.; van Suylen, R.J.; Steele, C.; Aratani, Y.; O’Donoghue, R.J.; Mutsaers, S.E.; Mossman, B.T.; Wouters, E.F.; Vernooy, J.H. Myeloperoxidase deficiency attenuates lipopolysaccharide-induced acute lung inflammation and subsequent cytokine and chemokine production. J. Immunol. 2009, 182, 7990–7996. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Bio-Medica Atenei Parm. 2020, 91, 157–160. [Google Scholar] [CrossRef]
- Yuan, Y.; Jiao, B.; Qu, L.; Yang, D.; Liu, R. The development of COVID-19 treatment. Front. Immunol. 2023, 14, 1125246. [Google Scholar] [CrossRef]
- Jamali, E.; Abbasi, M.; Tayer, A.H.; Monfared, A.A.; Tandel, P.; Tamaddon, G.; Kazerooni, E.S.; Rakhshandehroo, S.; Ranjbaran, R. The significance of surface neutrophilic MPO expression level in NETosis and NETosis-associated coagulopathies in COVID-19 infected patients. Blood Cells Mol. Dis. 2022, 96, 102676. [Google Scholar] [CrossRef]
- Huckriede, J.; de Vries, F.; Hultström, M.; Wichapong, K.; Reutelingsperger, C.; Lipcsey, M.; Garcia de Frutos, P.; Frithiof, R.; Nicolaes, G.A.F. Histone H3 Cleavage in Severe COVID-19 ICU Patients. Front. Cell. Infect. Microbiol. 2021, 11, 694186. [Google Scholar] [CrossRef]
- Saheb Sharif-Askari, N.; Saheb Sharif-Askari, F.; Mdkhana, B.; Hussain Alsayed, H.A.; Alsafar, H.; Alrais, Z.F.; Hamid, Q.; Halwani, R. Upregulation of oxidative stress gene markers during SARS-CoV-2 viral infection. Free Radic. Biol. Med. 2021, 172, 688–698. [Google Scholar] [CrossRef]
- Bonini, M.G.; Siraki, A.G.; Atanassov, B.S.; Mason, R.P. Immunolocalization of hypochlorite-induced, catalase-bound free radical formation in mouse hepatocytes. Free Radic. Biol. Med. 2007, 42, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tecson, K.M.; McCullough, P.A. Endothelial dysfunction contributes to COVID-19-associated vascular inflammation and coagulopathy. Rev. Cardiovasc. Med. 2020, 21, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Park, H.H.; Park, W.; Lee, Y.Y.; Kim, H.; Seo, H.S.; Choi, D.W.; Kwon, H.K.; Na, D.H.; Kim, T.H.; Choy, Y.B.; et al. Bioinspired DNase-I-Coated Melanin-Like Nanospheres for Modulation of Infection-Associated NETosis Dysregulation. Adv. Sci. 2020, 7, 2001940. [Google Scholar] [CrossRef]
- Twaddell, S.H.; Baines, K.J.; Grainge, C.; Gibson, P.G. The Emerging Role of Neutrophil Extracellular Traps in Respiratory Disease. Chest 2019, 156, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Holliday, Z.M.; Earhart, A.P.; Alnijoumi, M.M.; Krvavac, A.; Allen, L.H.; Schrum, A.G. Non-Randomized Trial of Dornase Alfa for Acute Respiratory Distress Syndrome Secondary to COVID-19. Front. Immunol. 2021, 12, 714833. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef]
- Cao, Z.; Cheng, G. Recombinant Myeloperoxidase as a New Class of Antimicrobial Agents. Microbiol. Spectr. 2022, 10, e0052221. [Google Scholar] [CrossRef]
- Soubhye, J.; Van Antwerpen, P.; Dufrasne, F. A patent review of myeloperoxidase inhibitors for treating chronic inflammatory syndromes (focus on cardiovascular diseases, 2013-2019). Expert Opin. Ther. Pat. 2020, 30, 595–608. [Google Scholar] [CrossRef]
- Kettle, A.J.; Gedye, C.A.; Hampton, M.B.; Winterbourn, C.C. Inhibition of myeloperoxidase by benzoic acid hydrazides. Biochem. J. 1995, 308 Pt 2, 559–563. [Google Scholar] [CrossRef]
- Stamp, L.K.; Turner, R.; Khalilova, I.S.; Zhang, M.; Drake, J.; Forbes, L.V.; Kettle, A.J. Myeloperoxidase and oxidation of uric acid in gout: Implications for the clinical consequences of hyperuricaemia. Rheumatology 2014, 53, 1958–1965. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, Y.; Deng, M. New developments and opportunities in drugs being trialed for amyotrophic lateral sclerosis from 2020 to 2022. Front. Pharmacol. 2022, 13, 1054006. [Google Scholar] [CrossRef]
- Ren, R.; Xu, Z.; Wang, X.; Jiang, W.; Yu, P. Verdiperstat attenuates acute lung injury by modulating MPO/μ-calpain/β-catenin signaling. Eur. J. Pharmacol. 2022, 924, 174940. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.W.; Gammon, S.T.; Yang, P.; Ma, W.; Wang, J.; Piwnica-Worms, D. Inhibition of myeloperoxidase enhances immune checkpoint therapy for melanoma. J. Immunother. Cancer 2023, 11. [Google Scholar] [CrossRef]
- Michaëlsson, E.; Lund, L.H.; Hage, C.; Shah, S.J.; Voors, A.A.; Saraste, A.; Redfors, B.; Grove, E.L.; Barasa, A.; Richards, A.M.; et al. Myeloperoxidase Inhibition Reverses Biomarker Profiles Associated With Clinical Outcomes in HFpEF. JACC Heart Fail. 2023, 11, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Lund, L.H.; Lam, C.S.P.; Pizzato, P.E.; Gabrielsen, A.; Michaëlsson, E.; Nelander, K.; Ericsson, H.; Holden, J.; Folkvaljon, F.; Mattsson, A.; et al. Rationale and design of ENDEAVOR: A sequential phase 2b-3 randomized clinical trial to evaluate the effect of myeloperoxidase inhibition on symptoms and exercise capacity in heart failure with preserved or mildly reduced ejection fraction. Eur. J. Heart Fail. 2023, 25, 1696–1707. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, R.B.; Buckbinder, L.; Bagley, S.W.; Carpino, P.A.; Conn, E.L.; Dowling, M.S.; Fernando, D.P.; Jiao, W.; Kung, D.W.; Orr, S.T.; et al. Discovery of 2-(6-(5-Chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamide (PF-06282999): A Highly Selective Mechanism-Based Myeloperoxidase Inhibitor for the Treatment of Cardiovascular Diseases. J. Med. Chem. 2015, 58, 8513–8528. [Google Scholar] [CrossRef]
- Soubhye, J.; Chikh Alard, I.; Aldib, I.; Prévost, M.; Gelbcke, M.; De Carvalho, A.; Furtmüller, P.G.; Obinger, C.; Flemmig, J.; Tadrent, S.; et al. Discovery of Novel Potent Reversible and Irreversible Myeloperoxidase Inhibitors Using Virtual Screening Procedure. J. Med. Chem. 2017, 60, 6563–6586. [Google Scholar] [CrossRef]
- Hori, H.; Fenna, R.E.; Kimura, S.; Ikeda-Saito, M. Aromatic substrate molecules bind at the distal heme pocket of myeloperoxidase. J. Biol. Chem. 1994, 269, 8388–8392. [Google Scholar] [CrossRef]
- Forbes, L.V.; Sjögren, T.; Auchère, F.; Jenkins, D.W.; Thong, B.; Laughton, D.; Hemsley, P.; Pairaudeau, G.; Turner, R.; Eriksson, H.; et al. Potent reversible inhibition of myeloperoxidase by aromatic hydroxamates. J. Biol. Chem. 2013, 288, 36636–36647. [Google Scholar] [CrossRef]
- Zhang, H.; Jing, X.; Shi, Y.; Xu, H.; Du, J.; Guan, T.; Weihrauch, D.; Jones, D.W.; Wang, W.; Gourlay, D.; et al. N-acetyl lysyltyrosylcysteine amide inhibits myeloperoxidase, a novel tripeptide inhibitor. J. Lipid Res. 2013, 54, 3016–3029. [Google Scholar] [CrossRef]
- Boufadi, Y.M.; Soubhye, J.; Riazi, A.; Rousseau, A.; Vanhaeverbeek, M.; Nève, J.; Boudjeltia, K.Z.; Van Antwerpen, P. Characterization and antioxidant properties of six Algerian propolis extracts: Ethyl acetate extracts inhibit myeloperoxidase activity. Int. J. Mol. Sci. 2014, 15, 2327–2345. [Google Scholar] [CrossRef] [PubMed]
- Tiyerili, V.; Camara, B.; Becher, M.U.; Schrickel, J.W.; Lütjohann, D.; Mollenhauer, M.; Baldus, S.; Nickenig, G.; Andrié, R.P. Neutrophil-derived myeloperoxidase promotes atherogenesis and neointima formation in mice. Int. J. Cardiol. 2016, 204, 29–36. [Google Scholar] [CrossRef]
- Jurva, U.; Weidolf, L.; Sandinge, A.S.; Leandersson, C.; Ekdahl, A.; Li, X.Q.; Antonsson, T.; Sundell, J.; Westerlund, K.; Amilon, C.; et al. Biotransformation of the Novel Myeloperoxidase Inhibitor AZD4831 in Preclinical Species and Humans. Drug Metab. Dispos. 2023, 51, 464–479. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.M.; Lagerström-Fermér, M.; Ericsson, H.; Nelander, K.; Lindstedt, E.L.; Michaëlsson, E.; Kjaer, M.; Heijer, M.; Whatling, C.; Fuhr, R. Safety, tolerability, pharmacokinetics and effect on serum uric acid of the myeloperoxidase inhibitor AZD4831 in a randomized, placebo-controlled, phase I study in healthy volunteers. Br. J. Clin. Pharmacol. 2019, 85, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.J.; Sucquart, I.E.; Whittaker, A.; Sanganee, H.J.; Barratt, C.L.R.; Martins da Silva, S.J. Myeloperoxidase inhibitor AZD5904 enhances human sperm function in vitro. Hum. Reprod. 2021, 36, 560–570. [Google Scholar] [CrossRef]
- Ramachandra, C.J.A.; Kp, M.M.J.; Chua, J.; Hernandez-Resendiz, S.; Liehn, E.A.; Knöll, R.; Gan, L.M.; Michaëlsson, E.; Jonsson, M.K.B.; Ryden-Markinhuhta, K.; et al. Inhibiting cardiac myeloperoxidase alleviates the relaxation defect in hypertrophic cardiomyocytes. Cardiovasc. Res. 2022, 118, 517–530. [Google Scholar] [CrossRef]
- Roth Flach, R.J.; Su, C.; Bollinger, E.; Cortes, C.; Robertson, A.W.; Opsahl, A.C.; Coskran, T.M.; Maresca, K.P.; Keliher, E.J.; Yates, P.D.; et al. Myeloperoxidase inhibition in mice alters atherosclerotic lesion composition. PLoS ONE 2019, 14, e0214150. [Google Scholar] [CrossRef] [PubMed]
- Teo, A.; Chan, L.L.Y.; Cheung, C.; Chia, P.Y.; Ong, S.W.X.; Fong, S.W.; Ng, L.F.P.; Renia, L.; Lye, D.C.; Young, B.E.; et al. Myeloperoxidase inhibition may protect against endothelial glycocalyx shedding induced by COVID-19 plasma. Commun. Med. 2023, 3, 62. [Google Scholar] [CrossRef]
- Teng, R.J.; Jing, X.; Martin, D.P.; Hogg, N.; Haefke, A.; Konduri, G.G.; Day, B.W.; Naylor, S.; Pritchard, K.A., Jr. N-acetyl-lysyltyrosylcysteine amide, a novel systems pharmacology agent, reduces bronchopulmonary dysplasia in hyperoxic neonatal rat pups. Free Radic. Biol. Med. 2021, 166, 73–89. [Google Scholar] [CrossRef]
- Yu, G.; Zheng, S.; Zhang, H. Inhibition of myeloperoxidase by N-acetyl lysyltyrosylcysteine amide reduces experimental autoimmune encephalomyelitis-induced injury and promotes oligodendrocyte regeneration and neurogenesis in a murine model of progressive multiple sclerosis. Neuroreport 2018, 29, 208–213. [Google Scholar] [CrossRef]
- Yu, G.; Liang, Y.; Zheng, S.; Zhang, H. Inhibition of Myeloperoxidase by N-Acetyl Lysyltyrosylcysteine Amide Reduces Oxidative Stress-Mediated Inflammation, Neuronal Damage, and Neural Stem Cell Injury in a Murine Model of Stroke. J. Pharmacol. Exp. Ther. 2018, 364, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, H.; Weihrauch, D.; Jones, D.W.; Jing, X.; Shi, Y.; Gourlay, D.; Oldham, K.T.; Hillery, C.A.; Pritchard, K.A., Jr. Inhibition of myeloperoxidase decreases vascular oxidative stress and increases vasodilatation in sickle cell disease mice. J. Lipid Res. 2013, 54, 3009–3015. [Google Scholar] [CrossRef] [PubMed]
- Forbes, L.V.; Kettle, A.J. A multi-substrate assay for finding physiologically effective inhibitors of myeloperoxidase. Anal. Biochem. 2018, 544, 13–21. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, W.; Chen, H.; Chen, X.; Guo, C. The Roles of Neutrophil-Derived Myeloperoxidase (MPO) in Diseases: The New Progress. Antioxidants 2024, 13, 132. https://doi.org/10.3390/antiox13010132
Lin W, Chen H, Chen X, Guo C. The Roles of Neutrophil-Derived Myeloperoxidase (MPO) in Diseases: The New Progress. Antioxidants. 2024; 13(1):132. https://doi.org/10.3390/antiox13010132
Chicago/Turabian StyleLin, Wei, Huili Chen, Xijing Chen, and Chaorui Guo. 2024. "The Roles of Neutrophil-Derived Myeloperoxidase (MPO) in Diseases: The New Progress" Antioxidants 13, no. 1: 132. https://doi.org/10.3390/antiox13010132
APA StyleLin, W., Chen, H., Chen, X., & Guo, C. (2024). The Roles of Neutrophil-Derived Myeloperoxidase (MPO) in Diseases: The New Progress. Antioxidants, 13(1), 132. https://doi.org/10.3390/antiox13010132