
Thioredoxin/Glutaredoxin Systems and Gut Microbiota in NAFLD: Interplay, Mechanism, and Therapeutical Potential



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Disruption of Thiol Homeostasis in NAFLD
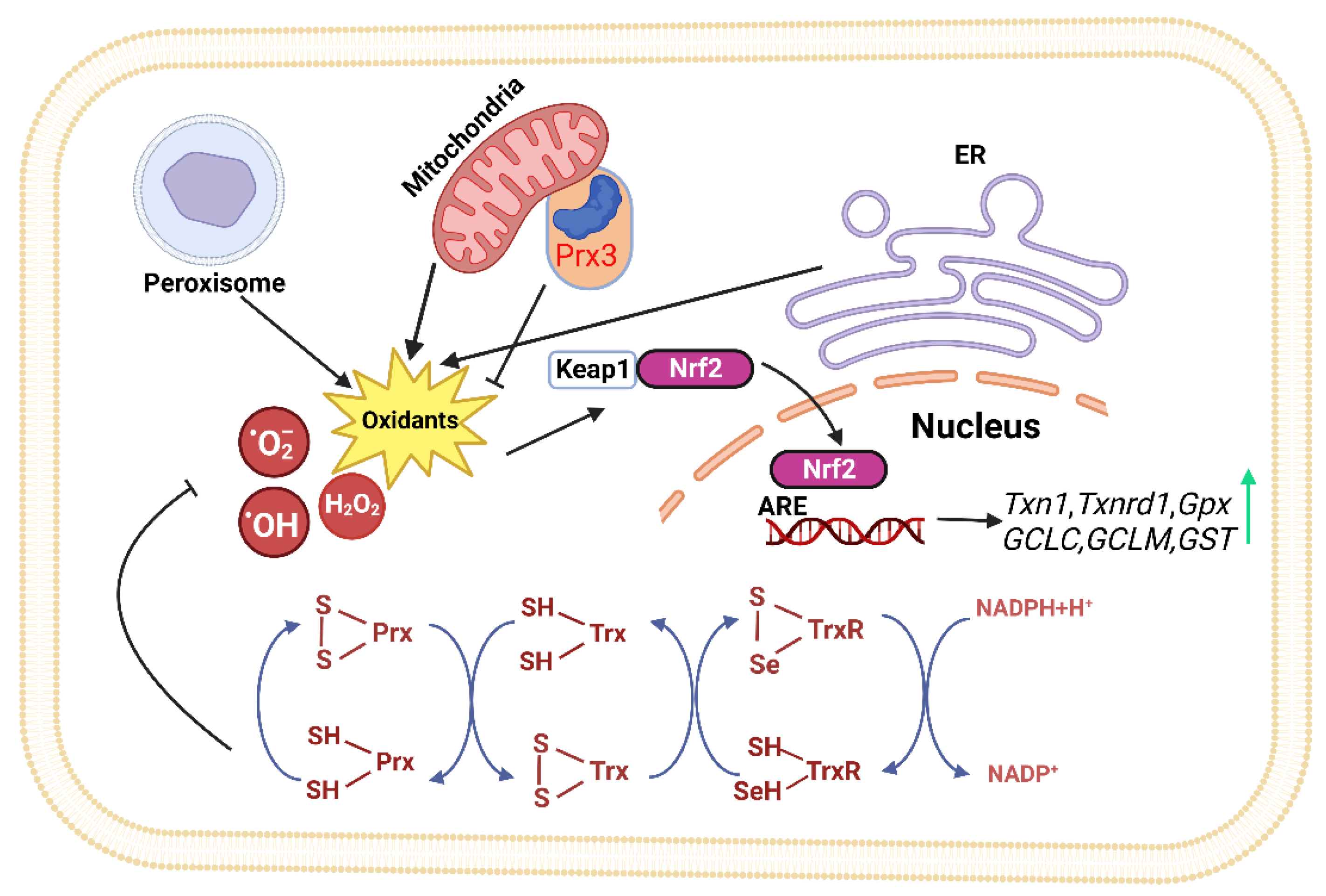
2.1. Sites of ROS Production
2.2. Thioredoxin (Trx) System
2.3. Glutathione (GSH)-Grx System
2.4. Nrf2 Signaling Pathway
3. Gut Microbiota in NAFLD
3.1. Dysbiosis and Oxidative Stress in NAFLD
3.2. Impact of Intestinal Barrier Dysfunction and Gut Microbiota Metabolites on NAFLD
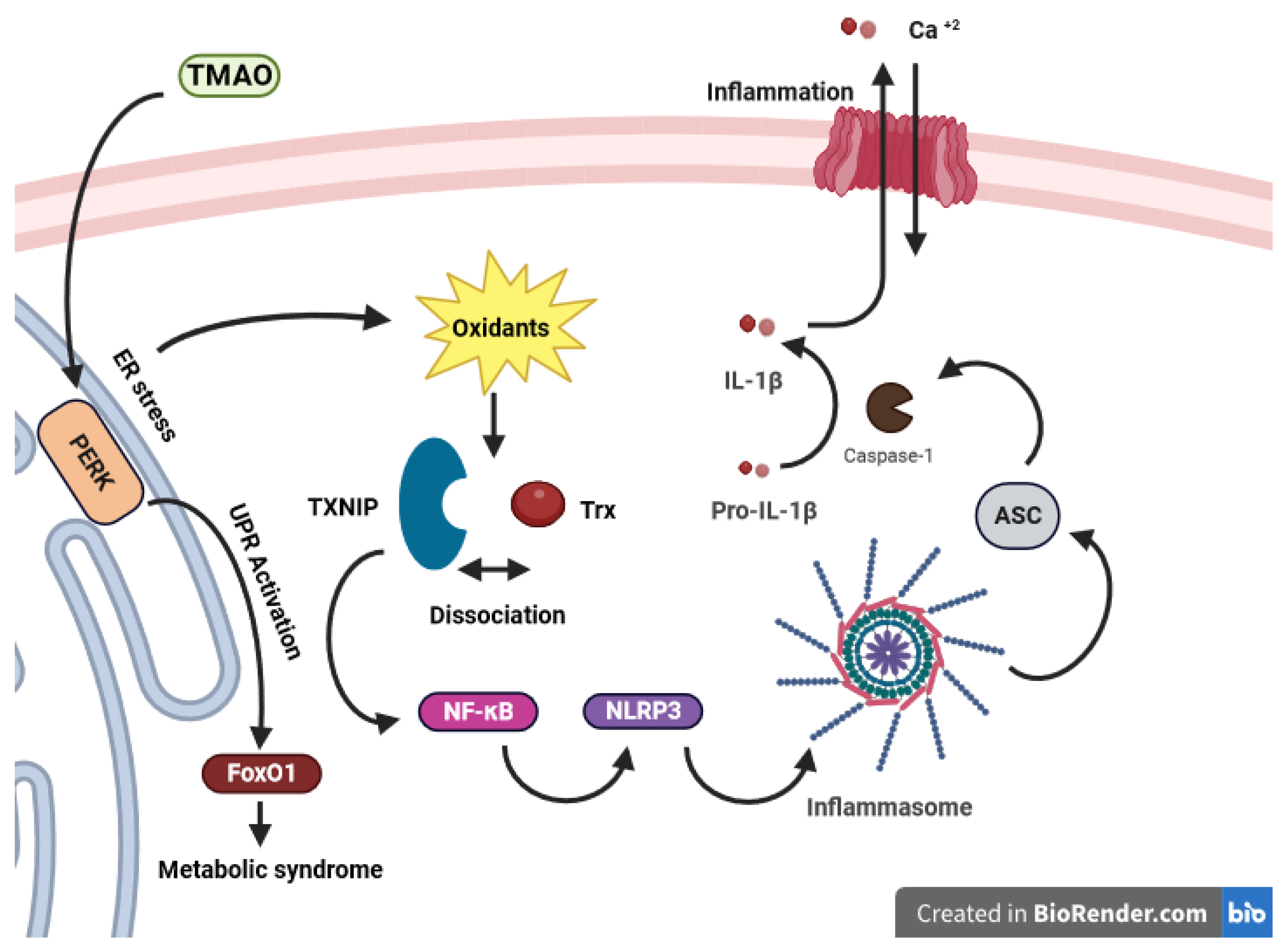
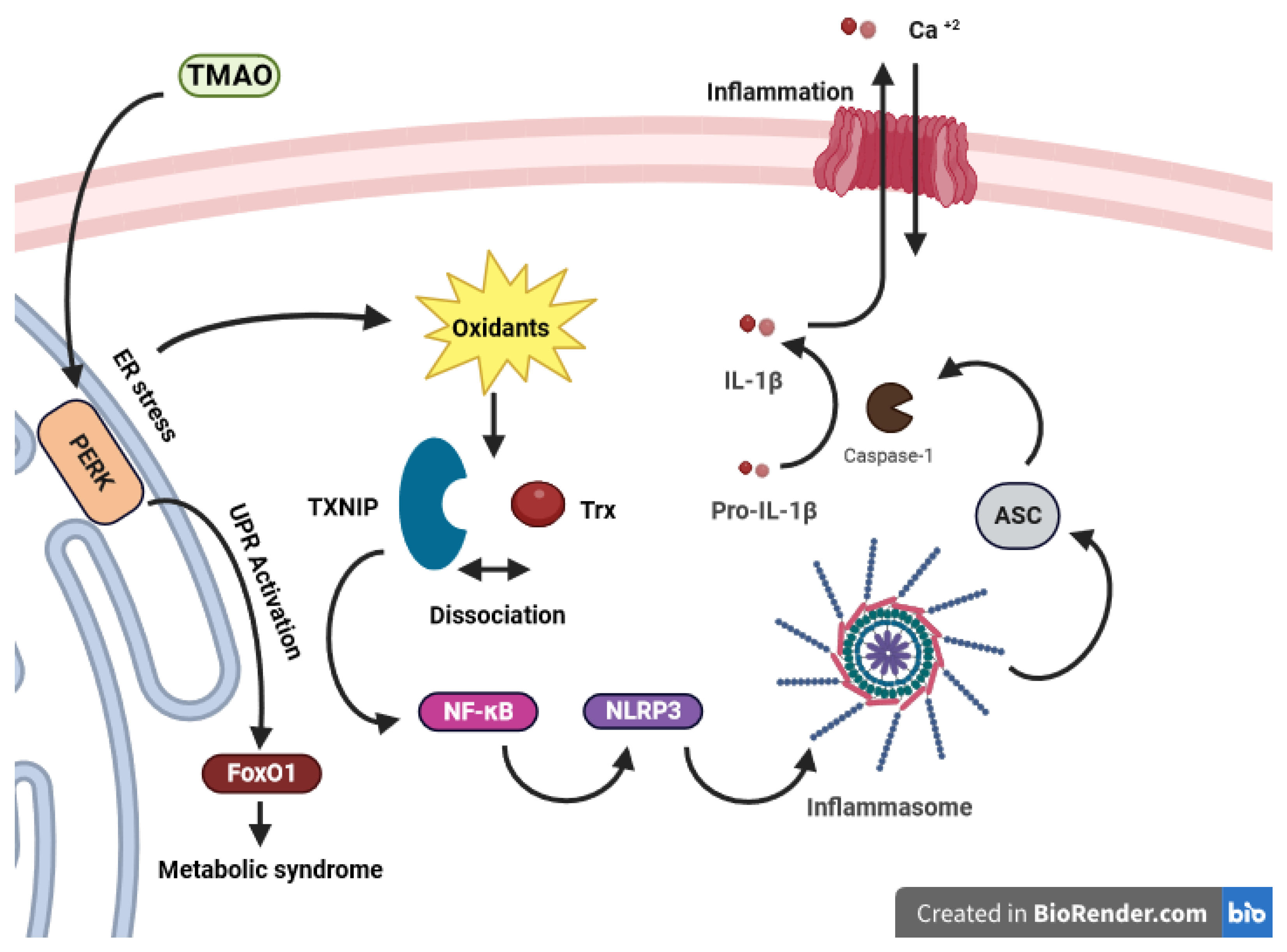
3.2.1. Trimethylamine N-oxide (TMAO)
3.2.2. Lipopolysaccharides (LPS)
3.2.3. Short-Chain Fatty Acids (SCFAs)
3.2.4. Amino Acids
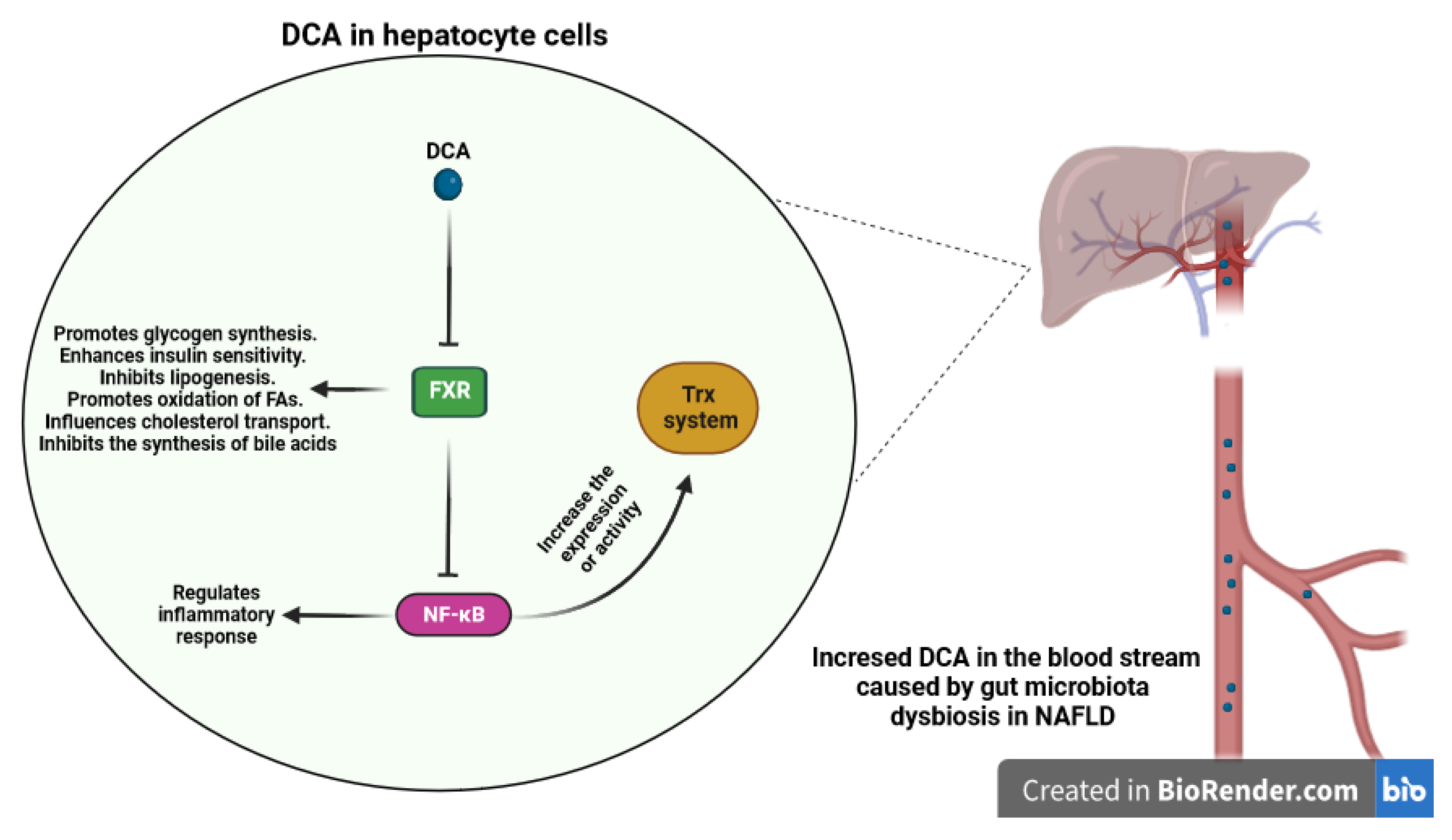
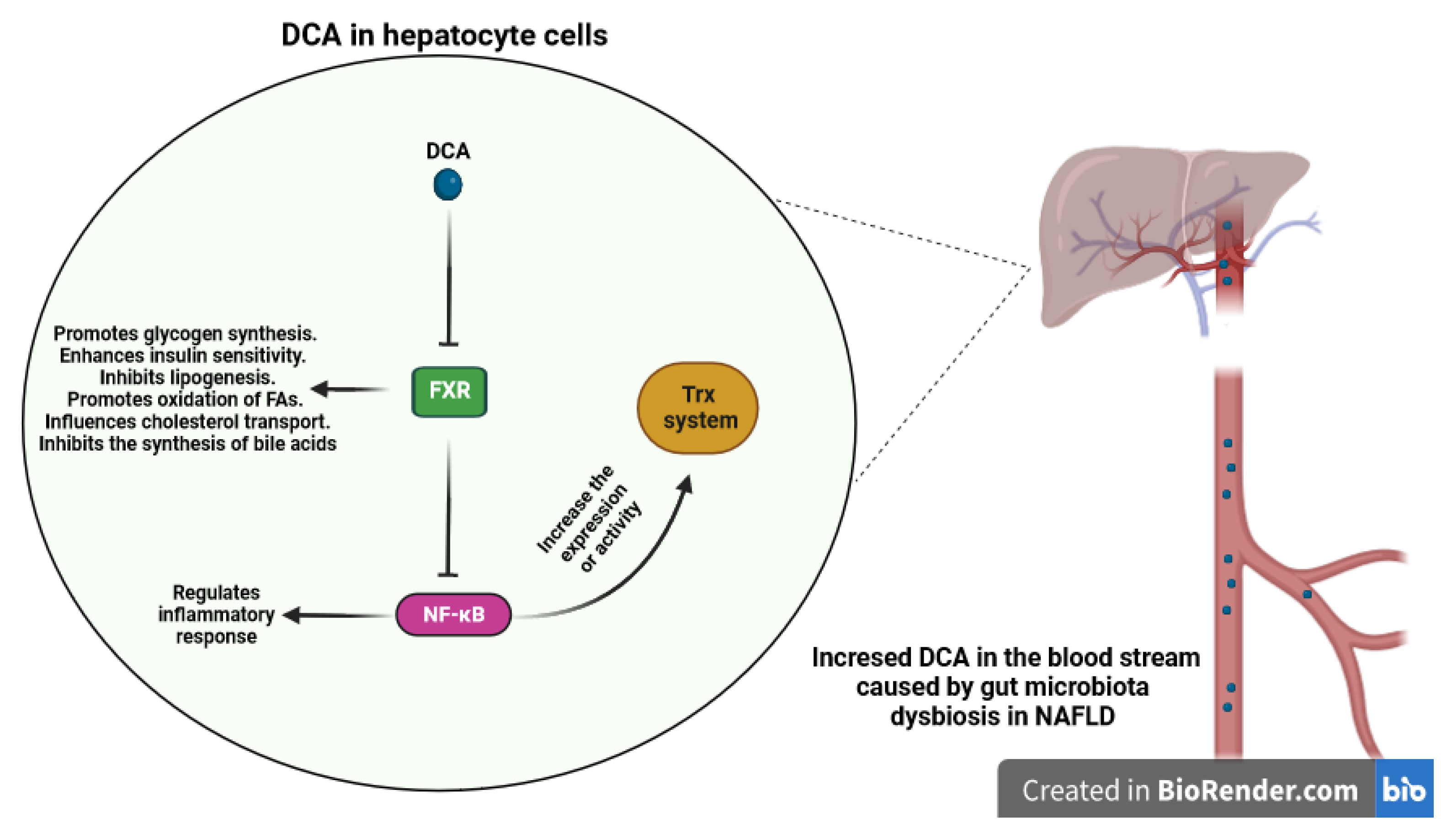
3.2.5. Bile Acids Metabolites
3.2.6. Ethanol
4. Therapeutical Potential for NAFLD/NASH with Specifically Thiol Redox Regulation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stefan, N.; Häring, H.U.; Cusi, K. Non-alcoholic fatty liver disease: Causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet. Diabetes Endocrinol. 2019, 7, 313–324. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014. [Google Scholar] [CrossRef] [PubMed]
- Seghieri, M.; Christensen, A.S.; Andersen, A.; Solini, A.; Knop, F.K.; Vilsbøll, T. Future Perspectives on GLP-1 Receptor Agonists and GLP-1/glucagon Receptor Co-agonists in the Treatment of NAFLD. Front. Endocrinol. 2018, 9, 649. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metab. Clin. Exp. 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Zarkovic, N. Roles and Functions of ROS and RNS in Cellular Physiology and Pathology. Cells 2020, 9, 767. [Google Scholar] [CrossRef]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef]
- Nordberg, J.; Arnér, E.S. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free. Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Imlay, J.A.; Chin, S.M.; Linn, S. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science 1988, 240, 640–642. [Google Scholar] [CrossRef]
- Shabalina, I.G.; Nedergaard, J. Mitochondrial (‘mild’) uncoupling and ROS production: Physiologically relevant or not? Biochem. Soc. Trans. 2011, 39, 1305–1309. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, J.; Ming, Y.; Chen, X.; Zeng, M.; Mao, Y. The aggravation of mitochondrial dysfunction in nonalcoholic fatty liver disease accompanied with type 2 diabetes mellitus. Scand. J. Gastroenterol. 2015, 50, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- García-Berumen, C.I.; Ortiz-Avila, O.; Vargas-Vargas, M.A.; Del Rosario-Tamayo, B.A.; Guajardo-López, C.; Saavedra-Molina, A.; Rodríguez-Orozco, A.R.; Cortés-Rojo, C. The severity of rat liver injury by fructose and high fat depends on the degree of respiratory dysfunction and oxidative stress induced in mitochondria. Lipids Health Dis. 2019, 18, 78. [Google Scholar] [CrossRef]
- Okumura, M.; Noi, K.; Kanemura, S.; Kinoshita, M.; Saio, T.; Inoue, Y.; Hikima, T.; Akiyama, S.; Ogura, T.; Inaba, K. Dynamic assembly of protein disulfide isomerase in catalysis of oxidative folding. Nat. Chem. Biol. 2019, 15, 499–509. [Google Scholar] [CrossRef]
- Rashdan, N.A.; Pattillo, C.B. Hydrogen peroxide in the ER: A tale of triage. Redox Biol. 2020, 28, 101358. [Google Scholar] [CrossRef]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Sun, W.; Balaz, M.; He, A.; Klug, M.; Wieland, S.; Caiazzo, R.; Raverdy, V.; Pattou, F.; Lefebvre, P.; et al. Peroxisomal β-oxidation acts as a sensor for intracellular fatty acids and regulates lipolysis. Nat. Metab. 2021, 3, 1648–1661. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Jeong, J.M.; Kim, S.J.; Seo, W.; Kim, M.H.; Choi, W.M.; Yoo, W.; Lee, J.H.; Shim, Y.R.; Yi, H.S.; et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat. Commun. 2017, 8, 2247. [Google Scholar] [CrossRef]
- Thakur, V.; Pritchard, M.T.; McMullen, M.R.; Wang, Q.; Nagy, L.E. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: Role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J. Leukoc. Biol. 2006, 79, 1348–1356. [Google Scholar] [CrossRef]
- Meng, T.; Yu, J.; Lei, Z.; Wu, J.; Wang, S.; Bo, Q.; Zhang, X.; Ma, Z.; Yu, J. Propofol reduces lipopolysaccharide-induced, NADPH oxidase (NOX 2) mediated TNF-α and IL-6 production in macrophages. Clin. Dev. Immunol. 2013, 2013, 325481. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.S.; Lee, J.H.; Choi, S.H.; Kim, S.; Almazan, F.; Witztum, J.L.; Miller, Y.I. Macrophages generate reactive oxygen species in response to minimally oxidized low-density lipoprotein: Toll-like receptor 4- and spleen tyrosine kinase-dependent activation of NADPH oxidase 2. Circ. Res. 2009, 104, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-S.; Kim, J.-J.; Lee, S.J.; Hwang, J.H.; Lee, C.-H.; Lee, M.-S.; Jo, E.-K. TLR3-Triggered Reactive Oxygen Species Contribute to Inflammatory Responses by Activating Signal Transducer and Activator of Transcription-1. J. Immunol. 2013, 190, 6368–6377. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Bioactive Lipid Species and Metabolic Pathways in Progression and Resolution of Nonalcoholic Steatohepatitis. Gastroenterology 2018, 155, 282–302.e8. [Google Scholar] [CrossRef]
- Loffredo, L.; Zicari, A.M.; Perri, L.; Carnevale, R.; Nocella, C.; Angelico, F.; Del Ben, M.; Mosca, A.; Zaffina, S.; Panera, N.; et al. Does Nox2 Overactivate in Children with Nonalcoholic Fatty Liver Disease? Antioxid. Redox Signal. 2019, 30, 1325–1330. [Google Scholar] [CrossRef]
- Grossini, E.; Garhwal, D.P.; Calamita, G.; Romito, R.; Rigamonti, C.; Minisini, R.; Smirne, C.; Surico, D.; Bellan, M.; Pirisi, M. Exposure to Plasma From Non-alcoholic Fatty Liver Disease Patients Affects Hepatocyte Viability, Generates Mitochondrial Dysfunction, and Modulates Pathways Involved in Fat Accumulation and Inflammation. Front. Med. 2021, 8, 693997. [Google Scholar] [CrossRef]
- Wang, J.; He, W.; Tsai, P.J.; Chen, P.H.; Ye, M.; Guo, J.; Su, Z. Mutual interaction between endoplasmic reticulum and mitochondria in nonalcoholic fatty liver disease. Lipids Health Dis. 2020, 19, 72. [Google Scholar] [CrossRef]
- Pessayre, D.; Berson, A.; Fromenty, B.; Mansouri, A. Mitochondria in steatohepatitis. Semin. Liver Dis. 2001, 21, 57–69. [Google Scholar] [CrossRef]
- Du, X.; Edelstein, D.; Obici, S.; Higham, N.; Zou, M.H.; Brownlee, M. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J. Clin. Investig. 2006, 116, 1071–1080. [Google Scholar] [CrossRef]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. 2018, 13, 321–350. [Google Scholar] [CrossRef]
- Abe, Y.; Hines, I.N.; Zibari, G.; Pavlick, K.; Gray, L.; Kitagawa, Y.; Grisham, M.B. Mouse model of liver ischemia and reperfusion injury: Method for studying reactive oxygen and nitrogen metabolites in vivo. Free. Radic. Biol. Med. 2009, 46, 1–7. [Google Scholar] [CrossRef]
- Holmgren, A. Thioredoxin. Annu. Rev. Biochem. 1985, 54, 237–271. [Google Scholar] [CrossRef] [PubMed]
- Arnér, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. Thioredoxin system in cell death progression. Antioxid. Redox Signal. 2012, 17, 1738–1747. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Arnér, E.S.; Holmgren, A. Structure and mechanism of mammalian thioredoxin reductase: The active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proc. Natl. Acad. Sci. USA 2000, 97, 5854–5859. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. Selenoproteins. J. Biol. Chem. 2009, 284, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Lillig, C.H.; Holmgren, A. Thioredoxin and related molecules–from biology to health and disease. Antioxid. Redox Signal. 2007, 9, 25–47. [Google Scholar] [CrossRef]
- Gladyshev, V.N.; Jeang, K.-T.; Stadtman, T.C. Selenocysteine, identified as the penultimate C-terminal residue in human T-cell thioredoxin reductase, corresponds to TGA in the human placental gene. Proc. Natl. Acad. Sci. USA 1996, 93, 6146–6151. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Arnér, E.S.; Ljung, J.; Åslund, F.; Holmgren, A. Rat and calf thioredoxin reductase are homologous to glutathione reductase with a carboxyl-terminal elongation containing a conserved catalytically active penultimate selenocysteine residue. J. Biol. Chem. 1998, 273, 8581–8591. [Google Scholar] [CrossRef]
- Lee, S.-R.; Kim, J.-R.; Kwon, K.-S.; Yoon, H.W.; Levine, R.L.; Ginsburg, A.; Rhee, S.G. Molecular cloning and characterization of a mitochondrial selenocysteine-containing thioredoxin reductase from rat liver. J. Biol. Chem. 1999, 274, 4722–4734. [Google Scholar] [CrossRef] [PubMed]
- Rigobello, M.P.; Callegaro, M.T.; Barzon, E.; Benetti, M.; Bindoli, A. Purification of mitochondrial thioredoxin reductase and its involvement in the redox regulation of membrane permeability. Free Radic. Biol. Med. 1998, 24, 370–376. [Google Scholar] [CrossRef]
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef]
- Morel, Y.; Barouki, R. Repression of gene expression by oxidative stress. Biochem. J. 1999, 342, 481–496. [Google Scholar] [CrossRef]
- Welsh, S.J.; Bellamy, W.T.; Briehl, M.M.; Powis, G. The redox protein thioredoxin-1 (Trx-1) increases hypoxia-inducible factor 1α protein expression: Trx-1 overexpression results in increased vascular endothelial growth factor production and enhanced tumor angiogenesis. Cancer Res. 2002, 62, 5089–5095. [Google Scholar] [PubMed]
- Makino, Y.; Yoshikawa, N.; Okamoto, K.; Hirota, K.; Yodoi, J.; Makino, I.; Tanaka, H. Direct association with thioredoxin allows redox regulation of glucocorticoid receptor function. J. Biol. Chem. 1999, 274, 3182–3188. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Masutani, H.; Arai, R.J.; Yamauchi, A.; Hirota, K.; Sakai, T.; Inamoto, T.; Yamaoka, Y.; Yodoi, J.; Nikaido, T. Thioredoxin-dependent redox regulation of p53-mediated p21 activation. J. Biol. Chem. 1999, 274, 35809–35815. [Google Scholar] [CrossRef]
- Crunkhorn, S. Cardiovascular disease: Thioredoxin lowers hypertension. Nat. Rev. Drug Discov. 2017, 16, 240. [Google Scholar] [CrossRef]
- Kaimul, A.M.; Nakamura, H.; Masutani, H.; Yodoi, J. Thioredoxin and thioredoxin-binding protein-2 in cancer and metabolic syndrome. Free Radic. Biol. Med. 2007, 43, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Peña-Orihuela, P.; Camargo, A.; Rangel-Zuñiga, O.A.; Perez-Martinez, P.; Cruz-Teno, C.; Delgado-Lista, J.; Yubero-Serrano, E.M.; Paniagua, J.A.; Tinahones, F.J.; Malagon, M.M.; et al. Antioxidant system response is modified by dietary fat in adipose tissue of metabolic syndrome patients. J. Nutr. Biochem. 2013, 24, 1717–1723. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, A.; Korac, A.; Buzadzic, B.; Otasevic, V.; Stancic, A.; Daiber, A.; Korac, B. Redox implications in adipose tissue (dys)function--A new look at old acquaintances. Redox Biol. 2015, 6, 19–32. [Google Scholar] [CrossRef]
- Jankovic, A.; Korac, A.; Srdic-Galic, B.; Buzadzic, B.; Otasevic, V.; Stancic, A.; Vucetic, M.; Markelic, M.; Velickovic, K.; Golic, I.; et al. Differences in the redox status of human visceral and subcutaneous adipose tissues--relationships to obesity and metabolic risk. Metab. Clin. Exp. 2014, 63, 661–671. [Google Scholar] [CrossRef]
- Bouwman, F.G.; Claessens, M.; van Baak, M.A.; Noben, J.P.; Wang, P.; Saris, W.H.; Mariman, E.C. The physiologic effects of caloric restriction are reflected in the in vivo adipocyte-enriched proteome of overweight/obese subjects. J. Proteome Res. 2009, 8, 5532–5540. [Google Scholar] [CrossRef]
- Huh, J.Y.; Kim, Y.; Jeong, J.; Park, J.; Kim, I.; Huh, K.H.; Kim, Y.S.; Woo, H.A.; Rhee, S.G.; Lee, K.J.; et al. Peroxiredoxin 3 is a key molecule regulating adipocyte oxidative stress, mitochondrial biogenesis, and adipokine expression. Antioxid. Redox Signal. 2012, 16, 229–243. [Google Scholar] [CrossRef]
- Okuyama, H.; Son, A.; Ahsan, M.K.; Masutani, H.; Nakamura, H.; Yodoi, J. Thioredoxin and thioredoxin binding protein 2 in the liver. IUBMB life 2008, 60, 656–660. [Google Scholar] [CrossRef]
- Kim, C.H.; Younossi, Z.M. Nonalcoholic fatty liver disease: A manifestation of the metabolic syndrome. Clevel. Clin. J. Med. 2008, 75, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Sumida, Y.; Nakashima, T.; Yoh, T.; Furutani, M.; Hirohama, A.; Kakisaka, Y.; Nakajima, Y.; Ishikawa, H.; Mitsuyoshi, H.; Okanoue, T.; et al. Serum thioredoxin levels as a predictor of steatohepatitis in patients with nonalcoholic fatty liver disease. J. Hepatol. 2003, 38, 32–38. [Google Scholar] [CrossRef]
- Grattagliano, I.; Caraceni, P.; Calamita, G.; Ferri, D.; Gargano, I.; Palasciano, G.; Portincasa, P. Severe liver steatosis correlates with nitrosative and oxidative stress in rats. Eur. J. Clin. Investig. 2008, 38, 523–530. [Google Scholar] [CrossRef]
- Park, B.J.; Cha, M.K.; Kim, I.H. Thioredoxin 1 as a serum marker for ovarian cancer and its use in combination with CA125 for improving the sensitivity of ovarian cancer diagnoses. Biomark. Biochem. Indic. Expo. Response Susceptibility Chem. 2014, 19, 604–610. [Google Scholar] [CrossRef]
- Gornicka, A.; Morris-Stiff, G.; Thapaliya, S.; Papouchado, B.G.; Berk, M.; Feldstein, A.E. Transcriptional profile of genes involved in oxidative stress and antioxidant defense in a dietary murine model of steatohepatitis. Antioxid. Redox Signal. 2011, 15, 437–445. [Google Scholar] [CrossRef]
- Holmgren, A. Thioredoxin and Glutaredoxin Systems. J. Biol. Chem. 1989, 264, 13963–13966. [Google Scholar] [CrossRef]
- Morgan, B.; Ezerina, D.; Amoako, T.N.E.; Riemer, J.; Seedorf, M.; Dick, T.P. Multiple glutathione disulfide removal pathways mediate cytosolic redox homeostasis. Nat. Chem. Biol. 2013, 9, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Vivancos, P.; de Simone, A.; Kiddie, G.; Foyer, C.H. Glutathione—Linking cell proliferation to oxidative stress. Free Radic. Biol. Med. 2015, 89, 1154–1164. [Google Scholar] [CrossRef] [PubMed]
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Bba-Gen. Subj. 2008, 1780, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Han, J.; Hou, X.; Fry, J.; Behring, J.B.; Seta, F.; Long, M.T.; Roy, H.K.; Cohen, R.A.; Matsui, R.; et al. Glutaredoxin-1 Deficiency Causes Fatty Liver and Dyslipidemia by Inhibiting Sirtuin-1. Antioxid. Redox Signal. 2017, 27, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Ponugoti, B.; Kim, D.H.; Xiao, Z.; Smith, Z.; Miao, J.; Zang, M.; Wu, S.Y.; Chiang, C.M.; Veenstra, T.D.; Kemper, J.K. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J. Biol. Chem. 2010, 285, 33959–33970. [Google Scholar] [CrossRef] [PubMed]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado De Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef]
- Pfluger, P.T.; Herranz, D.; Velasco-Miguel, S.; Serrano, M.; Tschöp, M.H. Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. USA 2008, 105, 9793–9798. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Fry, J.L.; Han, J.; Hou, X.; Pimentel, D.R.; Matsui, R.; Cohen, R.A.; Bachschmid, M.M. A redox-resistant sirtuin-1 mutant protects against hepatic metabolic and oxidant stress. J. Biol. Chem. 2014, 289, 7293–7306. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.Q.; Chen, L.L.; Li, N.X. The expression of SIRT1 in nonalcoholic fatty liver disease induced by high-fat diet in rats. Liver Int. Off. J. Int. Assoc. Study Liver 2007, 27, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Hu, M.; Liang, X.; Ajmo, J.M.; Li, X.; Bataller, R.; Odena, G.; Stevens, S.M., Jr.; You, M. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology 2014, 146, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Hudemann, C.; Lönn, M.E.; Godoy, J.R.; Zahedi Avval, F.; Capani, F.; Holmgren, A.; Lillig, C.H. Identification, expression pattern, and characterization of mouse glutaredoxin 2 isoforms. Antioxid. Redox Signal. 2009, 11, 1–14. [Google Scholar] [CrossRef]
- Lönn, M.E.; Hudemann, C.; Berndt, C.; Cherkasov, V.; Capani, F.; Holmgren, A.; Lillig, C.H. Expression pattern of human glutaredoxin 2 isoforms: Identification and characterization of two testis/cancer cell-specific isoforms. Antioxid. Redox Signal. 2008, 10, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Scalcon, V.; Folda, A.; Lupo, M.G.; Tonolo, F.; Pei, N.; Battisti, I.; Ferri, N.; Arrigoni, G.; Bindoli, A.; Holmgren, A.; et al. Mitochondrial depletion of glutaredoxin 2 induces metabolic dysfunction-associated fatty liver disease in mice. Redox Biol. 2022, 51, 102277. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef]
- Galicia-Moreno, M.; Lucano-Landeros, S.; Monroy-Ramirez, H.C.; Silva-Gomez, J.; Gutierrez-Cuevas, J.; Santos, A.; Armendariz-Borunda, J. Roles of Nrf2 in Liver Diseases: Molecular, Pharmacological, and Epigenetic Aspects. Antioxidants 2020, 9, 980. [Google Scholar] [CrossRef]
- Higgins, L.G.; Kelleher, M.O.; Eggleston, I.M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Transcription factor Nrf2 mediates an adaptive response to sulforaphane that protects fibroblasts in vitro against the cytotoxic effects of electrophiles, peroxides and redox-cycling agents. Toxicol. Appl. Pharmacol. 2009, 237, 267–280. [Google Scholar] [CrossRef]
- Hawkes, H.J.; Karlenius, T.C.; Tonissen, K.F. Regulation of the human thioredoxin gene promoter and its key substrates: A study of functional and putative regulatory elements. Biochim. Biophys. Acta 2014, 1840, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 2009, 46, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Masutani, H.; Yamaguchi, Y.; Itoh, K.; Yamamoto, M.; Yodoi, J. Hemin-induced activation of the thioredoxin gene by Nrf2. A differential regulation of the antioxidant responsive element by a switch of its binding factors. J. Biol. Chem. 2001, 276, 18399–18406. [Google Scholar] [CrossRef] [PubMed]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef]
- Yodoi, J.; Nakamura, H.; Masutani, H. Redox regulation of stress signals: Possible roles of dendritic stellate TRX producer cells (DST cell types). Biol. Chem. 2002, 383, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Charlot, C.; Dubois-Pot, H.; Serchov, T.; Tourrette, Y.; Wasylyk, B. A review of post-translational modifications and subcellular localization of Ets transcription factors: Possible connection with cancer and involvement in the hypoxic response. Methods Mol. Biol. 2010, 647, 3–30. [Google Scholar] [CrossRef]
- Shiota, M.; Izumi, H.; Miyamoto, N.; Onitsuka, T.; Kashiwagi, E.; Kidani, A.; Hirano, G.; Takahashi, M.; Ono, M.; Kuwano, M.; et al. Ets regulates peroxiredoxin1 and 5 expressions through their interaction with the high-mobility group protein B1. Cancer Sci. 2008, 99, 1950–1959. [Google Scholar] [CrossRef]
- Osborne, S.A.; Hawkes, H.J.; Baldwin, B.L.; Alexander, K.A.; Svingen, T.; Clarke, F.M.; Tonissen, K.F. The tert-butylhydroquinone-mediated activation of the human thioredoxin gene reveals a novel promoter structure. Biochem. J. 2006, 398, 269–277. [Google Scholar] [CrossRef]
- Bai, J.; Nakamura, H.; Kwon, Y.W.; Hattori, I.; Yamaguchi, Y.; Kim, Y.C.; Kondo, N.; Oka, S.; Ueda, S.; Masutani, H.; et al. Critical roles of thioredoxin in nerve growth factor-mediated signal transduction and neurite outgrowth in PC12 cells. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 503–509. [Google Scholar] [CrossRef]
- Rundlöf, A.K.; Carlsten, M.; Arnér, E.S. The core promoter of human thioredoxin reductase 1: Cloning, transcriptional activity, and Oct-1, Sp1, and Sp3 binding reveal a housekeeping-type promoter for the AU-rich element-regulated gene. J. Biol. Chem. 2001, 276, 30542–30551. [Google Scholar] [CrossRef]
- Canfora, E.E.; Meex, R.C.; Venema, K.; Blaak, E.E. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat. Rev. Endocrinol. 2019, 15, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Coker, O.O.; Chu, E.S.; Fu, K.; Lau, H.C.; Wang, Y.-X.; Chan, A.W.; Wei, H.; Yang, X.; Sung, J.J. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut 2021, 70, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Schoeler, M.; Caesar, R. Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, Y.; Yang, S.; Lu, J.; Jin, X.; Wu, M. Diet-gut microbiota-epigenetics in metabolic diseases: From mechanisms to therapeutics. Biomed. Pharmacother. 2022, 153, 113290. [Google Scholar] [CrossRef] [PubMed]
- Makri, E.; Goulas, A.; Polyzos, S.A. Epidemiology, pathogenesis, diagnosis and emerging treatment of nonalcoholic fatty liver disease. Arch. Med. Res. 2021, 52, 25–37. [Google Scholar] [CrossRef]
- Milosevic, I.; Vujovic, A.; Barac, A.; Djelic, M.; Korac, M.; Radovanovic Spurnic, A.; Gmizic, I.; Stevanovic, O.; Djordjevic, V.; Lekic, N. Gut-liver axis, gut microbiota, and its modulation in the management of liver diseases: A review of the literature. Int. J. Mol. Sci. 2019, 20, 395. [Google Scholar] [CrossRef] [PubMed]
- Delli Bovi, A.P.; Marciano, F.; Mandato, C.; Siano, M.A.; Savoia, M.; Vajro, P. Oxidative stress in non-alcoholic fatty liver disease. An updated mini review. Front. Med. 2021, 8, 595371. [Google Scholar] [CrossRef]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of oxidative stress in the pathogenesis of non-alcoholic fatty liver disease: Implications for prevention and therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef]
- Ma, Y.; Lee, G.; Heo, S.-Y.; Roh, Y.-S. Oxidative stress is a key modulator in the development of nonalcoholic fatty liver disease. Antioxidants 2021, 11, 91. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Xu, M.; Jeong, S.; Qian, Y.; Wu, H.; Xia, Q.; Kong, X. The role of Nrf2 in liver disease: Novel molecular mechanisms and therapeutic approaches. Front. Pharmacol. 2019, 9, 1428. [Google Scholar] [CrossRef] [PubMed]
- Okanoue, T.; Yamauchi, N.; Furutani, M.; Hirohama, A.; Sumida, Y.; Nakashima, T. Predictors of nonalcoholic steatohepatitis in Japanese patients: Thioredoxin and NASH. In NASH and Nutritional Therapy; Springer: Berlin/Heidelberg, Germany, 2005; pp. 64–72. [Google Scholar]
- Nakashima, T.; Sumida, Y.; Furutani, M.; Hirohama, A.; Okita, M.; Mitsuyoshi, H.; Itoh, Y.; Okanoue, T. Elevation of serum thioredoxin levels in patients with nonalcoholic steatohepatitis. Hepatol. Res. 2005, 33, 135–137. [Google Scholar] [CrossRef] [PubMed]
- Xiong, F.; Zhou, Q.; Huang, X.; Cao, P.; Wang, Y. Ferroptosis plays a novel role in nonalcoholic steatohepatitis pathogenesis. Front. Pharmacol. 2022, 13, 1055793. [Google Scholar] [CrossRef] [PubMed]
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Yuet-Yin Kok, C.; Okochi, H.; Nakano, H. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019, 10, 449. [Google Scholar] [CrossRef] [PubMed]
- Seco-Cervera, M.; González-Cabo, P.; Pallardó, F.V.; Romá-Mateo, C.; García-Giménez, J.L. Thioredoxin and glutaredoxin systems as potential targets for the development of new treatments in Friedreich’s ataxia. Antioxidants 2020, 9, 1257. [Google Scholar] [CrossRef]
- Pan, M.; Zhang, F.; Qu, K.; Liu, C.; Zhang, J. TXNIP: A double-edged sword in disease and therapeutic outlook. Oxidative Med. Cell. Longev. 2022, 2022, 7805115. [Google Scholar] [CrossRef]
- Lee, S.; Kim, S.M.; Lee, R.T. Thioredoxin and thioredoxin target proteins: From molecular mechanisms to functional significance. Antioxid. Redox Signal. 2013, 18, 1165–1207. [Google Scholar] [CrossRef]
- Le Roy, T.; Llopis, M.; Lepage, P.; Bruneau, A.; Rabot, S.; Bevilacqua, C.; Martin, P.; Philippe, C.; Walker, F.; Bado, A. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2013, 62, 1787–1794. [Google Scholar] [CrossRef]
- Del Chierico, F.; Nobili, V.; Vernocchi, P.; Russo, A.; De Stefanis, C.; Gnani, D.; Furlanello, C.; Zandonà, A.; Paci, P.; Capuani, G. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology 2017, 65, 451–464. [Google Scholar] [CrossRef]
- Shen, F.; Zheng, R.-D.; Sun, X.-Q.; Ding, W.-J.; Wang, X.-Y.; Fan, J.-G. Gut microbiota dysbiosis in patients with non-alcoholic fatty liver disease. Hepatobiliary Pancreat. Dis. Int. 2017, 16, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Zmora, N.; Adolph, T.E.; Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 2020, 20, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Ohnishi, H. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 7381. [Google Scholar] [CrossRef]
- Chen, J.; Vitetta, L. Gut microbiota metabolites in NAFLD pathogenesis and therapeutic implications. Int. J. Mol. Sci. 2020, 21, 5214. [Google Scholar] [CrossRef]
- Vallianou, N.; Stratigou, T.; Christodoulatos, G.S.; Dalamaga, M. Understanding the role of the gut microbiome and microbial metabolites in obesity and obesity-associated metabolic disorders: Current evidence and perspectives. Curr. Obes. Rep. 2019, 8, 317–332. [Google Scholar] [CrossRef]
- Carpino, G.; Del Ben, M.; Pastori, D.; Carnevale, R.; Baratta, F.; Overi, D.; Francis, H.; Cardinale, V.; Onori, P.; Safarikia, S. Increased liver localization of lipopolysaccharides in human and experimental NAFLD. Hepatology 2020, 72, 470–485. [Google Scholar] [CrossRef]
- Nocito, A.; Dahm, F.; Jochum, W.; Jang, J.H.; Georgiev, P.; Bader, M.; Renner, E.L.; Clavien, P.A. Serotonin mediates oxidative stress and mitochondrial toxicity in a murine model of nonalcoholic steatohepatitis. Gastroenterology 2007, 133, 608–618. [Google Scholar] [CrossRef]
- Hoyles, L.; Fernandez-Real, J.-M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1070–1080. [Google Scholar] [CrossRef]
- Molinaro, A.; Wahlström, A.; Marschall, H.-U. Role of bile acids in metabolic control. Trends Endocrinol. Metab. 2018, 29, 31–41. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G. Fatty liver disease caused by high-alcohol-producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 675–688.e7. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Z.; Li, H.; Zhao, J.; Wei, X.; Lin, W.; Zhao, X.; Jiang, A.; Yuan, J. Endogenous ethanol produced by intestinal bacteria induces mitochondrial dysfunction in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2020, 35, 2009–2019. [Google Scholar] [CrossRef] [PubMed]
- Jameson, E.; Quareshy, M.; Chen, Y. Methodological considerations for the identification of choline and carnitine-degrading bacteria in the gut. Methods 2018, 149, 42–48. [Google Scholar] [CrossRef]
- Rath, S.; Heidrich, B.; Pieper, D.H.; Vital, M. Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome 2017, 5, 54. [Google Scholar] [CrossRef]
- Hu, Y.; Zhao, Y.; Yuan, L.; Yang, X. Protective effects of tartary buckwheat flavonoids on high TMAO diet-induced vascular dysfunction and liver injury in mice. Food Funct. 2015, 6, 3359–3372. [Google Scholar] [CrossRef]
- Ren, D.; Liu, Y.; Zhao, Y.; Yang, X. Hepatotoxicity and endothelial dysfunction induced by high choline diet and the protective effects of phloretin in mice. Food Chem. Toxicol. 2016, 94, 203–212. [Google Scholar] [CrossRef]
- Wu, Q.; Zhang, X.; Zhao, Y.; Yang, X. High l-carnitine ingestion impairs liver function by disordering gut bacteria composition in mice. J. Agric. Food Chem. 2020, 68, 5707–5714. [Google Scholar] [CrossRef]
- Yang, C.; Zhao, Y.; Ren, D.; Yang, X. Protective effect of saponins-enriched fraction of gynostemma pentaphyllum against high choline-induced vascular endothelial dysfunction and hepatic damage in mice. Biol. Pharm. Bull. 2020, 43, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, Q.; Wang, N.; Zhang, L.; Yang, X.; Zhao, Y. Quercetin inhibits hepatotoxic effects by reducing trimethylamine-N-oxide formation in C57BL/6J mice fed with a high l-carnitine diet. Food Funct. 2023, 14, 206–214. [Google Scholar] [CrossRef]
- Chen, S.; Henderson, A.; Petriello, M.C.; Romano, K.A.; Gearing, M.; Miao, J.; Schell, M.; Sandoval-Espinola, W.J.; Tao, J.; Sha, B. Trimethylamine N-oxide binds and activates PERK to promote metabolic dysfunction. Cell Metab. 2019, 30, 1141–1151.e5. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.-L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef]
- Zhou, Y.; Tong, Z.; Jiang, S.; Zheng, W.; Zhao, J.; Zhou, X. The roles of endoplasmic reticulum in NLRP3 inflammasome activation. Cells 2020, 9, 1219. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.G.; Upton, J.-P.; Praveen, P.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Sun, X.; Jiao, X.; Ma, Y.; Liu, Y.; Zhang, L.; He, Y.; Chen, Y. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 2016, 481, 63–70. [Google Scholar] [CrossRef]
- Kim, S.K.; Choe, J.Y.; Park, K.Y. TXNIP-mediated nuclear factor-κB signaling pathway and intracellular shifting of TXNIP in uric acid-induced NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 2019, 511, 725–731. [Google Scholar] [CrossRef]
- Ferro, D.; Baratta, F.; Pastori, D.; Cocomello, N.; Colantoni, A.; Angelico, F.; Del Ben, M. New insights into the pathogenesis of non-alcoholic fatty liver disease: Gut-derived lipopolysaccharides and oxidative stress. Nutrients 2020, 12, 2762. [Google Scholar] [CrossRef] [PubMed]
- Suliman, H.B.; Carraway, M.S.; Welty-Wolf, K.E.; Whorton, A.R.; Piantadosi, C.A. Lipopolysaccharide stimulates mitochondrial biogenesis via activation of nuclear respiratory factor-1. J. Biol. Chem. 2003, 278, 41510–41518. [Google Scholar] [CrossRef] [PubMed]
- Baratta, F.; Pastori, D.; Bartimoccia, S.; Cammisotto, V.; Cocomello, N.; Colantoni, A.; Nocella, C.; Carnevale, R.; Ferro, D.; Angelico, F. Poor adherence to mediterranean diet and serum lipopolysaccharide are associated with oxidative stress in patients with non-alcoholic fatty liver disease. Nutrients 2020, 12, 1732. [Google Scholar] [CrossRef] [PubMed]
- Fukunishi, S.; Sujishi, T.; Takeshita, A.; Ohama, H.; Tsuchimoto, Y.; Asai, A.; Tsuda, Y.; Higuchi, K. Lipopolysaccharides accelerate hepatic steatosis in the development of nonalcoholic fatty liver disease in Zucker rats. J. Clin. Biochem. Nutr. 2014, 54, 39–44. [Google Scholar] [CrossRef]
- Park, H.S.; Jung, H.Y.; Park, E.Y.; Kim, J.; Lee, W.J.; Bae, Y.S. Cutting edge: Direct interaction of TLR4 with NAD (P) H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-κB. J. Immunol. 2004, 173, 3589–3593. [Google Scholar] [CrossRef]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef]
- Lee, U.E.; Friedman, S.L. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 195–206. [Google Scholar] [CrossRef]
- Iwaisako, K.; Brenner, D.A.; Kisseleva, T. What’s new in liver fibrosis? The origin of myofibroblasts in liver fibrosis. J. Gastroenterol. Hepatol. 2012, 27, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.L.; Gorin, Y. Myofibroblast differentiation during fibrosis: Role of NAD(P)H oxidases. Kidney Int. 2011, 79, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Bocca, C.; Protopapa, F.; Foglia, B.; Maggiora, M.; Cannito, S.; Parola, M.; Novo, E. Hepatic Myofibroblasts: A Heterogeneous and Redox-Modulated Cell Population in Liver Fibrogenesis. Antioxidants 2022, 11, 1278. [Google Scholar] [CrossRef]
- Xu, J.; Liu, X.; Koyama, Y.; Wang, P.; Lan, T.; Kim, I.G.; Kim, I.H.; Ma, H.Y.; Kisseleva, T. The types of hepatic myofibroblasts contributing to liver fibrosis of different etiologies. Front. Pharmacol. 2014, 5, 167. [Google Scholar] [CrossRef]
- Okuyama, H.; Nakamura, H.; Shimahara, Y.; Araya, S.; Kawada, N.; Yamaoka, Y.; Yodoi, Y. Overexpression of thioredoxin prevents acute hepatitis caused by thioacetamide or lipopolysaccharide in mice. Hepatology 2003, 37, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, H.; Nakamura, H.; Shimahara, Y.; Uyama, N.; Kwon, Y.-W.; Kawada, N.; Yamaoka, Y.; Yodoi, J. Overexpression of thioredoxin prevents thioacetamide-induced hepatic fibrosis in mice. J. Hepatol. 2005, 42, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xing, Y.; Tang, Z.; Tang, Y.; Shen, J.; Zhang, F. Thioredoxin-2 impacts the inflammatory response via suppression of NF-κB and MAPK signaling in sepsis shock. Biochem. Biophys. Res. Commun. 2020, 524, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Asehnoune, K.; Strassheim, D.; Mitra, S.; Kim, J.Y.; Abraham, E. Involvement of reactive oxygen species in Toll-like receptor 4-dependent activation of NF-κB. J. Immunol. 2004, 172, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Lee, S.-M. NLRP3 inflammasome activation in D-galactosamine and lipopolysaccharide-induced acute liver failure: Role of heme oxygenase-1. Free Radic. Biol. Med. 2013, 65, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Vallianou, N.; Christodoulatos, G.S.; Karampela, I.; Tsilingiris, D.; Magkos, F.; Stratigou, T.; Kounatidis, D.; Dalamaga, M. Understanding the role of the gut microbiome and microbial metabolites in non-alcoholic fatty liver disease: Current evidence and perspectives. Biomolecules 2021, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Kim, H.Y.; Kim, M.G.; Jeong, S.; Yun, C.-H.; Han, S.H. Short-chain fatty acids inhibit staphylococcal lipoprotein-induced nitric oxide production in murine macrophages. Immune Netw. 2019, 19, e9. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wang, Y.; Wang, P.; Huang, Y.; Wang, F. Short-chain fatty acids manifest stimulative and protective effects on intestinal barrier function through the inhibition of NLRP3 inflammasome and autophagy. Cell. Physiol. Biochem. 2018, 49, 190–205. [Google Scholar] [CrossRef]
- Kobayashi, M.; Mikami, D.; Kimura, H.; Kamiyama, K.; Morikawa, Y.; Yokoi, S.; Kasuno, K.; Takahashi, N.; Taniguchi, T.; Iwano, M. Short-chain fatty acids, GPR41 and GPR43 ligands, inhibit TNF-α-induced MCP-1 expression by modulating p38 and JNK signaling pathways in human renal cortical epithelial cells. Biochem. Biophys. Res. Commun. 2017, 486, 499–505. [Google Scholar] [CrossRef]
- Deng, M.; Qu, F.; Chen, L.; Liu, C.; Zhang, M.; Ren, F.; Guo, H.; Zhang, H.; Ge, S.; Wu, C. SCFAs alleviated steatosis and inflammation in mice with NASH induced by MCD. J. Endocrinol. 2020, 245, 425–437. [Google Scholar] [CrossRef]
- Araújo, J.R.; Tazi, A.; Burlen-Defranoux, O.; Vichier-Guerre, S.; Nigro, G.; Licandro, H.; Demignot, S.; Sansonetti, P.J. Fermentation products of commensal bacteria alter enterocyte lipid metabolism. Cell Host Microbe 2020, 27, 358–375.e357. [Google Scholar] [CrossRef]
- Den Besten, G.; Bleeker, A.; Gerding, A.; van Eunen, K.; Havinga, R.; van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngoud, D.-J. Short-chain fatty acids protect against high-fat diet–induced obesity via a PPARγ-dependent switch from lipogenesis to fat oxidation. Diabetes 2015, 64, 2398–2408. [Google Scholar] [CrossRef]
- van der Beek, C.M.; Canfora, E.E.; Lenaerts, K.; Troost, F.J.; Olde Damink, S.W.; Holst, J.J.; Masclee, A.A.; Dejong, C.H.; Blaak, E.E. Distal, not proximal, colonic acetate infusions promote fat oxidation and improve metabolic markers in overweight/obese men. Clin. Sci. 2016, 130, 2073–2082. [Google Scholar] [CrossRef]
- Canfora, E.E.; van der Beek, C.M.; Jocken, J.W.; Goossens, G.H.; Holst, J.J.; Olde Damink, S.W.; Lenaerts, K.; Dejong, C.H.; Blaak, E.E. Colonic infusions of short-chain fatty acid mixtures promote energy metabolism in overweight/obese men: A randomized crossover trial. Sci. Rep. 2017, 7, 2360. [Google Scholar] [CrossRef]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef]
- Murugesan, S.; Nirmalkar, K.; Hoyo-Vadillo, C.; García-Espitia, M.; Ramírez-Sánchez, D.; García-Mena, J. Gut microbiome production of short-chain fatty acids and obesity in children. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 621–625. [Google Scholar] [CrossRef]
- Park, G.; Jung, S.; Wellen, K.E.; Jang, C. The interaction between the gut microbiota and dietary carbohydrates in nonalcoholic fatty liver disease. Exp. Mol. Med. 2021, 53, 809–822. [Google Scholar] [CrossRef]
- Quesada-Vázquez, S.; Bone, C.; Saha, S.; Triguero, I.; Colom-Pellicer, M.; Aragonès, G.; Hildebrand, F.; Del Bas, J.M.; Caimari, A.; Beraza, N.; et al. Microbiota Dysbiosis and Gut Barrier Dysfunction Associated with Non-Alcoholic Fatty Liver Disease Are Modulated by a Specific Metabolic Cofactors’ Combination. Int. J. Mol. Sci. 2022, 23, 13675. [Google Scholar] [CrossRef]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef]
- Zhou, D.; Chen, Y.-W.; Zhao, Z.-H.; Yang, R.-X.; Xin, F.-Z.; Liu, X.-L.; Pan, Q.; Zhou, H.; Fan, J.-G. Sodium butyrate reduces high-fat diet-induced non-alcoholic steatohepatitis through upregulation of hepatic GLP-1R expression. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A.; Jin, C.J.; Brandt, A.; Sellmann, C.; Nier, A.; Burkard, M.; Venturelli, S.; Bergheim, I. Oral supplementation of sodium butyrate attenuates the progression of non-alcoholic steatohepatitis. Nutrients 2020, 12, 951. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef]
- Kelly, C.J.; Colgan, S.P. Breathless in the gut: Implications of luminal O2 for microbial pathogenicity. Cell Host Microbe 2016, 19, 427–428. [Google Scholar] [CrossRef] [PubMed]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and butyrate-producing colon bacteria: Importance and strategies for their stimulation in the human gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef]
- Derrien, M.; Vaughan, E.E.; Plugge, C.M.; de Vos, W.M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 2004, 54, 1469–1476. [Google Scholar] [CrossRef]
- Li, S.; Liu, J.; Wang, Z.; Duan, F.; Jia, Z.; Chen, X.; Li, S. The promising role of probiotics/prebiotics/synbiotics in energy metabolism biomarkers in patients with NAFLD: A systematic review and meta-analysis. Front. Public Health 2022, 10, 862266. [Google Scholar] [CrossRef]
- Carpi, R.Z.; Barbalho, S.M.; Sloan, K.P.; Laurindo, L.F.; Gonzaga, H.F.; Grippa, P.C.; Zutin, T.L.M.; Girio, R.J.; Repetti, C.S.F.; Detregiachi, C.R.P. The effects of probiotics, prebiotics and synbiotics in non-alcoholic fat liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH): A systematic review. Int. J. Mol. Sci. 2022, 23, 8805. [Google Scholar] [CrossRef]
- Agus, A.; Planchais, J.; Sokol, H. Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Shimada, Y.; Kinoshita, M.; Harada, K.; Mizutani, M.; Masahata, K.; Kayama, H.; Takeda, K. Commensal bacteria-dependent indole production enhances epithelial barrier function in the colon. PLoS ONE 2013, 8, e80604. [Google Scholar] [CrossRef]
- Ma, L.; Li, H.; Hu, J.; Zheng, J.; Zhou, J.; Botchlett, R.; Matthews, D.; Zeng, T.; Chen, L.; Xiao, X. Indole alleviates diet-induced hepatic steatosis and inflammation in a manner involving myeloid cell 6-Phosphofructo-2-kinase/Fructose-2, 6-Biphosphatase 3. Hepatology 2020, 72, 1191–1203. [Google Scholar] [CrossRef]
- Ji, Y.; Gao, Y.; Chen, H.; Yin, Y.; Zhang, W. Indole-3-acetic acid alleviates nonalcoholic fatty liver disease in mice via attenuation of hepatic lipogenesis, and oxidative and inflammatory stress. Nutrients 2019, 11, 2062. [Google Scholar] [CrossRef]
- Crane, J.D.; Palanivel, R.; Mottillo, E.P.; Bujak, A.L.; Wang, H.; Ford, R.J.; Collins, A.; Blümer, R.M.; Fullerton, M.D.; Yabut, J.M. Inhibiting peripheral serotonin synthesis reduces obesity and metabolic dysfunction by promoting brown adipose tissue thermogenesis. Nat. Med. 2015, 21, 166–172. [Google Scholar] [CrossRef]
- Choi, W.; Namkung, J.; Hwang, I.; Kim, H.; Lim, A.; Park, H.J.; Lee, H.W.; Han, K.-H.; Park, S.; Jeong, J.-S. Serotonin signals through a gut-liver axis to regulate hepatic steatosis. Nat. Commun. 2018, 9, 4824. [Google Scholar] [CrossRef]
- Bharti, V.; Tan, H.; Deol, J.; Wu, Z.; Wang, J.-F. Upregulation of antioxidant thioredoxin by antidepressants fluoxetine and venlafaxine. Psychopharmacology 2020, 237, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Ritze, Y.; Bárdos, G.; Hubert, A.; Böhle, M.; Bischoff, S.C. Effect of tryptophan supplementation on diet-induced non-alcoholic fatty liver disease in mice. Br. J. Nutr. 2014, 112, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sunny, N.E.; Kalavalapalli, S.; Bril, F.; Garrett, T.J.; Nautiyal, M.; Mathew, J.T.; Williams, C.M.; Cusi, K. Cross-talk between branched-chain amino acids and hepatic mitochondria is compromised in nonalcoholic fatty liver disease. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E311–E319. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.J.; Adams, S.H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 2014, 10, 723–736. [Google Scholar] [CrossRef]
- Adeva, M.M.; Calviño, J.; Souto, G.; Donapetry, C. Insulin resistance and the metabolism of branched-chain amino acids in humans. Amino Acids 2012, 43, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lee, F.Y.; Barrera, G.; Lee, H.; Vales, C.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- De Fabiani, E.; Mitro, N.; Gilardi, F.; Caruso, D.; Galli, G.; Crestani, M. Coordinated control of cholesterol catabolism to bile acids and of gluconeogenesis via a novel mechanism of transcription regulation linked to the fasted-to-fed cycle. J. Biol. Chem. 2003, 278, 39124–39132. [Google Scholar] [CrossRef]
- Cyphert, H.A.; Ge, X.; Kohan, A.B.; Salati, L.M.; Zhang, Y.; Hillgartner, F.B. Activation of the farnesoid X receptor induces hepatic expression and secretion of fibroblast growth factor 21. J. Biol. Chem. 2012, 287, 25123–25138. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef]
- Gruenbacher, G.; Gander, H.; Rahm, A.; Dobler, G.; Drasche, A.; Troppmair, J.; Nussbaumer, W.; Thurnher, M. The human G protein-coupled ATP receptor P2Y11 is associated with IL-10 driven macrophage differentiation. Front. Immunol. 2019, 10, 1870. [Google Scholar] [CrossRef]
- Keitel, V.; Donner, M.; Winandy, S.; Kubitz, R.; Häussinger, D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem. Biophys. Res. Commun. 2008, 372, 78–84. [Google Scholar] [CrossRef]
- Jiao, N.; Baker, S.S.; Chapa-Rodriguez, A.; Liu, W.; Nugent, C.A.; Tsompana, M.; Mastrandrea, L.; Buck, M.J.; Baker, R.D.; Genco, R.J. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 2018, 67, 1881–1891. [Google Scholar] [CrossRef]
- Nomoto, M.; Miyata, M.; Yin, S.; Kurata, Y.; Shimada, M.; Yoshinari, K.; Gonzalez, F.J.; Suzuki, K.; Shibasaki, S.; Kurosawa, T. Bile acid-induced elevated oxidative stress in the absence of farnesoid X receptor. Biol. Pharm. Bull. 2009, 32, 172–178. [Google Scholar] [CrossRef]
- Wang, Y.D.; Chen, W.D.; Wang, M.; Yu, D.; Forman, B.M.; Huang, W. Farnesoid X receptor antagonizes nuclear factor κB in hepatic inflammatory response. Hepatology 2008, 48, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Huang, G.; Gong, W.; Zhou, P.; Zhao, Y.; Zhang, Y.; Zeng, Y.; Gao, M.; Pan, Z.; He, F. FXR ligands protect against hepatocellular inflammation via SOCS3 induction. Cell. Signal. 2012, 24, 1658–1664. [Google Scholar] [CrossRef]
- Djavaheri-Mergny, M.; Javelaud, D.; Wietzerbin, J.; Besançon, F. NF-κB activation prevents apoptotic oxidative stress via an increase of both thioredoxin and MnSOD levels in TNFα-treated Ewing sarcoma cells. FEBS Lett. 2004, 578, 111–115. [Google Scholar] [CrossRef]
- Kairisalo, M.; Korhonen, L.; Blomgren, K.; Lindholm, D. X-linked inhibitor of apoptosis protein increases mitochondrial antioxidants through NF-κB activation. Biochem. Biophys. Res. Commun. 2007, 364, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Engstler, A.J.; Aumiller, T.; Degen, C.; Dürr, M.; Weiss, E.; Maier, I.B.; Schattenberg, J.M.; Jin, C.J.; Sellmann, C.; Bergheim, I. Insulin resistance alters hepatic ethanol metabolism: Studies in mice and children with non-alcoholic fatty liver disease. Gut 2016, 65, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- Ambade, A.; Mandrekar, P. Oxidative stress and inflammation: Essential partners in alcoholic liver disease. Int. J. Hepatol. 2012, 2012, 853175. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I.; Roychowdhury, S.; DiBello, P.M.; Jacobsen, D.W.; Nagy, L.E. Exogenous thioredoxin prevents ethanol-induced oxidative damage and apoptosis in mouse liver. Hepatology 2009, 49, 1709–1717. [Google Scholar] [CrossRef]
- Pfaff, A.R.; Beltz, J.; King, E.; Ercal, N. Medicinal Thiols: Current Status and New Perspectives. Mini Rev. Med. Chem. 2020, 20, 513–529. [Google Scholar] [CrossRef]
- Honda, Y.; Kessoku, T.; Sumida, Y.; Kobayashi, T.; Kato, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K.; Yoneda, M.; et al. Efficacy of glutathione for the treatment of nonalcoholic fatty liver disease: An open-label, single-arm, multicenter, pilot study. BMC Gastroenterol. 2017, 17, 96. [Google Scholar] [CrossRef]
- Pedre, B.; Barayeu, U.; Ezerina, D.; Dick, T.P. The mechanism of action of N-acetylcysteine (NAC): The emerging role of H(2)S and sulfane sulfur species. Pharmacol. Ther. 2021, 228, 107916. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Guo, R.; Pei, L.; Lai, S.; Li, J.; Yin, Y.; Xu, T.; Yang, W.; Song, Q.; Han, Q.; et al. N-Acetylcysteine alleviates high fat diet-induced hepatic steatosis and liver injury via regulating the intestinal microecology in mice. Food Funct. 2022, 13, 3368–3380. [Google Scholar] [CrossRef]
- Li, J.; Guo, C.; Wu, J. Astaxanthin in Liver Health and Disease: A Potential Therapeutic Agent. Drug Des. Dev. Ther. 2020, 14, 2275–2285. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, M.; Dagah, O.M.A.; Silaa, B.B.; Lu, J. Thioredoxin/Glutaredoxin Systems and Gut Microbiota in NAFLD: Interplay, Mechanism, and Therapeutical Potential. Antioxidants 2023, 12, 1680. https://doi.org/10.3390/antiox12091680
Zhu M, Dagah OMA, Silaa BB, Lu J. Thioredoxin/Glutaredoxin Systems and Gut Microbiota in NAFLD: Interplay, Mechanism, and Therapeutical Potential. Antioxidants. 2023; 12(9):1680. https://doi.org/10.3390/antiox12091680
Chicago/Turabian StyleZhu, Minghui, Omer M. A. Dagah, Billton Bryson Silaa, and Jun Lu. 2023. "Thioredoxin/Glutaredoxin Systems and Gut Microbiota in NAFLD: Interplay, Mechanism, and Therapeutical Potential" Antioxidants 12, no. 9: 1680. https://doi.org/10.3390/antiox12091680
APA StyleZhu, M., Dagah, O. M. A., Silaa, B. B., & Lu, J. (2023). Thioredoxin/Glutaredoxin Systems and Gut Microbiota in NAFLD: Interplay, Mechanism, and Therapeutical Potential. Antioxidants, 12(9), 1680. https://doi.org/10.3390/antiox12091680