
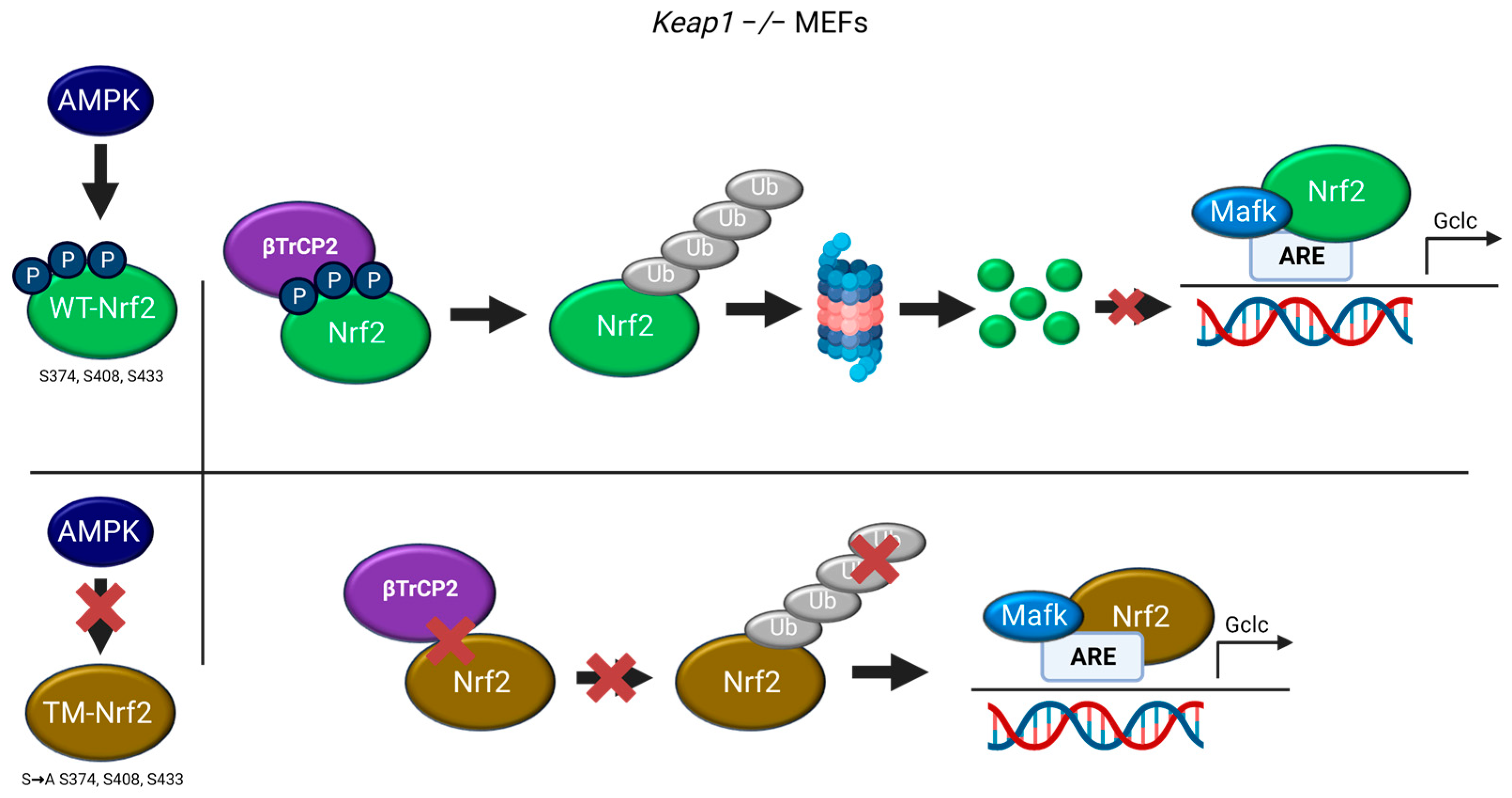
AMPK-Mediated Phosphorylation of Nrf2 at S374/S408/S433 Favors Its βTrCP2-Mediated Degradation in KEAP1-Deficient Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents and Compound Treatment
2.3. Plasmids
2.4. siRNA Sequences
2.5. Transient Transfections
2.6. Immunoblotting
2.7. Immunoprecipitation
2.8. Proximity Ligation Assay (PLA) and Indirect Immunofluorescence
2.9. Quantitative Chain Polymerase Reaction (qPCR)
2.10. In Vitro Ubiquitinylation Assay
2.11. Statistical Analysis
3. Results
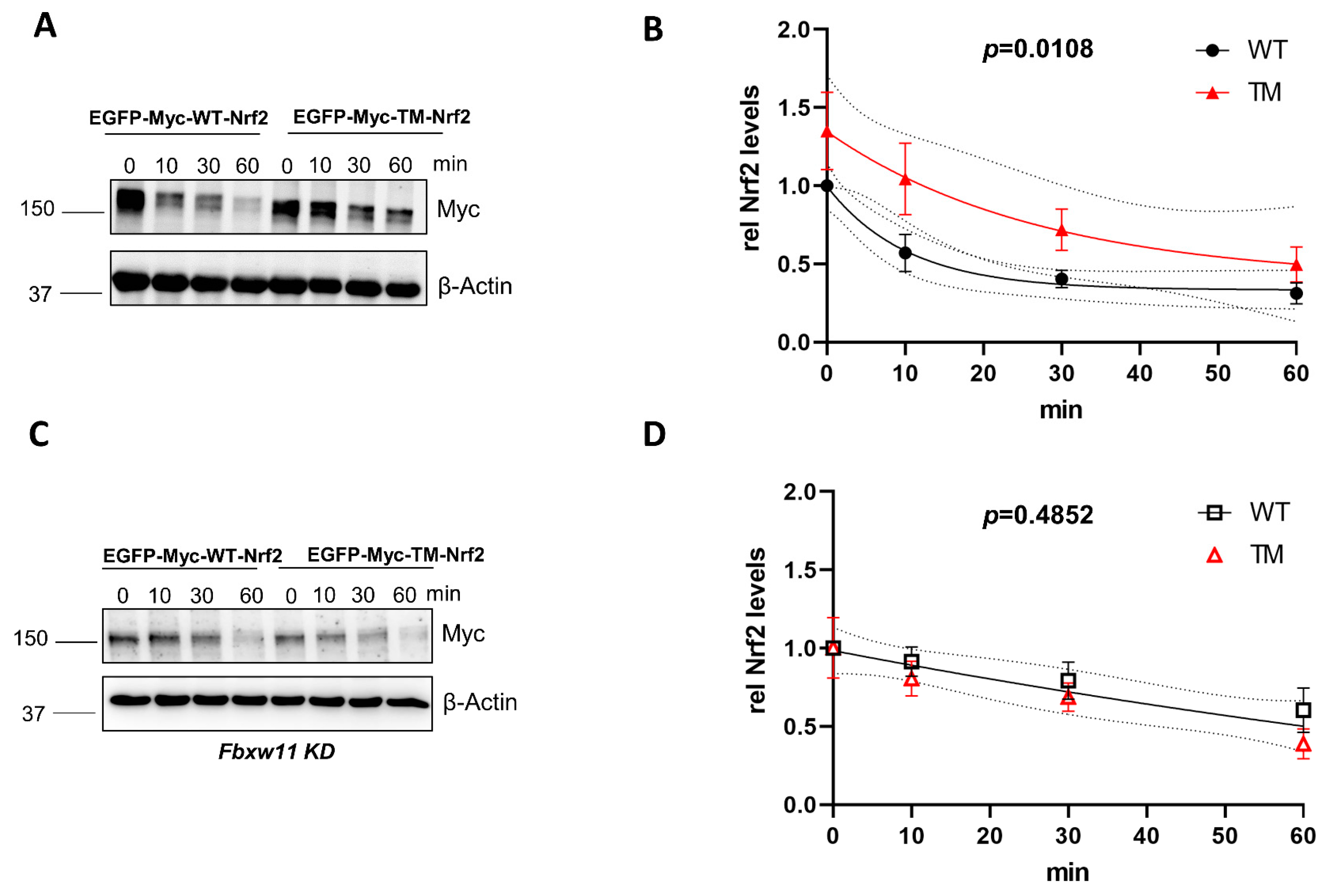
3.1. Mutation of the AMPK-Dependent Phospho-Sites S374, S408, and S433 to Alanine Stabilizes Nrf2 in Keap1−/− Cells in a βTrCP2-Dependent Manner
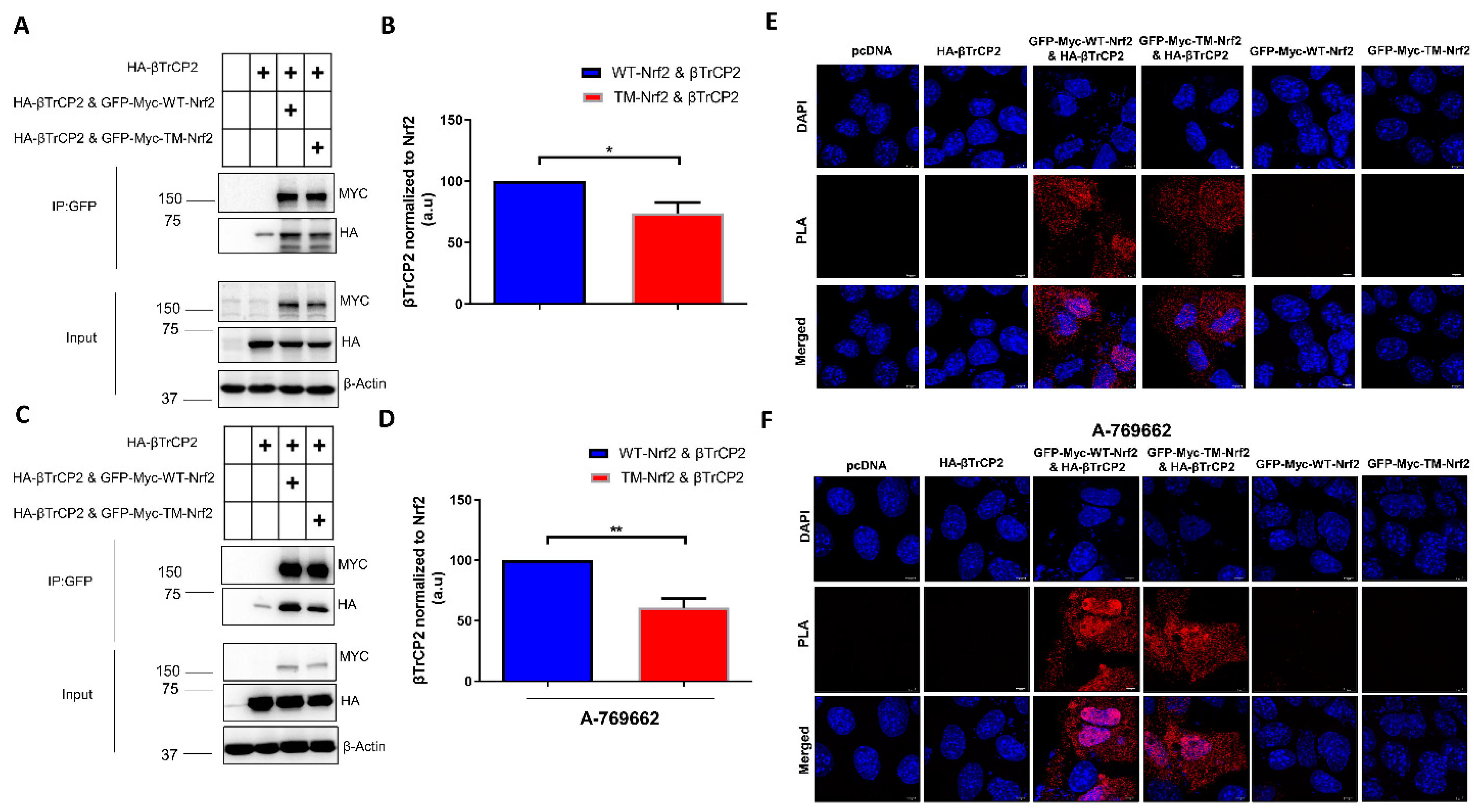
3.2. Mutation of the AMPK-Dependent Phospho-Sites to Alanine Impedes the Interaction of Nrf2 with β-TrCP2 in Keap1−/− Cells
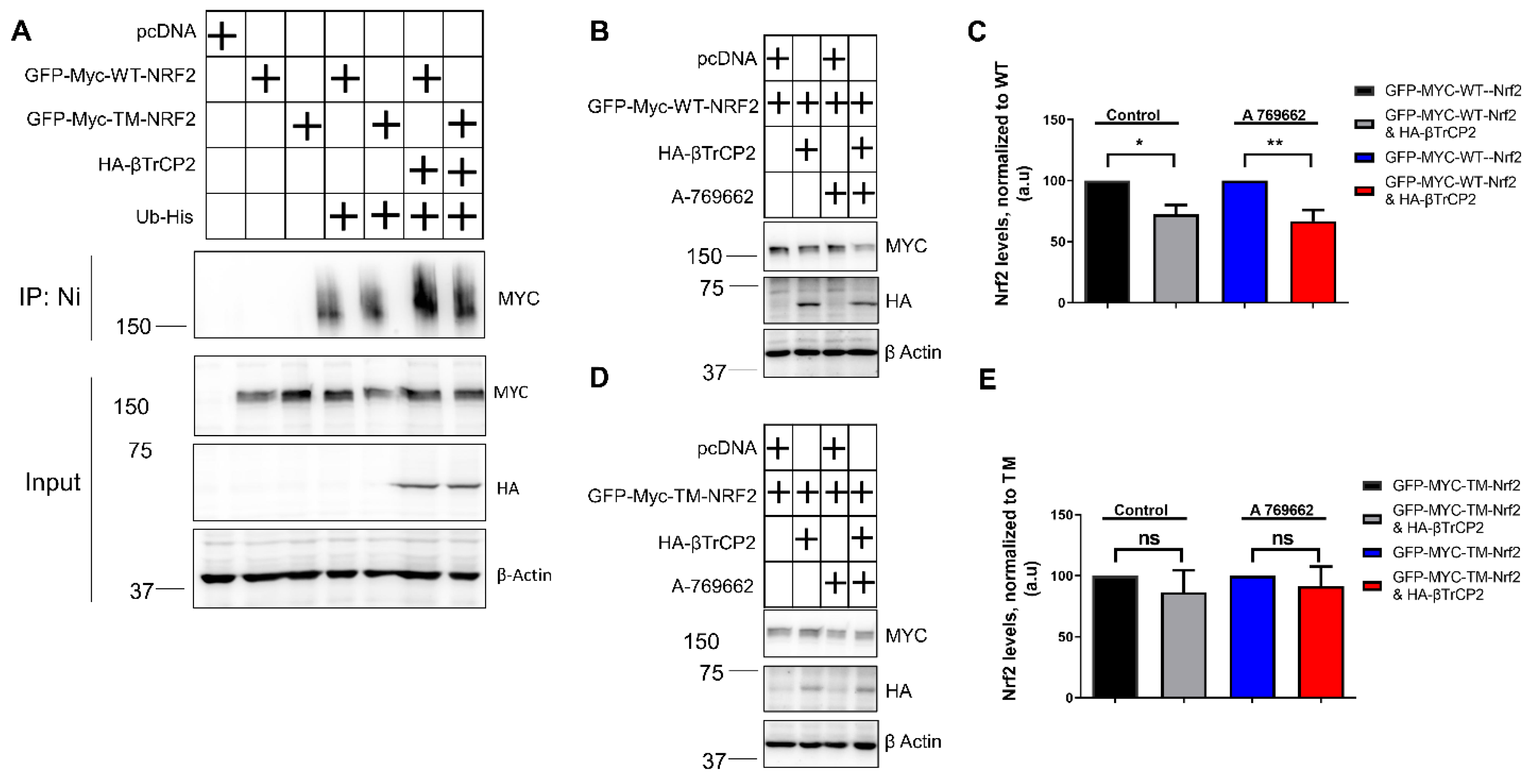
3.3. Mutation of the AMPK-Dependent Phospho-Sites to Alanine on Nrf2 Impedes Its βTrcP2-Mediated Ubiquitination and Degradation in Keap1−/− Cells
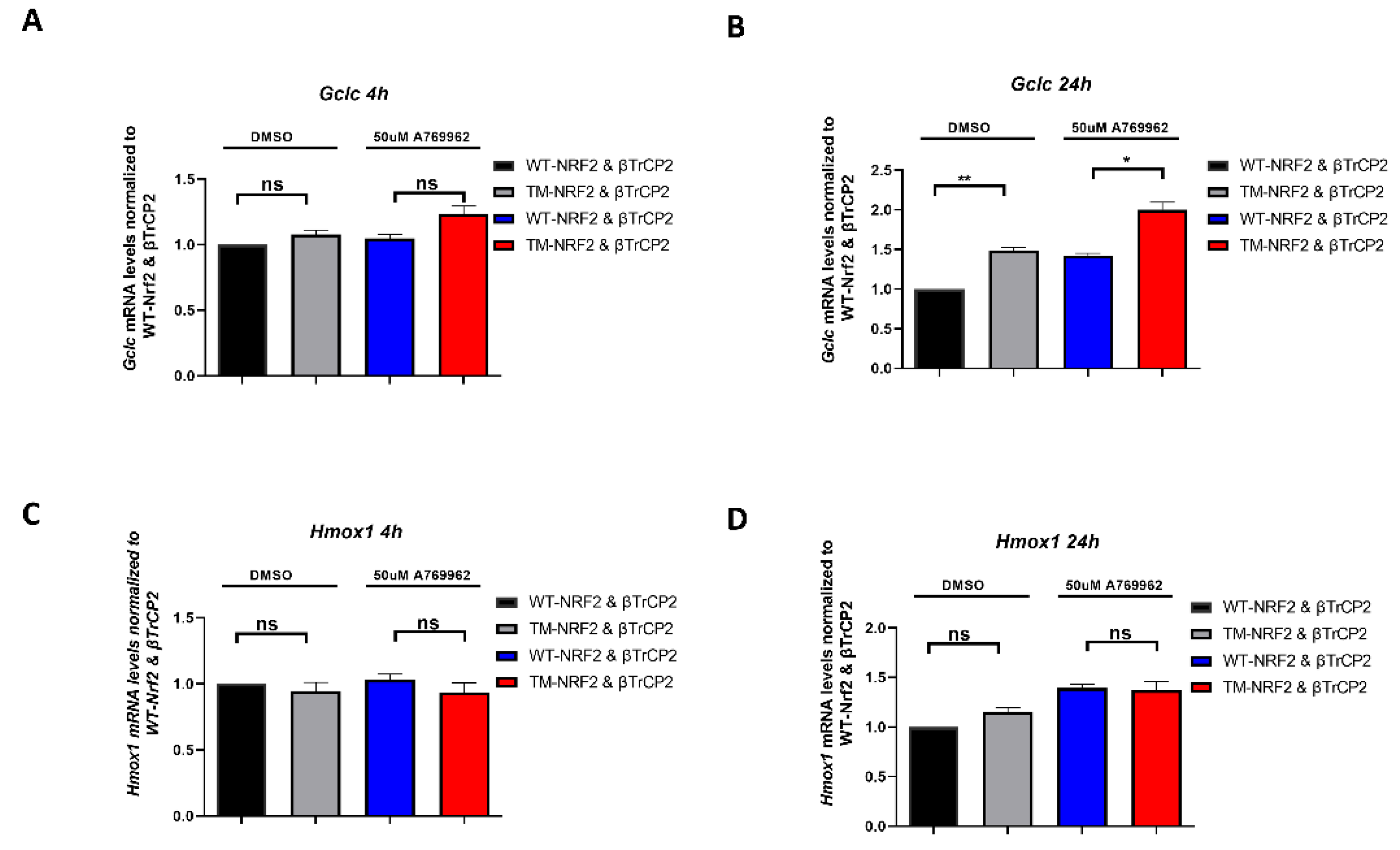
3.4. The Identified AMPK-Dependent Phospho-Sites in Nrf2 Affect the Expression of Endogenous Nrf2 Target Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Matzinger, M.; Fischhuber, K.; Heiss, E.H. Activation of Nrf2 signaling by natural products-can it alleviate diabetes? Biotechnol. Adv. 2018, 36, 1738–1767. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Hayes, J.D.; Chowdhry, S.; Dinkova-Kostova, A.T.; Sutherland, C. Dual regulation of transcription factor Nrf2 by Keap1 and by the combined actions of beta-TrCP and GSK-3. Biochem. Soc. Trans. 2015, 43, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; Leon, R.; Lopez, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/beta-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Nguyen, T.; Sherratt, P.J.; Huang, H.C.; Yang, C.S.; Pickett, C.B. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26 S proteasome. J. Biol. Chem. 2003, 278, 4536–4541. [Google Scholar] [CrossRef]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Li, L.; Iwamoto, N.; Nakajima-Takagi, Y.; Kaneko, H.; Nakayama, Y.; Eguchi, M.; Wada, Y.; Kumagai, Y.; Yamamoto, M. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol. Cell. Biol. 2009, 29, 493–502. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Lamont, D.J.; Beattie, K.A.; Hayes, J.D. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc. Natl. Acad. Sci. USA 2010, 107, 18838–18843. [Google Scholar] [CrossRef]
- He, F.; Antonucci, L.; Karin, M. NRF2 as a regulator of cell metabolism and inflammation in cancer. Carcinogenesis 2020, 41, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Petsouki, E.; Cabrera, S.N.S.; Heiss, E.H. AMPK and NRF2: Interactive players in the same team for cellular homeostasis? Free Radic. Biol. Med. 2022, 190, 75–93. [Google Scholar] [CrossRef]
- Liu, J.; Xu, Y.; Yan, M.; Yu, Y.; Guo, Y. 18beta-Glycyrrhetinic acid suppresses allergic airway inflammation through NF-kappaB and Nrf2/HO-1 signaling pathways in asthma mice. Sci. Rep. 2022, 12, 3121. [Google Scholar] [CrossRef]
- Zhao, Y.Y.; Wang, H.L.; Cheng, X.L.; Wei, F.; Bai, X.; Lin, R.C.; Vaziri, N.D. Metabolomics analysis reveals the association between lipid abnormalities and oxidative stress, inflammation, fibrosis, and Nrf2 dysfunction in aristolochic acid-induced nephropathy. Sci. Rep. 2015, 5, 12936. [Google Scholar] [CrossRef]
- Wang, P.; Geng, J.; Gao, J.; Zhao, H.; Li, J.; Shi, Y.; Yang, B.; Xiao, C.; Linghu, Y.; Sun, X.; et al. Macrophage achieves self-protection against oxidative stress-induced ageing through the Mst-Nrf2 axis. Nat. Commun. 2019, 10, 755. [Google Scholar] [CrossRef]
- Zhang, H.X.; Chen, Y.; Xu, R.; He, Q.Y. Nrf2 mediates the resistance of human A549 and HepG2 cancer cells to boningmycin, a new antitumor antibiotic, in vitro through regulation of glutathione levels. Acta Pharmacol. Sin. 2018, 39, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, S.; Patinen, T.; Jawahar Deen, A.; Pitkanen, S.; Harkonen, J.; Kansanen, E.; Kublbeck, J.; Levonen, A.L. The KEAP1-NRF2 pathway: Targets for therapy and role in cancer. Redox Biol. 2023, 63, 102726. [Google Scholar] [CrossRef]
- Sayin, V.I.; LeBoeuf, S.E.; Singh, S.X.; Davidson, S.M.; Biancur, D.; Guzelhan, B.S.; Alvarez, S.W.; Wu, W.L.; Karakousi, T.R.; Zavitsanou, A.M.; et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife 2017, 6, e28083. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.; Hayashi, M.; Zavitsanou, A.M.; Papagiannakopoulos, T. NRF2: KEAPing Tumors Protected. Cancer Discov. 2022, 12, 625–643. [Google Scholar] [CrossRef] [PubMed]
- Quinti, L.; Dayalan Naidu, S.; Trager, U.; Chen, X.; Kegel-Gleason, K.; Lleres, D.; Connolly, C.; Chopra, V.; Low, C.; Moniot, S.; et al. KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington’s disease patients. Proc. Natl. Acad. Sci. USA 2017, 114, E4676–E4685. [Google Scholar] [CrossRef]
- Uruno, A.; Yamamoto, M. The KEAP1-NRF2 System and Neurodegenerative Diseases. Antioxid. Redox Signal. 2023, 38, 974–988. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sanchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Iijima, K.; Miyamoto, M.; Nakahara, I.; Tanaka, H.; Ohtsuji, M.; Suzuki, T.; Kobayashi, A.; Yokota, J.; Sakiyama, T.; et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008, 68, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.J.; Cheng, X.D.; Zhang, J.; Zhang, W.D. Dual roles and therapeutic potential of Keap1-Nrf2 pathway in pancreatic cancer: A systematic review. Cell Commun. Signal. 2019, 17, 121. [Google Scholar] [CrossRef]
- Purohit, V.; Wang, L.; Yang, H.; Li, J.; Ney, G.M.; Gumkowski, E.R.; Vaidya, A.J.; Wang, A.; Bhardwaj, A.; Zhao, E.; et al. ATDC binds to KEAP1 to drive NRF2-mediated tumorigenesis and chemoresistance in pancreatic cancer. Genes Dev. 2021, 35, 218–233. [Google Scholar] [CrossRef]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Best, S.A.; De Souza, D.P.; Kersbergen, A.; Policheni, A.N.; Dayalan, S.; Tull, D.; Rathi, V.; Gray, D.H.; Ritchie, M.E.; McConville, M.J.; et al. Synergy between the KEAP1/NRF2 and PI3K Pathways Drives Non-Small-Cell Lung Cancer with an Altered Immune Microenvironment. Cell Metab. 2018, 27, 935–943.e934. [Google Scholar] [CrossRef]
- Scalera, S.; Mazzotta, M.; Cortile, C.; Krasniqi, E.; De Maria, R.; Cappuzzo, F.; Ciliberto, G.; Maugeri-Sacca, M. KEAP1-Mutant NSCLC: The Catastrophic Failure of a Cell-Protecting Hub. J. Thorac. Oncol. 2022, 17, 751–757. [Google Scholar] [CrossRef]
- Ghareghomi, S.; Moosavi-Movahedi, F.; Saso, L.; Habibi-Rezaei, M.; Khatibi, A.; Hong, J.; Moosavi-Movahedi, A.A. Modulation of Nrf2/HO-1 by Natural Compounds in Lung Cancer. Antioxidants 2023, 12, 735. [Google Scholar] [CrossRef]
- Lv, Y.; Lv, X.; Zhang, J.; Cao, G.; Xu, C.; Zhang, B.; Lin, W. BRD4 Targets the KEAP1-Nrf2-G6PD Axis and Suppresses Redox Metabolism in Small Cell Lung Cancer. Antioxidants 2022, 11, 661. [Google Scholar] [CrossRef]
- Young, L.H. A crystallized view of AMPK activation. Cell Metab. 2009, 10, 5–6. [Google Scholar] [CrossRef][Green Version]
- Steinberg, G.R.; Carling, D. AMP-activated protein kinase: The current landscape for drug development. Nat. Rev. Drug Discov. 2019, 18, 527–551. [Google Scholar] [CrossRef]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef]
- Jansen, T.; Kvandova, M.; Daiber, A.; Stamm, P.; Frenis, K.; Schulz, E.; Munzel, T.; Kroller-Schon, S. The AMP-Activated Protein Kinase Plays a Role in Antioxidant Defense and Regulation of Vascular Inflammation. Antioxidants 2020, 9, 525. [Google Scholar] [CrossRef]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- Salminen, A.; Hyttinen, J.M.; Kaarniranta, K. AMP-activated protein kinase inhibits NF-kappaB signaling and inflammation: Impact on healthspan and lifespan. J. Mol. Med. 2011, 89, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Cheng, Y.C.; Nicol, C.J.; Lin, K.H.; Yen, C.H.; Chiang, M.C. Activation of AMPK is neuroprotective in the oxidative stress by advanced glycosylation end products in human neural stem cells. Exp. Cell Res. 2017, 359, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Bujak, A.L.; Crane, J.D.; Lally, J.S.; Ford, R.J.; Kang, S.J.; Rebalka, I.A.; Green, A.E.; Kemp, B.E.; Hawke, T.J.; Schertzer, J.D.; et al. AMPK activation of muscle autophagy prevents fasting-induced hypoglycemia and myopathy during aging. Cell Metab. 2015, 21, 883–890. [Google Scholar] [CrossRef]
- Zhao, M.; Klionsky, D.J. AMPK-dependent phosphorylation of ULK1 induces autophagy. Cell Metab. 2011, 13, 119–120. [Google Scholar] [CrossRef]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, B.; Chhipa, R.R. Evolving Lessons on the Complex Role of AMPK in Normal Physiology and Cancer. Trends Pharmacol. Sci. 2016, 37, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.B.; Zhou, G.; Li, C. AMPK: An emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009, 9, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Eichner, L.J.; Brun, S.N.; Herzig, S.; Young, N.P.; Curtis, S.D.; Shackelford, D.B.; Shokhirev, M.N.; Leblanc, M.; Vera, L.I.; Hutchins, A.; et al. Genetic Analysis Reveals AMPK Is Required to Support Tumor Growth in Murine Kras-Dependent Lung Cancer Models. Cell Metab. 2019, 29, 285–302.e287. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Oka, S.; Liu, T.; Zhai, P.; Ago, T.; Sciarretta, S.; Li, H.; Sadoshima, J. A redox-dependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation. Cell Metab. 2014, 19, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Habib, S.L.; Yadav, A.; Kidane, D.; Weiss, R.H.; Liang, S. Novel protective mechanism of reducing renal cell damage in diabetes: Activation AMPK by AICAR increased NRF2/OGG1 proteins and reduced oxidative DNA damage. Cell Cycle 2016, 15, 3048–3059. [Google Scholar] [CrossRef]
- Kukidome, D.; Nishikawa, T.; Sonoda, K.; Imoto, K.; Fujisawa, K.; Yano, M.; Motoshima, H.; Taguchi, T.; Matsumura, T.; Araki, E. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes 2006, 55, 120–127. [Google Scholar] [CrossRef]
- Mo, C.; Wang, L.; Zhang, J.; Numazawa, S.; Tang, H.; Tang, X.; Han, X.; Li, J.; Yang, M.; Wang, Z.; et al. The crosstalk between Nrf2 and AMPK signal pathways is important for the anti-inflammatory effect of berberine in LPS-stimulated macrophages and endotoxin-shocked mice. Antioxid. Redox Signal. 2014, 20, 574–588. [Google Scholar] [CrossRef]
- Zimmermann, K.; Baldinger, J.; Mayerhofer, B.; Atanasov, A.G.; Dirsch, V.M.; Heiss, E.H. Activated AMPK boosts the Nrf2/HO-1 signaling axis—A role for the unfolded protein response. Free Radic. Biol. Med. 2015, 88, 417–426. [Google Scholar] [CrossRef]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell. Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef]
- Lv, H.; Liu, Q.; Wen, Z.; Feng, H.; Deng, X.; Ci, X. Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute lung injury via induction of AMPK/GSK3beta-Nrf2 signal axis. Redox Biol. 2017, 12, 311–324. [Google Scholar] [CrossRef]
- Cuadrado, A. Structural and functional characterization of Nrf2 degradation by glycogen synthase kinase 3/beta-TrCP. Free Radic. Biol. Med. 2015, 88, 147–157. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobon-Velasco, J.C.; Devijver, H.; Garcia-Mayoral, M.F.; Van Leuven, F.; et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol. Cell. Biol. 2012, 32, 3486–3499. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, M.; Fischhuber, K.; Poloske, D.; Mechtler, K.; Heiss, E.H. AMPK leads to phosphorylation of the transcription factor Nrf2, tuning transactivation of selected target genes. Redox Biol. 2020, 29, 101393. [Google Scholar] [CrossRef] [PubMed]
- Hellyer, J.A.; Padda, S.K.; Diehn, M.; Wakelee, H.A. Clinical Implications of KEAP1-NFE2L2 Mutations in NSCLC. J. Thorac. Oncol. 2021, 16, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.I.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 2004, 101, 2040–2045. [Google Scholar] [CrossRef] [PubMed]
- Heiss, E.H.; Schachner, D.; Zimmermann, K.; Dirsch, V.M. Glucose availability is a decisive factor for Nrf2-mediated gene expression. Redox Biol. 2013, 1, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Bahiraii, S.; Brenner, M.; Yan, F.; Weckwerth, W.; Heiss, E.H. Sulforaphane diminishes moonlighting of pyruvate kinase M2 and interleukin 1beta expression in M1 (LPS) macrophages. Front. Immunol. 2022, 13, 935692. [Google Scholar] [CrossRef]
- Petsouki, E.; Gerakopoulos, V.; Szeto, N.; Chang, W.; Humphrey, M.B.; Tsiokas, L. FBW7 couples structural integrity with functional output of primary cilia. Commun. Biol. 2021, 4, 1066. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- Kassovska-Bratinova, S.; Yang, G.; Igarashi, K.; Dennery, P.A. Bach1 modulates heme oxygenase-1 expression in the neonatal mouse lung. Pediatr. Res. 2009, 65, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, C.; Leighton, I.A.; Cohen, P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem. J. 1993, 296 Pt 1, 15–19. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petsouki, E.; Ender, S.; Sosa Cabrera, S.N.; Heiss, E.H. AMPK-Mediated Phosphorylation of Nrf2 at S374/S408/S433 Favors Its βTrCP2-Mediated Degradation in KEAP1-Deficient Cells. Antioxidants 2023, 12, 1586. https://doi.org/10.3390/antiox12081586
Petsouki E, Ender S, Sosa Cabrera SN, Heiss EH. AMPK-Mediated Phosphorylation of Nrf2 at S374/S408/S433 Favors Its βTrCP2-Mediated Degradation in KEAP1-Deficient Cells. Antioxidants. 2023; 12(8):1586. https://doi.org/10.3390/antiox12081586
Chicago/Turabian StylePetsouki, Eleni, Sylvia Ender, Shara Natalia Sosa Cabrera, and Elke H. Heiss. 2023. "AMPK-Mediated Phosphorylation of Nrf2 at S374/S408/S433 Favors Its βTrCP2-Mediated Degradation in KEAP1-Deficient Cells" Antioxidants 12, no. 8: 1586. https://doi.org/10.3390/antiox12081586
APA StylePetsouki, E., Ender, S., Sosa Cabrera, S. N., & Heiss, E. H. (2023). AMPK-Mediated Phosphorylation of Nrf2 at S374/S408/S433 Favors Its βTrCP2-Mediated Degradation in KEAP1-Deficient Cells. Antioxidants, 12(8), 1586. https://doi.org/10.3390/antiox12081586