Abstract
Oxidative stress is the subject of numerous studies, most of them focusing on the negative effects exerted at both molecular and cellular levels, ignoring the possible benefits of free radicals. More and more people admit to having heard of the term “oxidative stress”, but few of them understand the meaning of it. We summarized and analyzed the published literature data in order to emphasize the importance and adaptation mechanisms of basal oxidative stress. This review aims to provide an overview of the mechanisms underlying the positive effects of oxidative stress, highlighting these effects, as well as the risks for the population consuming higher doses than the recommended daily intake of antioxidants. The biological dose–response curve in oxidative stress is unpredictable as reactive species are clearly responsible for cellular degradation, whereas antioxidant therapies can alleviate senescence by maintaining redox balance; nevertheless, excessive doses of the latter can modify the redox balance of the cell, leading to a negative outcome. It can be stated that the presence of oxidative status or oxidative stress is a physiological condition with well-defined roles, yet these have been insufficiently researched and explored. The involvement of reactive oxygen species in the pathophysiology of some associated diseases is well-known and the involvement of antioxidant therapies in the processes of senescence, apoptosis, autophagy, and the maintenance of cellular homeostasis cannot be denied. All data in this review support the idea that oxidative stress is an undesirable phenomenon in high and long-term concentrations, but regular exposure is consistent with the hormetic theory.
1. Introduction
Contemporary living standards and advances in the medical sciences promote the idea of acquiring long and healthy life by any means possible. A considerable percentage of the population is familiar with the term “oxidative stress” in association with various pathological conditions, but few people understand its beneficial role. Because of a desire to lower their oxidative status, many individuals use excessive amounts of dietary supplements containing antioxidant compounds. Therefore, the consumption of vitamins considered to be free from side effects and, at the same time, ignoring the existence of reductive stress, is one of the problems affecting the modern world. Some of the positive effects of oxidative stress and, respectively, the negative effects of reductive stress will be discussed in the following paragraphs.
In the human body there are two main types of sources for reactive oxygen species (ROS), specifically the mitochondria and nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase/NOX). Thus, ROS generation is oxidative phosphorylation-dependent, which is a process that ensures electron flow, and which takes place in the mitochondria involving four protein complexes (I–IV) [1,2]. This process does not occur perfectly, as electron leakage can also occurs, in which electrons attach to oxygen molecules (O2) and thus ROS are obtained [3]. Consequently, during aerobic metabolism, the complexes that form the electron transport chain (ETC) transfer the electrons from NADH and dihydroflavin adenine dinucleotide (FADH2) towards mitochondrial complex IV. This complex then donates these electrons to O2 in order to obtain water (H2O). As stated previously, this process is not perfect, causing a slight percentage (<0.5%) of the electrons to react via a non-enzymatic pathway with the O2, forming the superoxide anion . Finally, through complexes I and III, is released into the mitochondrial matrix and into the cytoplasm, respectively, mediated by voltage-dependent anion channels (VDACs). Concomitantly, the production rate of ROS of mitochondrial origin depends on kinetic and thermodynamic factors, on O2 availability, on electron transporters and, most importantly, on the membrane potential of the mitochondria [4,5]. An interesting fact is that, due to their dynamics, the mitochondria can travel across the cell interior and create areas of increased concentrations of ROS, which are made available for the transduction of cellular signaling [6].
The second source of ROS is represented by the NADPH oxidase, belonging to the NOX enzyme family. These proteins transfer the electrons from NADPH to O2, generating . ROS production, mediated by NOX. This transfer is regulated by flavin adenine dinucleotide (FAD), by phosphorylated proteins, and by the Ca2+ ion. The factors that can activate the NOX protein complex are insulin, growth factors, angiotensin, and tumor necrosis factor (TNF) [7].
Regarding the mechanisms that are supposed to limit the excess of reactive species, the human body possesses a set of proteins with an antioxidant role. Therefore, in the case of the accumulation of increased levels of , superoxid dismutase (SOD) can convert into the peroxide H2O2. Subsequently, this set of antioxidant proteins, which can involve the peroxiredoxin (PRX)/thioredoxin (TRX) system and the glutathione peroxidase (GPX)/glutathione (GSH), system can act to reduce the previously generated H2O2 to H2O. This process includes the inactivation of PRX at the moment that H2O2 is reduced, the oxidation of cysteine (Cys) residues from the TRX structure, and consequently the reduction and reactivation of PRX. The oxidized, inactive TRX is then reduced by thioredoxin reductase, which has NADPH as a co-factor. The second system, which is based on glutathione, functions similarly to the one described above, except that the reduction of H2O2 is performed by GSH, which becomes oxidized glutathione (GSSG). In both systems, TRX and GSSG are reduced by NADPH, which is the result of the enzymatic activity of isocitrate dehydrogenase and malic enzymes from the pentose phosphate pathway. An important aspect is the large distribution of these antioxidant systems, granting the human organism antioxidant protection [8]. Along with these systems, nuclear factor erythroid 2-related factor (Nrf2) plays a key role, being one of the main regulators of antioxidant systems. Moreover, Nrf2 is involved in GSH regeneration by increasing glutathione reductase (GR) expression. Its molecular mechanism is discussed further in this paper [9,10].
The homeostasis of the body, tissue trophicity, and cellular signaling depend on the organic presence of ROS; therefore, the balance between oxidative stress, caused by the massive generation of reactive species, and reductive stress, caused by the excessive presence of antioxidants, must be maintained [11,12]. Cellular stress conditions, which generate sufficient ROS, promote cellular apoptosis [13] through external mitochondrial membrane permeabilization, membrane potential, and the release of cytochrome c. Cytochrome c then bonds to apoptotic protease activating factor 1 (APAF1) and forms an apoptosome, which initiates the intrinsic pathway of apoptosis via caspase 9. The intrinsic pathway involves the attachment of the cellular-death receptor Fas/CD95 and the activation of caspase-8 [14,15,16]. In certain situations, apoptosis is a beneficial process, because the abnormal cells which can no longer fulfill their function are destroyed, reducing the risk of these cells surviving and proliferating [16].
Oxidative metabolism of the cell generates reactive species, with very high energy, that can interact with susceptible endogenous molecules, such as proteins, unsaturated fatty acids, and DNA [17,18,19,20]. Overall, oxidative stress is described as a redox ratio between antioxidants and oxidizing agents, favoring the latter, which leads to the assumption that reactive oxygen and nitrogen species (RONS) [21] present only an intrinsic negative effect [22,23]. This definition is not completely accurate as a moderate level of ROS and/or reactive nitrogen species (RNS) is needed to maintain cellular homeostasis [24,25,26,27,28] due to their involvement in immune mechanisms, angiogenesis, adaptation, and muscle remodeling [29,30,31,32].
The presence of ROS in small or moderate doses is considered to be beneficial for physiological functions involved in the progression of the cellular cycle, such as differentiation, the development of cells, and cellular apoptosis. Reactive species are involved in different phases of cellular signaling, often via the reversible oxidation of thiol groups (-SH) on the proteins, which will be described in the following sections. One of these phases is the activation of some transcription factors, such as the phosphoinositide 3-kinase/protein kinase B system (PI3K/Akt system), mitogen-activated protein kinase (MAPK), nuclear factor erythroid 1- and 2-related factors (Nrf1, Nrf2), Kelch-like ECH-associated protein 1 (Keap-1), and nuclear factor kappa B (NF-kB). Moreover, the presence of ROS is essential for the activity of the immune system and for maintaining redox balance through the activation of antioxidant systems. The modification of this balance in favor of ROS production is often linked to pathological states, diabetes mellitus, atherosclerosis, cancer, or neurodegenerative diseases (such as Alzheimer’s and Parkinson’s). Despite previous beliefs, ROS is associated with these conditions, rather than being a direct cause of them. On the other hand, the inadequate use of supplements based on antioxidants can cause a paradoxical state of “antioxidant stress”. The efficiency of the antioxidant and homeostatic systems is dependent on ROS exposure, since the expression of the proteins involved in these processes is increased by the presence of oxidative stress. The improvement of antioxidant activity is therefore due to the presence of an oxidative state, given that some health aspects are linked to pro-oxidant factors such as physical exercise.
Based on the above considerations, it can be stated that the presence of an oxidative state is required for the proper functioning of the body, having well-defined roles, but to date it has been insufficiently researched and explored.
2. Influence of Oxidative Stress on Physical Activity
Physical activity, in addition to improving health status and preventing the development of pathological conditions, is responsible for producing reactive species. However, the human body is able to reduce free radicals through several mechanisms and they do not represent a threat under physiological conditions [33]. The correlation between the type of physical activity and oxidative stress, and its beneficial effects on health and redox balance are still subjects of controversy [34].
Since the energetic needs and oxygen consumption levels are different, the generation of ROS during physical activity depends on many factors, such as the intensity type of physical effort and its duration. In cases of physical activity of reduced intensity and duration, the body’s antioxidant systems are sufficient to maintain the redox balance. An increase in duration and intensity becomes challenging, and in this case oxidative lesions can be observed. However, chronic exposure to oxidative stress due to regular physical exercise does not lead to cellular lesions, but it is rather—in accordance with the theory of hormesis—an adaptive mechanism [35]. The type of physical exercise plays a crucial role regarding oxidative stress; illustrative examples include the cycling exercise in which the activity of the antioxidant systems is increased [36,37]. In contrast, for the same intensity of some anaerobic exercises (sprints, intermittent running, isometric exercise, jumps), oxidative injuries can be observed. [38]. The latter is due to the activation of xanthin-oxidase (XO), which produces ROS during ischemia reperfusion. XO reduces O2 to and H2O2, which is then reduced to hydroxyl (), which is considered to be responsible for these cellular lesions [39]. The underlying mechanisms of the increase in ROS could be moments of ischemia-reperfusion, as well as mechanical stress, muscular injury, and the migration of the inflammatory cells to the affected area. Following the same model, endurance physical exercise enhances antioxidant enzymatic activity, indicating that the type and intensity of physical activity are predictors of oxidative stress generation [40]. In summary, the abovementioned factors can either have negative effects or positive effects or both, depending also on the individual basal levels of oxidative stress. An important factor to be mentioned, which is involved in the adaptive mechanism and antioxidant protection, is gender, but this is outside of the scope of this paper and will not be further discussed.
Even though the role of oxidative stress in effort-induced adaptation remains to be elucidated, one thing is certain: an optimal yet undefined level of ROS is mandatory for the proper function of the adaptive mechanisms. Similarly, to aerobic activity, it is unclear if ROS generated during anaerobic exercises represent an adaptive/necessary event or are harmful, and studies in this area are rather scarce [41].
Striated muscular fibers react differently in the presence of ROS/ RNS, depending on the degree of stress adaptation. As such, for people who perform physical activity on a regular basis, an increase in the intensity of physical exercise is correlated with better glucose metabolism, an increase in the number of mitochondria, and muscular hypertrophy [42,43,44]. On the contrary, for physically inactive people, the trophicity of the muscular tissue is affected [45,46,47,48,49,50].
Many studies suggest that moderate-intensity exercise is responsible for generating a beneficial level of reactive species [28,51,52,53], as physical activity is associated with health and slowing down the process of aging [54,55,56] by increasing the expression of antioxidant enzymes [51,57].
The ability of the human body to adapt to oxidative stress produced during endurance exercise is a result of an increased mitochondrial volume and a decreased inflammatory response [23,58,59,60]. As the free radicals and have a short half-life and cannot be quantified, oxidative damage is determined indirectly through biomarkers such as oxidized low-density lipoprotein (oxLDL) [61], malondialdehyde (MDA) [62,63], the reduced and oxidized glutathione ratio (GSH/GSSG) [64], F2-isoprostane [65,66], carbonylated proteins [67,68], or 8-hydroxy-2′-deoxyguanosine (8-OHdG) [69,70], which are lower in physically active individuals than in sedentary and inactive ones [32]. Sedentarism is a risk factor for muscular atrophy, a process that paradoxically produces ROS and decreases the ability of the cells to trap free radicals [71]. Transitory oxidative status is required during physical effort to initiate the adaptive mechanisms of skeletal muscles. Based on these hypotheses, the hormesis theory was formulated [56,72]. This theory claims that continuous exposure to low level of stress factors improves the ability of the cells to respond to higher levels of stress [73] due to the phenomenon of the up regulation of endogenous antioxidant systems. The exact pathways through which reactive species that are produced during physical effort can induce adaptive mechanisms are not completely known, but it seems that enzymatic systems [74,75] that increase the expression of manganese-dependent superoxide dismutase (MnSOD) [51], catalase (CAT) [52], and glutathione peroxidase (GPx) are activated [76,77,78].
NF-kB is activated in the presence of pro-inflammatory cytokines, TNF-α, interleukins 1 and 6 (IL-1, IL-6) [79,80], ionizing radiation [81,82], and other stressors, with the signaling molecule being H2O2 [30] acting through the dissociation of kappa B inhibitory factor (I-kB) [83,84] and translocation into the nucleus [76]. Moreover, in order to improve the vascularization of striated muscles via NF-kB and hypoxia-inducible factor 1 (HIF-1)—which is associated with peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) [85,86,87] and ROS and is especially, RNS-dependent through nitric oxide (NO)—angiogenesis is favored. High concentrations of H2O2, together with , are considered risk factors due to their pro-inflammatory effect, resulting in a thickening of vascular walls and the occurrence of associated vascular diseases. Vascular endothelial growth factor (VEGF) activates ROS-generating NADPH oxidase/NOX [88] and triggers cell proliferation and migration [89], subsequent to the stimulation of endothelial cells by the tyrosine kinase receptor (RTK) VEGF 2 [90]. Several signaling pathways, such as MAPK, Akt, and endothelial nitric oxide synthase (eNOS), are activated in these chain reactions that are essential for the neovascularization process [91]. Regarding these considerations, regular exercise of moderate intensity respects the theory of hormesis due to the ability of the body to adapt to stronger oxidative stressors.
The elevated levels of ROS that occur in both performance athletes and individuals who practice sports for recreational purposes cause variable cellular adaptive mechanisms that depend on several factors such as the origin and the concentration of reactive species, on the exposure time to ROS and, especially, on the particular/individual oxidative status. Thus, RONS act as messengers, signaling physiological regulatory functions, through mechanisms involving post-translational redox changes of -SH groups on Cys residues [92]. Redox signaling is based on the structural modification of compounds such as Cys, although other protein molecules, as well as other signaling pathways that use various kinases and/or phosphatases producing phosphorylation and/or dephosphorylation reactions, are also involved [93,94,95], as shown in Figure 1. Therefore, by modulating the Cys residues, ROS can activate or inactivate other proteins. For RTKs, ROS generation is necessary for the downstream signaling. Phosphorylation of tyrosine (Tyr) residues requires the inactivation of tyrosine-phosphatase (PTPs). These enzymes are dependent on the Cys residues which are located in their active center, and the oxidation of the -SH groups inhibit enzymatic activity [96]. Moreover, ROS can also modulate the extracellular part of the RTKs, hence activating it in the same way that they inactivate the PTPs. [97]. Contrary to previous hypotheses, the main source of ROS is not mitochondria but rather enzymatic sources such as NOX(s), phospholipase A2 (PLA2), and/or XO [50,98,99]. ROS-based signaling pathways are involved in various acute and chronic skeletal muscle responses to exercise, from increased insulin sensitivity [100] to the modulation of endogenous enzymes with antioxidant activity [101], mitochondrial biogenesis [102], and the modification of muscle contraction and adaptation [103]. Regarding the generation of RNS during muscle contraction, NO is produced by neuronal nitric oxide synthase (nNOS). Post-translational reversible changes, such as S-nitrosylation, S-glutathionylation, sulfenylation, or the formation of disulfide bonds by Cys-reactive thiol groups, are in fact redox changes—the response of the cells to abnormal ROS and RNS levels [104,105,106], as shown in Figure 1.
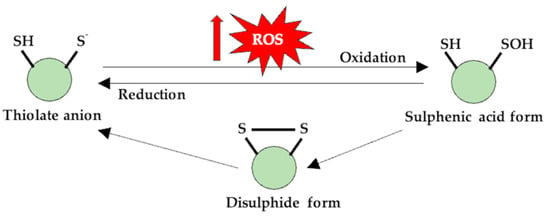
Figure 1.
Thiol (-SH) groups are susceptible to redox reactions, and at physiological pH values, they are found in an anionic form. The oxidation of this form generates a sulfenic derivative, which can reverse to its initial form through the disulfidic form, and, in this case, the protein can change its functionality.
For example, through the S-nitrosylation process, the activity of proteins involved in muscle contraction—ryanodynamic receptors (RyR1), myosin, cAMP response element-binding protein (CREB), proteins specific to insulin signaling—are altered [107,108,109,110], but at the same time, these reversible redox changes in the -SH groups of Cys also have a protective role for proteins as they make them less susceptible to oxidative processes [104].
For optimal muscle contraction, a certain level of reactive species is required, because depending on its level, the contraction of skeletal muscle can be improved by increasing the sensitivity of muscle fibers to the Ca2+ ion, whereas post-effort, when the ROS level is high in the muscle, the contraction force and sensitivity to Ca2+ is decreased [111]. For this reason, the administration of dietary supplements with antioxidant compounds may not be beneficial as it might decrease the effort capacity [111,112,113]. In a study performed by Reid et al. the hypothesis of a “redox break” was introduced, stating that fatigue and decreased muscle contraction is due to the generation of high levels of ROS, and it is actually a negative feedback mechanism through which the damage caused by ROS to the skeletal muscle is minimized [114,115]. In addition to altering the proteins involved in regulating the Ca2+ concentration or the sensitivity of the muscle fiber to this ion, they propose the hypothesis that ROS influences Na+/K+ pump activity and thus reduces muscle contraction force in the case of endurance-based physical effort [116]. Physical effort causes depletion of K+ ion sand a massive increase in the intracellular concentration of Na+ ions, which affects the membrane excitability, and therefore the force of muscle contractions is diminished. This idea is supported by the experimental data showing that the administration of an antioxidant, N-acetylcysteine (NAC), reduces muscle fatigue that occurs during exercise by regulating the intracellular concentration of K+ ions [117].
There are various communication pathways that use redox signaling and the most important ones that also have an impact on physical effort are NF-kB, MAPK, and PGC-1α, and their activity is influenced by the concentration of H2O2 [118]. Another important aspect to mention is that these communication pathways are interdependent and intersected, through a process known as “crosstalk” [119]. Studies also present data confirming the influence of PGC-1α on mitochondrial function and dynamics (fusion, fission) on muscle fiber differentiation and on the expression of antioxidant enzymes, which are present in oxygen-consuming tissues (brain, heart, skeletal muscle) [120,121].
The mitochondrion cellular organelle is of vital importance. It exhibits bioenergetic and functional roles in maintaining cell viability, throughout the many biochemical processes that are under its control (the Krebs cycle, β-oxidase, oxidative phosphorylation, OXPHOS). Concomitantly, the mitochondrion is a dynamic organelle as it can switch its morphology depending on intra- and extracellular conditions in order to preserve cellular homeostasis. Thus, the presence of a stressor, for example, food deprivation, activates the mitochondrial fusion process, controlled by optical atrophy 1 (Opa1) and mitofusin 1/2 (Mfn1/2), to grow and enhance the OXPHOS rhythm. It is believed that mitochondrial fusion increases activity and energy production, whereas fission precedes mitophagy.
On the opposite side, in the case of nutrient excess, fragmentation of mitochondria takes place, also known as fission [122,123], controlled by a pair of proteins, dynamin-related protein 1 (Drp-1) and mitochondrial fission 1 protein (Fis1) [98,124,125].
Mitochondrial homeostasis and biogenesis can be pharmacologically modulated by agonists (thiazolidinediones, fibrates) of peroxisome proliferator-activated receptor γ, and α (PPARγ, PPARα), respectively [126,127], as shown in Figure 2.
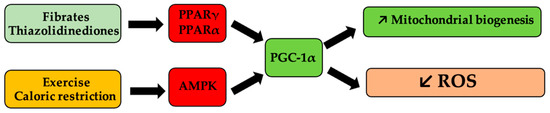
Figure 2.
Schematic representation of mitochondrial biogenesis pathways of influence and of reactive oxygen species (ROS). Antihyperlipidemic medication, such as fibrates and antihyperglycemic medication, such as thiazolidinedione derivatives, activate peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) via peroxisome proliferator-activated receptor γ and α (PPARα, PPARγ), respectively, and determine the onset of mitogenesis and the decrease in ROS levels. Similarly, caloric restriction or physical effort, via activation of the same receptors, following the activation of 5′ protein kinase by the AMP (AMPK) pathway, lead to the same processes.
During physical activity, PGC-1α expression is elevated as a result of the increase in the concentration of Ca2+ and activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII), CREB, and myocyte enhancer factor-2 (Mef2) [128,129,130,131,132]. Meanwhile, under the action of PGC-1α, the number of mitochondria is high within striated muscle, and as such, energetic resources are more efficiently used [133,134,135,136,137]. Numerous animal and human studies demonstrate that moderate-intensity physical effort increases the values of mitochondrial biogenesis markers and the expression of antioxidant systems, demonstrating the appearance of the adaptative response [138,139]. More and more data suggest that physical effort has an overall antioxidant role, depending on the type of exercise [140], as well as age and sex, which is a cause of controversy in the hormesis theory. Thus, endurance physical effort is a massive consumer of energy, which increases the level of adenosine monophosphate (AMP) with the activation of the 5′ protein kinase activated by AMP (AMPK) [141], MAPK, CAMKII, and Mef2 [124], which triggers the activation processes of PGC-1α, which is necessary for the adaptative mechanism of muscle tissue and the transition of glycolytic fibers to oxidative fibers [141,142,143,144].
At the same time, the skeletal muscle is a consumer of adenosine triphosphate (ATP), so it creates an energy deficit with the generation of high concentrations of AMP, which activates AMPK, another determining factor in increasing PGC-1α expression [136,137], as shown in Figure 3. On the other hand, for the proper functioning of the muscles during physical effort and in order achieve optimal muscle contraction, the presence of NO, produced during physical movement, is necessary. NO increases blood flow and improves the antioxidant status of skeletal muscle and is synthesized via inducible NO synthase (iNOS), an enzyme of which the expression is induced by physical movement via NF-kB [131]. Although the NF-kB pathway is important for maintaining normal oxidative status, chronic activation may have adverse effects such as amplifying the synthesis of pro-inflammatory cytokines that generate ROS, inhibit PGC-1α activity, and promote apoptotic processes [138]. All these processes are not universal, but are influenced by certain variable factors, such as age, sex, eating habits, and even the physiological condition of an individual.
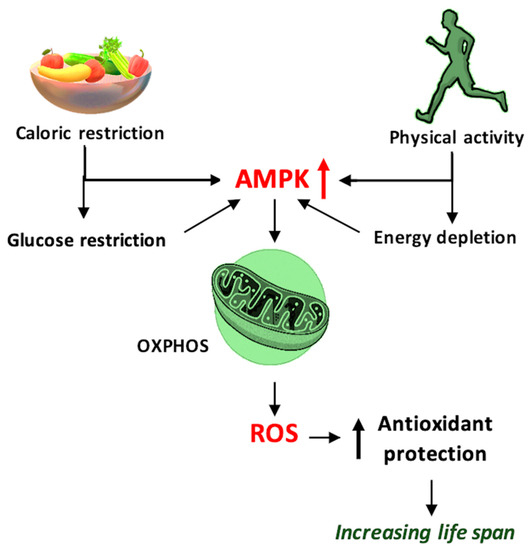
Figure 3.
Schematic representation of the mechanism by which caloric restriction and physical activity prolong the lifespan and improve health, using 5′ adenosine monophosphate-activated protein kinase (AMPK) and reactive oxygen species (ROS) as mediators, and ROS generated through oxidative phosphorylation (OXPHOS)—schematic mechanism.
In patients with type 2 diabetes, physical effort improves glycemic status [139,140], one of the mechanisms involving oxidative stress, which is correlated with the expression of the glucose transporter type 4 (GLUT4) [141,142] and the involvement of proteins such as AMPK, PGC-1α [143], and MAPK [140,142]. The activation of PGC-1α as a result of aerobic effort [144,145] and an increased AMP/ATP ratio [146] decreases oxidative stress and inflammation and increases mitochondrial biogenesis [147,148] in response to the interaction with Nrf-1 [35] and Nrf-2 [88,147] and promotes muscle adaptation mechanisms following AMPK activation [31,148,149]. AMPK is an energy sensor with an important impact on metabolic status and on the mRNA expression of PGC-1α, requiring the presence of ROS. This fact was demonstrated using antioxidant compounds that suppressed the adaptive mechanisms and the formation of PGC-1α [132,150,151]. Regarding Nrf-2, in oxidative stress conditions, the oxidation of -SH groups from Kelch-like ECH-associated protein 1 (Keap-1) allows Nrf-2 to translocate in the nucleus [115,152], subsequent to the separation of Keap-1 [153]. In response, an increase in the expression of the antioxidant responsive element gene (ARE) occurs [154,155,156], which controls the level of glutathione reductase (GR) and glutathione synthase (GSS) [157], acting as a mediator between oxidative and reductive stress [158], as shown in Figure 4.
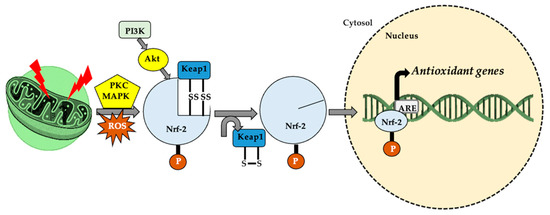
Figure 4.
In cellular homeostasis, nuclear factor erythroid 2-related factor (Nrf-2) stabilizes by attaching to Keap-1 in the cytoplasm. The appearance of a stressor/reactive oxygen species (ROS) generator, under the action of protein kinase C (PKC), mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase–protein kinase B (PI3K–Akt system), the thiol (-SH) groups belonging to Kelch-like ECH-associated protein 1 (Keap-1) are oxidized (S-S cross links). This leads to Nrf-2 dissociation and translocation in the nucleus, where it attaches to a specific sequence, antioxidant responsive element (ARE), thus increasing cellular antioxidant activity.
Thus, ROS are sources of oxidative stress, but they are necessary in small amounts in order to maintain physiological functions, as well as for muscle contraction. However, an excessive increase in ROS during intense physical activity can have detrimental effects on performance.
3. Impact of Oxidative Stress on Aging
It is known and generally accepted that mitochondria are responsible for generating reactive species and their excess causes oxidative lesions and cellular aging. However, as previously mentioned throughout this paper, ROS also play a role in cellular signaling. Thus, the existence of two types of ROS can be discussed: species with low reactivity (, H2O2) and species with high reactivity (, peroxynitrite, ). Although the species with low reactivity act as cellular messengers, the ones with high reactivity contribute to oxidative injuries, to the diminishing of cognitive capacities, brain aging, and finally to the onset of neurodegenerative diseases [6].
Studies carried out on Caenorhabditis elegans presented interesting data regarding aging processes. They indicated that glucose deprivation increased OXPHOS and mitochondrial ROS production (mtROS) but diminished cellular aging and increased life expectancy. This phenomenon, in which the adaptation of mitochondria takes place as a consequence of exposure to increased levels of ROS and the activation of antiaging mechanisms occurs, is referred to as mitohormesis [159,160]. For this phenomenon to take place, the properties of the mitochondria have to be intact. One can infer that with increased O2 availability, ROS production increases simultaneously. In fact, studies indicate that hypoxic states are correlated with increased or even accelerated production of ROS and also with increased aging processes. Thus, during hypoxia, inhibition of mitochondrial complex III generates ROS and alters the cellular response in this particular state, by impeding HIF-1α stabilization [161]. Another relevant example is the linkage between mtROS and uncoupling protein 2 (UCP2). During periods of low nutrient intake, mtROS is generated via the β-oxidation process. This increases the activity of UPC2, which preserves a low level of ROS, but maintains the processes responsible for the generation of energy. In cases where this redox balance, maintained with the active participation of mitochondria, is altered, the activation of UCP2 and the production of energy are impeded. This can partly explain the ineffectiveness of antioxidant therapies in neurodegenerative diseases. Based on this fact, treatments dedicated to these particular diseases should aim to reach or maintain redox homeostasis, rather than aiming at the reduction of the total levels of ROS [162].
In the opposite situation, the altered and non-functional mitochondria are eliminated via mitophagy and the proteasome system [163]. On this note, studies state that an increase in the activity and effectiveness of mitophagy and the proteasome system can increase life expectancy and slow down the aging rate, at least in some model organisms (rodents, flies, and worms) [164,165,166]. However, the notion that the mere presence of functional mitochondria would prevent aging or the manifestation of already-present neurodegenerative diseases remains unclear.
Diseases with neurodegenerative components are characterized by affected functions as well as progressive destruction of neuronal cells. The modification of redox homeostasis is a specific attribute of neurodegenerative conditions and despite the fact that in some experimental models of Alzheimer’s, Parkinson’s, or other neurological conditions antioxidant agents demonstrated beneficial effects, in clinical studies, this neuroprotective effect could not be demonstrated [167,168].
From a morphological perspective, the destruction of neuronal mass is linked to protein misfolding and the accumulation of misfolded protein aggregates. An interesting fact is that some neuronal populations are resistant, whereas some areas are sensitive to oxidative stress, a phenomenon termed selective vulnerability. Finally, the association between protein aggregates and oxidative stress triggers a chain reaction, which leads to neuronal death [169].
In the case of the dysfunction of the proteasome system, which is responsible for the disposal of the altered proteins, and which is therefore indispensable for maintaining cellular homeostasis and neuronal survival, the protein aggregates can trigger the generation of ROS. This phenomenon is often encountered in diseases with an oxidative component, especially the neurodegenerative ones. It is known that the brain does not possess a remarkable antioxidant capacity, which makes it susceptible to oxidative damage, particularly during aging, when the antioxidant systems are less active and favor the accumulation of reactive species and the progression of diseases such as Alzheimer’s and Parkinson’s [170,171,172].
Alzheimer’s disease is a widespread neurodegenerative condition, involving the progressive loss of memory, cognitive capacities, and personality disorders as the most important symptoms. Morphological particularities in Alzheimer’s are represented by amyloid plaques, dystrophic neurites, cerebral amyloid angiopathy, reactive astrogliosis, as well as microglial activation [173]. The absence of the protein Nrf-2 from the nucleus of the hippocampal neurons is implicated in this disease, which can be confirmed through biochemical analysis of the patient’s brain. Concomitantly, it is likely that in these patients, the response of Nrf-2 to ROS and the mitochondrial dynamics are affected, which further leads to the progression of molecular mass loss [174]. The accumulation of β-amyloid and Tau proteins is age-dependent—it increases as one person gets older, and this accumulation disrupts Ca2+ ion homeostasis and the mitochondrial membrane potential is lost. Furthermore, the internal membrane develops pores and ATP synthesis decreases, but the ROS level increases as a result of electron drain.
Regarding Parkinson’s disease, which is the second most widespread neurodegenerative condition after Alzheimer’s disease [175], it mostly constitutes motor symptoms (tremor at rest, bradykinesia, muscular rigidity) and non-motor symptoms (constipation, depression, personality disorder). The symptomatology is the result of the degeneration of dopaminergic neurons in the substantia nigra [176,177]. In this case, the presence of ROS leads to Nrf-2 activation, which further disrupts α-synuclein (α-Syn) metabolism, which would thus prevent the loss of neuronal dopaminergic mass. However, in patients diagnosed with Parkinson’s disease, a deficit of the Nrf-2 protein is observed, and this deficit causes neuroinflammation and favors the progression of the disease [174]. In both neurodegenerative diseases, age is one of the most important determining factors of the onset and/or the progression of the disease.
It is generally accepted that oxidative stress is involved in various diseases, such as neurodegenerative diseases (Alzheimer’s, Parkinson’s), but it is possible that ROS are not actually the trigger for the disease but rather the cause of the exacerbation of symptoms within these neurological disorders [178,179]. Therefore, various studies have been performed to investigate the influence of oxidative stress and physical effort on central nervous system conditions. The data presented in the literature demonstrate an inversely proportional relationship between the incidence of neurodegenerative diseases and physical activity, and a slowdown of their progression in the case of physical activity [135,180,181,182,183].
Moreover, as neuronal metabolism increases, there is a consecutive increase in oxygen consumption and ROS production, triggering a positive feedback effect on antioxidant mechanisms and neurotrophin synthesis, which improves cognitive functions and offers protection against neuronal degradation [184]. Brain-derived neurotrophic factor (BDNF) is required for neurogenesis and neuronal plasticity [159,185], and its release is dependent on oxidative status [88] and caloric restriction (CR) [186,187]. Evidence to support these hypotheses was obtained through a study on Alzheimer’s disease [188], in which a decrease in the accumulation of β-amyloid [189] was observed, possibly due to the proteolysis and autophagy of denatured proteins [190], and the overexpression of neuroglobin as a protective factor of the brain from the neurotoxic action of β-amyloid [188]. The data presented in the literature suggest that the moderate level of reactive species formed during exercise favors the expression of 8-oxoG DNA glycosylase [190], an enzyme involved in the repair of nucleic acid chains due to the accumulation of 8-OHdG [52,184]. Preventing the accumulation of degraded DNA presents beneficial effects on senescence and other associated degenerative diseases [191]. A study conducted by Radák et al. confirmed the beneficial effect of moderate physical exertion on neurodegenerative diseases, mediated by proteasome activity and the degradation of altered proteins [192].
ROS are also involved in the formation and storage of memories via improving synaptic plasticity [192], suggesting the modulation of cognitive capacity, which entails the activation of hormetic processes at the neuronal level. The generation of free radicals associated with the influx of Ca2+ ions activate transcription factors CREB and NF-kB, causing a release of cytoprotective proteins [193]. Thus, it can be concluded that exposure of the brain to moderate levels of reactive species improves endurance and slows down the aging process of neuronal tissue [194]. Altogether, these data show a neurohormetic effect that can suppress the progression of neurodegenerative diseases [195].
This is a paradox—exercise-induced oxidative stress, by activating and improving adaptive mechanisms, is responsible for improving overall physiological function, reducing the prevalence of various diseases, increasing the quality of life, and, consequently, inducing the adaptability of the body to oxidative stress [196]. Thus, increasing the body’s capacity to trap and neutralize ROS can provide better protection and resistance during training and can also intervene in reducing the aging process. A study performed by Bouzid et al. supports the hypothesis that the aging process alters antioxidant activity regardless of the physical activity level, thus affecting both sedentary and active people. However, regular physical activity improves antioxidant capacity in the young population to a greater extent than in the elderly, suggesting that the beneficial effects of regular physical activity could be affected by the aging process. In addition, the study data support the idea that regular physical activity reduces the aging process [197].
Overall, physical activity must be performed throughout the lifetime in order to maintain the benefits of adaptive mechanisms following exercise-induced oxidative stress [197]. These data are supported by other studies concluding that with age, muscle mass decreases, a process known as sarcopenia. There are several factors involved in the development of sarcopenia, such as neuro-endocrine factors, nutritional factors, decreased physical activity, and especially oxidative stress caused by mitochondrial dysfunction [198,199,200]. The mitochondrial theory of sarcopenia claims that muscle weakness occurs due to decreased oxidative capacity of mitochondria and thus a reduced amount of generated ATP. This is due to the decreased ability to replace altered mitochondrial proteins involved in the ETC with age [201,202]. In addition, in senescent muscles, NF-kB is constitutively activated, which leads to increased production of pro-inflammatory cytokines and ROS. At the same time, the forkhead family of transcription factors (FoxO) is activated through ROS, which decreases the expression of PGC-1α and favors the degradation of the neuromuscular junction and mitochondrial trophicity [203].
Unlike the past decades, in which ROS were considered the main cause of the aging process, it has been confirmed that small amounts of free radicals prevent senescence and improve the lifespan and the quality of life. Another associated theory is related to CR, demonstrated by the ketogenic diet [54,194,204,205], as it improves antioxidant activity and reduces the amount of ROS to a level that slows down senescence. The exact mechanism through which CR reduces oxidative stress is not completely known and available data are ambiguous. A potential mechanism could involve increased degradation of the modified molecular structures with a consecutive increase in the synthesis of new molecules to replace them, as well as a simultaneous decrease in the rate of production of free radicals by mitochondria [123]. With age, carbonylated proteins tend to accumulate as their degradation mechanisms decrease, but in physically active people due to ROS produced during exertion, the expression of the proteasome complex [184,206], responsible for protein degradation, is potentiated [207].
Most frequently, the aging process is associated with the loss of tissue, the underlying theory being that of oxidative stress coupled with lesions caused by ROS. Thus, sustained physical activity is related to a better neutralization of ROS, which leads to protection against oxidative injuries and the prevention of age-related conditions.
4. A Link between Oxidative Stress and Immune Response
In a general sense, a compound with antioxidant properties is considered to be able to neutralize the RONS species either directly, indirectly, or by inhibiting their subsequent generation [208]. As a result, antioxidants can be classified according to their mechanism of action [209,210] into:
- enzymes (SOD, CAT, GPx), proteins (ceruloplasmin, ferritin), and minerals (zinc, copper, selenium) considered to be agents that prevent the formation of new free radicals;
- agents considered to be scavengers act by inhibiting the initiation and propagation of peroxidation reactions (vitamin C, vitamin E, β-carotenes, glutathione, flavonoids);
- enzymes involved in the reconsolidation of affected cell membranes (lipases, proteases, transferases, DNA repair enzymes).
Considering that the theory of hormesis is generally accepted and that RONS are necessary for cellular homeostasis, muscle regeneration, slowing down the process of aging, protein degradation, and cellular signaling, it is still not possible to determine if a reductive status of the body is beneficial or not, as it generates reductive stress [211] that causes cellular metabolism disorders and cytotoxicity [212]. Nonetheless, the literature emphasizes the involvement of mitochondrial oxidative metabolism in stem cell differentiation, such that complete differentiation is dependent on the presence of ROS [213,214,215,216,217,218]. This information does not exclude the fact that ROS or increased levels of ROS are not harmful, but there are circumstances and types of cells (adipocytes, muscles) of which the differentiation and renewal depend on the organic presence of these species [219,220]. In the meantime, ROS influence the successive phases that occur in the cellular cycle, and the administration of the compounds that donate -SH groups (NAC) apparently cause cycle arrest in the G1 phase, whereas in the absence of these compounds, the S phase is prolonged and the G1 phase is shortened [221,222]. Studies demonstrate that antioxidants have a wide range of beneficial effects, with most of these studies conducted in conditions of induced oxidative stress and with only a few studies questioning what happens under normal physiological conditions [223,224].
Therefore, in the absence of an oxidative status, the self-renewal capacity of cells and autophagy processes are disrupted, with the accumulation of chromosomal defects and abnormalities and the inability to repair DNA and induce apoptosis [225,226]. Highly active antioxidant systems induce reductive stress, influencing [130] cell growth and mitochondrial functions and are incriminated in the progression of pathological conditions such as Alzheimer’s disease [227] because of an impaired protein folding process [12], or cardiomyopathy, cancer, or metabolic syndrome [86,228,229,230]. Some examples of the negative influence of the most frequently used antioxidant substances are illustrated in Table 1.

Table 1.
Other effects related to excessive supplementation with different antioxidants.
It was initially considered that reductive stress is preceded by oxidative stress, but the relation between the two concepts is reciprocal [253]. High doses of antioxidants did not provide conclusive data [193,254] regarding their influence on lifespan [122,255] and, on the contrary, created a pro-oxidant status with the alteration of the redox balance. The accumulation of reduced forms of coenzymes (NADH and NADPH) and reduced glutathione (GSH) [256] can cause cellular dysfunction. Furthermore, mitochondria generate ROS in response to the accumulation of reductive stress. One example is vitamin C (an antioxidant and reducing agent at the same time), which reduces Fe3+ to Fe2+, and thus has the ability to re-enter the Fenton reaction, generating , followed by its transformation into HO* radicals [193].
In addition to vitamin C, other compounds, including resveratrol and lipoic acid (LA), can be mentioned, but the phenomenon is not limited to these examples. Thus, in the case of resveratrol, a series of beneficial effects correlated with its antioxidant properties was observed: cardioprotection, neuroprotection, anti-inflammatory, and antimicrobial activities, and it also mimics the effects of CR, which are correlated with lifespan [257]. Moreover, some studies demonstrated effects on ovarian and testicular activities, prolonging their viability as a result of a potential antiaging effect [258]. On the contrary, resveratrol can also present negative effects [259], associated with its pro-oxidant behavior. Depending on the enzymes involved, the compound can be oxidized and/or auto-oxidized, resulting in semiquinones and 4′-phenoxyl radicals, responsible for ROS generation [260], which can alter the normal functioning of lipid membranes and/or genetic material [261]. It is also important to mention that excessive doses of resveratrol can present other adverse effects which are not correlated with oxidative stress, such as impeding cellular growth and the activation of apoptosis in normal cells, atherogenesis, and phytoestrogen-like behavior [262,263,264].
LA, also known as α-lipoic acid and/or thioctic acid, displays biochemical roles and antioxidant properties. LA is reduced to dihydrolipoic acid (DHLA) by GR and TRX in the presence of NADPH. Both molecules, LA and DHLA, exhibit their antioxidant properties in hydrophilic as well as in lipophilic media because of their amphipathic character [265]. The racemic mixture of LA is used in therapy, especially in diabetic neuropathy. Its antioxidant properties are only evident in the interaction with other antioxidant systems. For example, the capacity of reducing GSSG to GSH is based on the initial reduction of LA to DHLA, which further reduces cystine to Cys, and the latter is used for GSH synthesis [266]. On a mitochondrial level, along with other antioxidants (GSH, vitamin C, and vitamin E), LA increases the activity of the enzymes involved in the Krebs cycle [267]. Furthermore, in a study conducted by Marangon et al., supplementation with LA resulted in diminishing urinary levels of F2-isoprostan and oxLDL [268]. A potential antiaging effect based on the protection of cardiac cells in aged rodents was also suggested [269].
However, its pro-oxidant properties, demonstrated in in vitro and in vivo studies, cannot be excluded. In vitro, DHLA forms chelates with both ferric and ferrous ions, which can extract the iron from ferritin and reduce it to Fe2+, at the same time increasing the possibility of oxidative processes via the Fenton reaction. In another study conducted by Çatakay et al., it was observed that the administration of LA in aged rats resulted in higher levels of carbonylated proteins compared to the control group [270].
The idea that substances/compounds with antioxidant activity are harmless is imprinted in popular belief and this conception is also supported by the media. However, there are questions as to how antioxidant compounds intervene in slowing down the aging process and the appearance of various diseases. The ability of antioxidants to prevent the accumulation of oxidizing molecules, thus helping to prevent aging, cancer, and even infections, has been also questioned.
The adaptation capacity of the immune system is essential for antimicrobial defense, as well as for the immunological memory. In this sense, the presence of mitochondria is mandatory, as it facilitates the aforementioned antimicrobial defense through ROS generation. At the same time, mitochondria are necessary for T-cell activation, which generates ROS and activates the nuclear factor of activated T cells (NFAT), which further enhances IL-2 synthesis [271,272]. This statement is supported by research studies that demonstrated that T-cells, for which antioxidant agents were made available, showed reduced levels of IL-2 and a reduction in the degree of proliferation was observed, sustaining an immunosuppressive effect resulting from the exposure to antioxidant agents. The same effects could also be noted in the case of T-cells that lacked mitochondrial complex III, supporting the importance of ROS in the immune response [273].
Macrophages and the neutrophils constrain the ability to phagocytize foreign compounds/microorganisms and subsequently to activate the immune system [274]. When macrophages recognize the antigen, the assembly of the protein complex of cytochrome I from the ETC is prevented, and mitochondrial respiration on complexes I and II is increased under the influence of phagosomal NOX and ROS-dependent tyrosine kinase Fgr. This process leads in increase in the ROS concentration [275]. The presence of ROS leads to the activation of MAPK, extracellular signal-regulated kinases (ERK), c-Jun N-terminal kinases (JNK) and NF-kB, which are key factors in the secretion of chemokines and pro-inflammatory cytokines (TNF-α, IL-1β, IL-6, IFN-γ) [276,277], as shown in Figure 5. Likewise, in this process of T-cell activation, NOX is also involved, which is activated by reactive species generated at the mitochondrial level, in order to maintain the proper ROS levels for this process [278]. Therefore, the presence of ROS is necessary for the destruction of the bacteria, which is contrast to the situation in chronic granulomatosis disease, in which ROS cannot be generated due to a genetic defect of NOX [279,280].
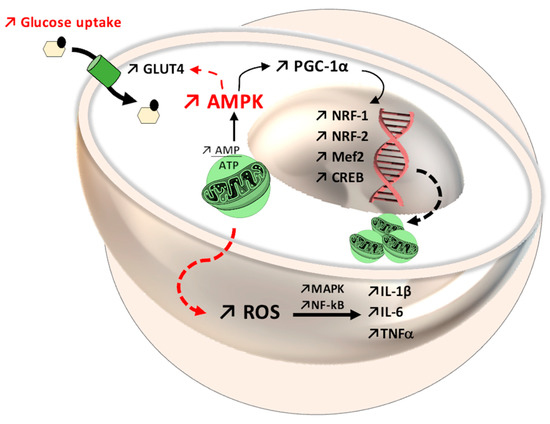
Figure 5.
Mitochondrial dynamics through the 5′ adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor gamma coactivator 1-alpha (AMPK-PGC-1α) system, with increased expression of nuclear factor erythroid 1-related factor (Nrf-1), nuclear factor erythroid 1-related factor (Nrf-2), myocyte enhancer factor-2 (Mef2), and cAMP response element-binding protein (CREB). At the same time, in the AMPK pathway, an improvement in glycemic status can be observed, through translocation of glucose transporter GLUT4 in the cellular membrane. Regarding the immune system, reactive oxygen species (ROS) enhance the activity of mitogen-activated protein kinase (MAPK) and nuclear Factor kappa B (NF-kB), which further favors interleukin and cytokine secretion (IL-1β, IL-6, tumor necrosis factor, TNF).
A certain basal level of ROS is essential for the proper functioning of the immune system, given that the destruction of pathogenic microorganisms by leukocytes is based on the generation of reactive species [281,282,283]. As previously mentioned in this review, ROS are intra- and intercellular communication pathways, along with MAPK, which uses these reactive species to generate cytokines and activate the immune system. Moreover, the activation of T lymphocytes and macrophages requires limited amounts of and H2O2; thus, neutralizing these physiological levels would lead to adverse effects on the immune system. Nevertheless, it is necessary that a balance of this oxidative stress is maintained since excess ROS can jeopardize the viability of T-cells. Thus, T lymphocyte functionality is dependent on ROS generation, as well as on the reduction of these species. Activated T-cells limit the permeability of the membrane and prevent ROS migration through the cytoplasm. At the same time, it has been discovered that antigen-presenting dendritic cells (APCs) release Cys in contact with T-cells. Cys is then captured and used by T-cells for GSH synthesis in order to protect them from excess ROS. Based on these considerations, antioxidant systems, as well as ROS, are essential for the proper functioning of the adaptive immune system mediated by T-cells [284,285].
Another area affected by the excessive use of antioxidant compounds is the biochemical one, in which interference with different metabolic pathways can occur. Given that the reactions of the Krebs cycle or in the ETC are redox phenomena, maintaining a long-term reduction status can have negative effects, such as the slowing down of metabolism, the accumulation of intermediates, and decreased ATP synthesis, with all these reactions being interconnected [286,287]. Another mechanism to control free radicals generated by ROS is based on heat shock proteins (HSPs) [288]. These proteins have properties to correct the misfolding of polypeptides and to promote the degradation of altered peptides. Under physiological conditions, the expression of HSPs is induced by various factors, including ROS, but the irresponsible use of antioxidant compounds can have negative effects on the activity and synthesis of HSPs [289,290,291].
The administration of antioxidants in viral infections has provided only evasive data, because in some studies, the benefits are not highlighted, and the antioxidants can favor the progression of the infection [292]. In the antioxidant therapeutic approach in cancer cells, the level of ROS must be considered because it can favor the situation in which, in the absence of a proper level of ROS, the cancer therapy can promote carcinogenesis via NF-kB, HIFs, and MAPK activation [27]. Paradoxically, cancer cells induce their own antioxidant systems with the purpose of reducing excess ROS, which would be detrimental through cytotoxic effects. In this context, there are two therapeutic approaches, one of which is based on antioxidant administration, which could reduce ROS levels required for the proliferation of cancer cells, making them susceptible to cellular apoptosis. However, this approach has raised numerous questions on this topic, and the results have been evasive [293].
The second approach refers to inducing oxidative stress to reach levels that cannot be neutralized by cancer cells. The therapy can be performed directly with therapeutic agents that promote ROS generation (radio- and chemotherapy) or indirectly, with agents that inhibit the antioxidant systems [294]. In this manner, the existence of the tumor cells depends on ROS levels; moderate levels activate NF-kB and promote survival mechanisms, whereas excessive levels degrade the cellular organelles, specifically the mitochondria, and alter the DNA and ARN molecules, favoring the initiation of apoptotic reactions [295].
For this reason, it is important to acknowledge the oxidative status of each individual, as antioxidants should not be administered to individuals without a proper immune response (see Table 2). However, a decrease in the activity of the immune system requires the antioxidant therapy approach.
Table 2.
Positive and negative effects related to the generation of oxidative stress on organs, systems, diseases, and physiological conditions.
5. Conclusions
The data presented in the literature suggest that ROS and RNS negatively influence the human body, but that this effect depends to a great extent on the individual’s oxidative status, as ROS are also involved in growth, differentiation, proliferation, and the processes of apoptosis and autophagy, which are highly important for maintaining cellular homeostasis. Medium-intensity aerobic exercise is responsible for generating ROS at levels that are beneficial and have the ability to induce counterregulatory and adaptation mechanisms, such as PGC-1α. In striated muscles, adaptation mechanisms are induced by exposure to ROS and RNS produced during resistance training and are mainly mediated through NF-kB and PGC-1α. With age, the incidence of neurodegenerative diseases increases, but a well-balanced CR seems to diminish this trend. Animal studies confirm the hormesis theory as CR is a form of stress itself, inducing adaptation mechanisms in the brain through the endocrine system. Thus, the expression of neurotrophic factors is dependent on CR and neurogenesis is favored.
In fact, the present article discusses an evolution paradox, the transition from anaerobic to aerobic metabolism and how, during evolution, the body manages to use ROS generated from aerobic metabolism, which is harmful, in purposeful ways to ensure adaptability, homeostasis, and cell signaling. Another interesting fact to emphasize is the duality of these reactive species, which are harmful yet indispensable for living.
In addition, given the delicate regulation mechanisms that dictate the generation and elimination rates, the duality of ROS (beneficial and negative effects) must be considered in therapy. In case of antitumor therapies, there are some limitations, i.e., toxic and/or immunosuppressive effects of agents generating ROS. Therefore, the dosage of anti/pro-oxidant agents has to target the maintenance of redox signaling capacity between the unaffected cells.
The biological dose–response curve in oxidative stress is unpredictable as reactive species are clearly responsible for cellular degradation, whereas antioxidant therapies can alleviate senescence by maintaining redox balance. Nevertheless, excessive doses of the latter can modify the redox balance of the cell, leading to a negative outcome. In order to avoid redox lesions, a balanced diet and moderate-intensity physical exercises are recommended to be performed regularly, especially for non-trained individuals.
The implication of ROS in many different processes, such as cell proliferation and differentiation, signal transduction, apoptosis, and autophagy, is not surprising, considering that these species have been present since the appearance of aerobic organisms, and have become indispensable for the abovementioned processes. It is supposed that cellular apoptosis does not occur directly due to oxidative stress, but due to ROS species that activate the physiological processes of apoptosis. It has also been observed that the reduction of ROS levels below a certain threshold would impede cell proliferation and differentiation and the immune response. Based on this, we can infer the essential role of the presence of ROS in the homeostasis of the body.
For this reason, it is important to practice responsible behavior when using antioxidant compounds, particularly for individuals who are known to have an affected antioxidant status.
Author Contributions
Writing—original draft preparation, G.J.; writing—review and editing, G.J., B.E.Ő., C.E.V., A.T.-V., C.-M.R., A.P.M. and M.-G.B.; visualization, G.J., B.E.Ő., C.E.V., A.T.-V. and C.-M.R. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by George Emil Palade University of Medicine, Pharmacy, Science, and Technology of Târgu Mureș Research Grant number 510/19/17.01.2022.
Acknowledgments
The authors would like to thank Adrian Năznean for the English language revision of the manuscript.
Conflicts of Interest
The authors declare no financial or other conflict of interest.
References
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Reyes, I.; Diebold, L.P.; Kong, H.; Schieber, M.; Huang, H.; Hensley, C.T.; Mehta, M.; Wang, T.; Santos, J.H.; Woychik, R.; et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol. Cell 2016, 61, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Chandel, N.S. Regulation of redox balance in cancer and T cells. J. Biol. Chem. 2018, 293, 7499–7507. [Google Scholar] [CrossRef]
- Yazdanpanah, B.; Wiegmann, K.; Tchikov, V.; Krut, O.; Pongratz, C.; Schramm, M.; Kleinridders, A.; Wunderlich, T.; Kashkar, H.; Utermöhlen, O.; et al. Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nature 2009, 460, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin Functions as a Peroxidase and a Regulator and Sensor of Local Peroxides. J. Biol. Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Egea, J.; Fabregat, I.; Frapart, Y.M.; Ghezzi, P.; Görlach, A.; Kietzmann, T.; Kubaichuk, K.; Knaus, U.G.; Lopez, M.G.; Olaso-Gonzalez, G.; et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 2017, 13, 94–162. [Google Scholar] [CrossRef] [PubMed]
- Poljsak, B.; Milisav, I. The Neglected Significance of “Antioxidative Stress”. Oxidative Med. Cell. Longev. 2012, 2012, 1–12. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef] [PubMed]
- Milisav, I.; Poljsak, B.; Šuput, D. Adaptive Response, Evidence of Cross-Resistance and Its Potential Clinical Use. Int. J. Mol. Sci. 2012, 13, 10771–10806. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Milisav, I.; Poljšak, B.; Ribarič, S. Reduced risk of apoptosis: Mechanisms of stress responses. Apoptosis 2016, 22, 265–283. [Google Scholar] [CrossRef] [PubMed]
- Al-Mehdi, A.-B.; Pastukh, V.M.; Swiger, B.M.; Reed, D.J.; Patel, M.R.; Bardwell, G.C.; Pastukh, V.V.; Alexeyev, M.F.; Gillespie, M.N. Perinuclear Mitochondrial Clustering Creates an Oxidant-Rich Nuclear Domain Required for Hypoxia-Induced Transcription. Sci. Signal. 2012, 5, ra47. [Google Scholar] [CrossRef] [PubMed]
- Bielski, B.H.; Arudi, R.L.; Sutherland, M.W. A study of the reactivity of HO2/O2-with unsaturated fatty acids. J. Biol. Chem. 1983, 258, 4759–4761. [Google Scholar] [CrossRef]
- Stanković, M.; Radovanović, D. Oxidative stress and physical activity. Sport Logia 2012, 8, 1–11. [Google Scholar]
- Wu, Q.; Ni, X. ROS-Mediated DNA Methylation Pattern Alterations in Carcinogenesis. Curr. Drug Targets 2015, 16, 13–19. [Google Scholar] [CrossRef]
- Hussein, S.A.; Omayma, A.R.A.; Elsenosy, Y.A. Antioxidants on oxidative stress and mitochondrial dysfunction in brain of experimentally induced hepatic failure in rats. Benha Vet. Med. J. 2012, 23, 1–12. [Google Scholar]
- Pellegrino, M.A.; Desaphy, J.-F.; Brocca, L.; Pierno, S.; Camerino, D.C.; Bottinelli, R. Redox homeostasis, oxidative stress and disuse muscle atrophy. J. Physiol. 2011, 589, 2147–2160. [Google Scholar] [CrossRef] [PubMed]
- Constantini, D. Understanding diversity in oxidative status and oxidative stress: The opportunities and challenges ahead. J. Exp. Biol. 2019, 222, 1–9. [Google Scholar]
- Schenk, B.; Fulda, S. Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene 2015, 34, 5796–5806. [Google Scholar] [CrossRef]
- Kluckova, K.; Sticha, M.; Černý, J.; Mracek, T.; Dong, L.; Drahota, Z.; Gottlieb, E.; Neuzil, J.; Rohlena, J. Ubiquinone-binding site mutagenesis reveals the role of mitochondrial complex II in cell death initiation. Cell Death Dis. 2015, 6, e1749. [Google Scholar] [CrossRef] [PubMed]
- Hara-Chikuma, M.; Watanabe, S.; Satooka, H. Involvement of aquaporin-3 in epidermal growth factor receptor signaling via hydrogen peroxide transport in cancer cells. Biochem. Biophys. Res. Commun. 2016, 471, 603–609. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Koyama, K. Exercise-induced oxidative stress: A tool for “hormesis” and “adaptive response”. The Journal of Physical Fitness and Sports. Medicine 2014, 3, 115–120. [Google Scholar]
- Lovlin, R.; Cottle, W.; Pyke, I.; Kavanagh, M.; Belcastro, A.N. Are indices of free radical damage related to exercise intensity. Graefe’s Arch. Clin. Exp. Ophthalmol. 1987, 56, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Abruzzo, P.M.; Esposito, F.; Marchionni, C.; di Tullio, S.; Belia, S.; Fulle, S.; Veicsteinas, A.; Marini, M. Moderate Exercise Training Induces ROS-Related Adaptations to Skeletal Muscles. Int. J. Sports Med. 2013, 34, 676–687. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Li, J.; Liu, Z.; Chuang, C.-C.; Yang, W.; Zuo, L. Redox Mechanism of Reactive Oxygen Species in Exercise. Front. Physiol. 2016, 7, 486. [Google Scholar] [CrossRef] [PubMed]
- Kozakiewicz, M.; Rowiński, R.; Kornatowski, M. Relation of Moderate Physical Activity to Blood Markers of Oxidative Stress and Antioxidant Defense in the Elderly. Oxidative Med. Cell. Longev. 2019, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Boccatonda, A.; Tripaldi, R.; Davì, G. Oxidative Stress Modulation through Habitual Physical Activity. Curr. Pharm. Des. 2016, 22, 3648–3680. [Google Scholar] [CrossRef] [PubMed]
- Yavari, A.; Javadi, M.; Mirmiran, P.; Bahadoran, Z. Exercise-Induced Oxidative Stress and Dietary antioxidants. Asian J. Sports Med. 2015, 6, e24898. [Google Scholar] [CrossRef]
- Thirupathi, A.; Pinho, R.A.; Ugbolue, U.C.; He, Y.; Meng, Y.; Gu, Y. Effect of Running Exercise on Oxidative Stress Biomarkers: A Systematic Review. Front. Physiol. 2021, 11, 610112. [Google Scholar] [CrossRef] [PubMed]
- Bogdanis, G.; Stavrinou, P.; Fatouros, I.; Philippou, A.; Chatzinikolaou, A.; Draganidis, D.; Ermidis, G.; Maridaki, M. Short-term high-intensity interval exercise training attenuates oxidative stress responses and improves antioxidant status in healthy humans. Food Chem. Toxicol. 2013, 61, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, A.; Chico, L.; Pasquali, L.; Bisordi, C.; Gerfo, A.L.; Fabbrini, M.; Petrozzi, L.; Marconi, L.; Ienco, E.C.; Mancuso, M.; et al. Gly482Ser PGC-1α Gene Polymorphism and Exercise-Related Oxidative Stress in Amyotrophic Lateral Sclerosis Patients. Front. Cell. Neurosci. 2016, 10, 102. [Google Scholar] [CrossRef]
- Bloomer, R.J.; Fry, A.C.; Falvo, M.; Moore, C.A. Protein carbonyls are acutely elevated following single set anaerobic exercise in resistance trained men. J. Sci. Med. Sport 2007, 10, 411–417. [Google Scholar] [CrossRef]
- Heunks, L.M.A.; Vina, J.; Van Herwaarden, C.L.A.; Folgering, H.T.M.; Gimeno, A.; Dekhuijzen, P.N.R. Xanthine oxidase is involved in exercise-induced oxidative stress in chronic obstructive pulmonary disease. Am. J. Physiol. Integr. Comp. Physiol. 1999, 277, R1697–R1704. [Google Scholar] [CrossRef]
- Thirupathi, A.; Wang, M.; Lin, J.K.; Fekete, G.; István, B.; Baker, J.S.; Gu, Y. Effect of Different Exercise Modalities on Oxidative Stress: A Systematic Review. Biol. Med. Res. Int. 2021, 2021, 1–10. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.; Bloomer, R.J. Acute exercise and oxidative stress: A 30 year history. Dyn. Med. 2009, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Warner, S.O.; Yao, M.V.; Cason, R.L.; Winnick, J.J. Exercise-Induced Improvements to Whole Body Glucose Metabolism in Type 2 Diabetes: The Essential Role of the Liver. Front. Endocrinol. 2020, 11, 567. [Google Scholar] [CrossRef] [PubMed]
- Sorriento, D.; Di Vaia, E.; Iaccarino, G. Physical Exercise: A Novel Tool to Protect Mitochondrial Health. Front. Physiol. 2021, 12, 660068. [Google Scholar] [CrossRef] [PubMed]
- Konopka, A.R.; Harber, M.P. Skeletal Muscle Hypertrophy after Aerobic Exercise Training. Exerc. Sport Sci. Rev. 2014, 42, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.C.; Liu, Y.; Hayworth, C.R.; Bhattacharya, A.; Lustgarten, M.S.; Muller, F.L.; Chaudhuri, A.; Qi, W.; Li, Y.; Huang, J.-Y.; et al. Dietary restriction attenuates age-associated muscle atrophy by lowering oxidative stress in mice even in complete absence of Cu, Zn, SOD. Aging Cell 2012, 11, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.P.; Pavlović, I.; Poljšak, B.; Šuput, D.; Milisav, I. Beneficial Role of ROS in Cell Survival: Moderate Increases in H2O2 Production Induced by Hepatocyte Isolation Mediate Stress Adaptation and Enhanced Survival. Antioxidants 2019, 8, 434. [Google Scholar] [CrossRef] [PubMed]
- López-Lluch, G.; Hunt, N.; Jones, B.; Zhu, M.; Jamieson, H.; Hilmer, S.; Cascajo, M.V.; Allard, J.; Ingram, D.K.; Navas, P.; et al. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc. Natl. Acad. Sci. USA 2006, 103, 1768–1773. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Adaptive response to oxidative stress: Bacteria, fungi, plants and animals. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2011, 153, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, E.; Sestili, P. Reactive Oxygen Species in Skeletal Muscle Signaling. J. Signal Transduct. 2012, 2012, 982794. [Google Scholar] [CrossRef] [PubMed]
- Mason, S.; Wadley, G.D. Skeletal muscle reactive oxygen species: A target of good cop/bad cop for exercise and disease. Redox Rep. 2014, 19, 97–106. [Google Scholar] [CrossRef]
- Ji, L.L.; Gomez-Cabrera, M.C.; Vina, J. Exercise and Hormesis: Activation of Cellular Antioxidant Signaling Pathway. Ann. N. Y. Acad. Sci. 2006, 1067, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Finkler, M.; Lichtenberg, D.; Pinchuk, I. The Relationship between Oxidative Stress and Exercise. J. Basic Clin. Physiol. Pharmacol. 2014, 25, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Morillas-Ruiz, J.M.; Hernández-Sánchez, P. Oxidative Stress and Antioxidant Defenses Induced by Physical Exercise. Basic Princ. Clin. Significance Oxidative Stress 2015, 2015, 222–241. [Google Scholar]
- Mesquita, A.; Weinberger, M.; Silva, A.; Sampaio-Marques, B.; Almeida, B.; Leão, C.; Costa, V.; Rodrigues, F.; Burhans, W.C.; Ludovico, P. Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Proc. Natl. Acad. Sci. USA 2010, 107, 15123–15128. [Google Scholar] [CrossRef] [PubMed]
- Warraich, U.-E.-A.; Hussain, F.; Kayani, H.U.R. Aging—Oxidative stress, antioxidants and computational modeling. Heliyon 2020, 6, e04107. [Google Scholar] [CrossRef] [PubMed]
- Wiggs, M.P. Can endurance exercise preconditioning prevention disuse muscle atrophy? Front. Physiol. 2015, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Domenech, E.; Viña, J. Moderate exercise is an antioxidant: Upregulation of antioxidant genes by training. Free Radic. Biol. Med. 2008, 44, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Mattusch, F.; Dufaux, B.; Heine, O.; Mertens, I.; Rost, R. Reduction of the Plasma Concentration of C-Reactive Protein Following Nine Months of Endurance Training. Int. J. Sports Med. 2000, 21, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Greiwe, J.S.; Cheng, B.; Rubin, D.C.; Yarasheski, K.; Semenkovich, C. Resistance exercise decreases skeletal muscle tumor necrosis factor α in frail elderly humans. FASEB J. 2001, 15, 475–482. [Google Scholar] [CrossRef]
- Gielen, S.; Adams, V.; Möbius-Winkler, S.; Linke, A.; Erbs, S.; Yu, J.; Kempf, W.; Schubert, A.; Schuler, G.; Hambrecht, R. Anti-inflammatory effects of exercise training in the skeletal muscle of patients with chronic heart failure. J. Am. Coll. Cardiol. 2003, 42, 861–868. [Google Scholar] [CrossRef]
- Levitan, I.; Volkov, S.; Subbaiah, P.V. Oxidized LDL: Diversity, Patterns of Recognition, and Pathophysiology. Antioxid. Redox Signal. 2010, 13, 39–75. [Google Scholar] [CrossRef] [PubMed]
- Fogarasi, E.; Croitoru, M.D.; Fülöp, I.; Nemes-Nagy, E.; Tripon, R.G.; Simon-Szabo, Z.; Muntean, D.-L. Malondialdehyde levels can be measured in serum and saliva by using a fast HPLC method with visible detection/Determinarea printr-o metodă HPLC-VIS rapidă a concentraţiilor serice şi salivare ale malondialdehidei. Rev. Romana Med. Lab. 2016, 24, 319–326. [Google Scholar] [CrossRef]
- Jîtcă, G.; Fogarasi, E.; Ősz, B.-E.; Vari, C.E.; Tero-Vescan, A.; Miklos, A.; Bătrînu, M.-G.; Rusz, C.M.; Croitoru, M.D.; Dogaru, M.T. A Simple HPLC/DAD Method Validation for the Quantification of Malondialdehyde in Rodent’s Brain. Molecules 2021, 26, 5066. [Google Scholar] [CrossRef] [PubMed]
- Jîtcă, G.; Fogarasi, E.; Ősz, B.-E.; Vari, C.E.; Fülöp, I.; Croitoru, M.D.; Rusz, C.M.; Dogaru, M.T. Profiling the Concentration of Reduced and Oxidized Glutathione in Rat Brain Using HPLC/DAD Chromatographic System. Molecules 2021, 26, 6590. [Google Scholar] [CrossRef] [PubMed]
- Milne, G.L.; Yin, H.; Brooks, J.D.; Sanchez, S.; Roberts, L.J.; Morrow, J.D. Quantification of F2-Isoprostanes in Biological Fluids and Tissues as a Measure of Oxidant Stress. Quinones Quinone Enzym. Part A 2007, 433, 113–126. [Google Scholar] [CrossRef]
- Labuschagne, C.F.; Broek, N.J.F.V.D.; Postma, P.; Berger, R.; Brenkman, A.B. A Protocol for Quantifying Lipid Peroxidation in Cellular Systems by F2-Isoprostane Analysis. PLoS ONE 2013, 8, e80935. [Google Scholar] [CrossRef] [PubMed]
- Madian, A.G.; Regnier, F.E. Proteomic Identification of Carbonylated Proteins and Their Oxidation Sites. J. Proteome Res. 2010, 9, 3766–3780. [Google Scholar] [CrossRef] [PubMed]
- Alomari, E.; Bruno, S.; Ronda, L.; Paredi, G.; Bettati, S.; Mozzarelli, A. Protein carbonylation detection methods: A comparison. Data Brief 2018, 19, 2215–2220. [Google Scholar] [CrossRef]
- Guo, C.; Li, X.; Wang, R.; Yu, J.; Ye, M.; Mao, L.; Zhang, S.; Zheng, S. Association between Oxidative DNA Damage and Risk of Colorectal Cancer: Sensitive Determination of Urinary 8-Hydroxy-2′-deoxyguanosine by UPLC-MS/MS Analysis. Sci. Rep. 2016, 6, 32581. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L.; Dickman, J.R.; Kang, C. Exercise-induced hormesis may help healthy aging. Dose Response 2010, 8, 73–79. [Google Scholar] [CrossRef]
- Abd-Elsameea, A.A.; Moustaf, A.A.; Mohamed, A.M. Modulation of the oxidative stress by metformin in the cerebrum of rats exposed to global cerebral ischemia and ischemia/reperfusion. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 2387–2392. [Google Scholar] [PubMed]
- Cunha, T.F.; Bacurau, A.V.N.; Jose, B.N. Exercise training prevents oxidative stress and ubiquitin-proteasome system overac-tivity and reverse skeletal muscle atrophy in heart failure. PLoS ONE 2012, 7, e41701. [Google Scholar] [CrossRef] [PubMed]
- Lambertucci, R.H.; Levada-Pires, A.C.; Rossoni, L.V. Effects of aerobic exercise training on antioxidant enzyme activities and m, RNA levels in soleus muscle from young and aged rats. Mech. Ageing Dev. 2007, 128, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Masoro, E.J. Overview of caloric restriction and ageing. Mech. Ageing Dev. 2005, 126, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Radák, Z.; Ji, L.L. Exercise-induced oxidative stress: Past, present and future. J. Physiol. 2016, 594, 5081–5092. [Google Scholar] [CrossRef]
- Radák, Z.; Chung, H.Y.; Koltai, E. Exercise, oxidative stress and hormesis. Ageing Res, Rev. 2008, 7, 34–42. [Google Scholar] [CrossRef]
- Ji, L.L. Antioxidant signaling in skeletal muscle: A brief review. Exp. Gerontol. 2007, 42, 582–593. [Google Scholar] [CrossRef]
- Lingappan, K. NF-k, B in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Flynn, M.G.; McFarlin, B.K.; Markofski, M.M. State of the Art Reviews: The Anti-Inflammatory Actions of Exercise Training. Am. J. Lifestyle Med. 2007, 1, 220–235. [Google Scholar] [CrossRef]
- Radák, Z.; Chung, H.Y.; Naito, H. Age-associated increases in oxidative stress and nuclear transcription factor κB activation are attenuated in rat liver by regular exercise. FASEB J. 2004, 18, 749–750. [Google Scholar] [CrossRef] [PubMed]
- Veuger, S.J.; E Hunter, J.; Durkacz, B.W. Ionizing radiation-induced NF-κB activation requires PARP-1 function to confer radioresistance. Oncogene 2008, 28, 832–842. [Google Scholar] [CrossRef]
- Terblanche, S.E. The effects of exhaustive exercise on the activity levels of catalase in various tissues of male and female rats. Cell Biol. Int. 2000, 23, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Mukhopadhyay, A.; Kundu, G.C.; Mahabeleshwar, G.H.; Singh, S.; Aggarwal, B.B. Hydrogen Peroxide Activates NF-κB through Tyrosine Phosphorylation of IκBα and Serine Phosphorylation of p65: Evidence for the involvement of IκBα kinase and Syk protein-tyrosine kinase. J. Biol. Chem. 2003, 278, 24233–24241. [Google Scholar] [CrossRef] [PubMed]
- Betteridge, D.J. What is oxidative stress? Metabolism 2000, 49, 3–8. [Google Scholar] [CrossRef]
- Chinsomboon, J.; Ruas, J.; Gupta, R.K.; Thom, R.; Shoag, J.; Rowe, G.C.; Sawada, N.; Raghuram, S.; Arany, Z. The transcriptional coactivator PGC-1α mediates exercise-induced angiogenesis in skeletal muscle. Proc. Natl. Acad. Sci. USA 2009, 106, 21401–21406. [Google Scholar] [CrossRef]
- Miura, Y. Oxidative Stress, Radiation-Adaptive Responses, and Aging. J. Radiat. Res. 2004, 45, 357–372. [Google Scholar] [CrossRef]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem cells and the impact of ROS signaling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef] [PubMed]
- Janssen-Heininger, Y.M.; Poynter, M.E.; Baeuerle, P.A. Recent advances towards understanding redox mechanisms in the acti-vation of nuclear factor κb. Free Radic Biol. Med. 2000, 28, 1317–1327. [Google Scholar] [CrossRef]
- Morino, Y.; Koga, H.; Tachibana, K.; Shoguchi, E.; Kiyomoto, M.; Wada, H. Heterochronic activation of VEGF signaling and the evolution of the skeleton in echinoderm pluteus larvae. Evol. Dev. 2012, 14, 428–436. [Google Scholar] [CrossRef]
- Patel, R.; Rinker, L.; Peng, J. Reactive Oxygen Species: The Good and the Bad. Reactive Oxygen Species (ROS) in Living Cells. Environ. Health Perspect. 2017, 2017, 7–28. [Google Scholar]
- Ushio-Fukai, M.; Wayne Alexander, R. Reactive oxygen species as mediators of angiogenesis signaling: Role of NAD(P)H oxidase. Mol. Cell Biochem. 2004, 264, 85–97. [Google Scholar] [CrossRef]
- Matias, C.; Bicho, M.; Laires, M.; Monteiro, C. Athletes have more susceptibility to oxidative stress: Truth or myth? A study in swimmers. Sci. Sports 2018, 35, 20–28. [Google Scholar] [CrossRef]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Pourova, J.; Kottova, M.; Voprsalova, M.; Pour, M. Reactive oxygen and nitrogen species in normal physiological processes. Acta Physiol. 2010, 198, 15–35. [Google Scholar] [CrossRef] [PubMed]
- Collins, Y.; Chouchani, E.; James, A.M.; Menger, K.; Cochemé, H.M.; Murphy, M.P. Mitochondrial redox signalling at a glance. J. Cell Sci. 2012, 125, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Denu, J.M.; Tanner, K.G. Specific and Reversible Inactivation of Protein Tyrosine Phosphatases by Hydrogen Peroxide: Evidence for a Sulfenic Acid Intermediate and Implications for Redox Regulation. Biochemistry 1998, 37, 5633–5642. [Google Scholar] [CrossRef]
- E Paulsen, C.; Truong, T.H.; Garcia, F.J.; Homann, A.; Gupta, V.; E Leonard, S.; Carroll, K.S. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 2011, 8, 57–64. [Google Scholar] [CrossRef]
- Powers, S.K.; Jackson, M.J. Exercise-Induced Oxidative Stress: Cellular Mechanisms and Impact on Muscle Force Production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef]
- Alves, J.O.; Pereira, L.M.; Monteiro, I.C.C.D.R.; Dos Santos, L.H.P.; Ferraz, A.S.M.; Loureiro, A.C.C.; Lima, C.C.; Leal-Cardoso, J.H.; Carvalho, D.P.; Fortunato, R.S.; et al. Strenuous Acute Exercise Induces Slow and Fast Twitch-Dependent NADPH Oxidase Expression in Rat Skeletal Muscle. Antioxidants 2020, 9, 57. [Google Scholar] [CrossRef]
- Trewin, A.J.; Lundell, L.S.; Perry, B.D.; Patil, K.V.; Chibalin, A.; Levinger, I.; McQuade, L.R.; Stepto, N.K. Effect of N-acetylcysteine infusion on exercise-induced modulation of insulin sensitivity and signaling pathways in human skeletal muscle. Am. J. Physiol. Metab. 2015, 309, E388–E397. [Google Scholar] [CrossRef]
- Vincent, H.K.; Powers, S.K.; Stewart, D.J.; Demirel, H.A.; Shanely, R.A.; Naito, H. Short-term exercise training improves diaphragm antioxidant capacity and endurance. Graefe’s Arch. Clin. Exp. Ophthalmol. 2000, 81, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.-C.; Domenech, E.; Romagnoli, M.; Arduini, A.; Borras, C.; Pallardo, F.V.; Sastre, J.; Viña, J. Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am. J. Clin. Nutr. 2008, 87, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, G.; Hamarsland, H.; Cumming, K.T.; Johansen, R.E.; Hulmi, J.J.; Børsheim, E.; Wiig, H.; Garthe, I.; Raastad, T. Vitamin C and E supplementation alters protein signalling after a strength training session, but not muscle growth during 10 weeks of training. J. Physiol. 2014, 592, 5391–5408. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Chida, A.S.; Rahman, I. Redox modifications of protein-thiols: Emerging roles in cell signaling. Biochem. Pharmacol. 2006, 71, 551–564. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Cycle or a Double-Edged Sword? Antioxid. Redox Signal. 2007, 9, 2277–2294. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J. Redox regulation of adaptive responses in skeletal muscle to contractile activity. Free Radic. Biol. Med. 2009, 47, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xin, C.; Eu, J.P.; Stamler, J.S.; Meissner, G. Cysteine-3635 is responsible for skeletal muscle ryanodine receptor modulation by NO. Proc. Natl. Acad. Sci. USA 2001, 98, 11158–11162. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, A.M.; Reiken, S.; Carlson, C.; Mongillo, M.; Liu, X.; Rothman, L.; Matecki, S.; Lacampagne, A.; Marks, A.R. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 2009, 15, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Carvalho-Filho, M.A.; Ueno, M.; Hirabara, S.M.; Seabra, A.B.; Carvalheira, J.B.; de Oliveira, M.G.; Velloso, L.A.; Curi, R.; Saad, M.J. S-Nitrosation of the Insulin Receptor, Insulin Receptor Substrate 1, and Protein Kinase B/Akt. Diabetes 2005, 54, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, L.; Figueiredo-Freitas, C.; Casimiro-Lopes, G.; Magdesian, M.H.; Assreuy, J.; Sorenson, M.M. Myosin is reversibly inhibited by S-nitrosylation. Biochem. J. 2009, 424, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Andrade, F.H.; Reid, M.B.; Allen, D.G.; Westerblad, H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J. Physiol. 1998, 509, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.B. Invited Review: Redox modulation of skeletal muscle contraction: What we know and what we don’t. J. Appl. Physiol. 2001, 90, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Moopanar, T.R.; Allen, D.G. Reactive oxygen species reduce myofibrillar Ca2+ sensitivity in fatiguing mouse skeletal muscle at 37 °C. J. Physiol. 2005, 564, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.B. Free radicals and muscle fatigue: Of ROS, canaries, and the IOC. Free Radic. Biol. Med. 2008, 44, 169–179. [Google Scholar] [CrossRef]
- Mason, S.; Morrison, D.; McConell, G.; Wadley, G. Muscle redox signalling pathways in exercise. Role of antioxidants. Free Radic. Biol. Med. 2016, 98, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Deminice, R.; Ozdemir, M.; Yoshihara, T.; Bomkamp, M.P.; Hyatt, H. Exercise-induced oxidative stress: Friend or foe? J. Sport Health Sci. 2020, 9, 415–425. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.J.; Medved, I.; Goodman, C.; Brown, M.J.; Bjorksten, A.R.; Murphy, K.; Petersen, A.C.; Sostaric, S.; Gong, X. N-acetylcysteine attenuates the decline in muscle Na+,K+-pump activity and delays fatigue during prolonged exercise in humans. J. Physiol. 2006, 576, 279–288. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Ji, L.L. Redox signaling in skeletal muscle: Role of aging and exercise. Adv. Physiol. Educ. 2015, 39, 352–359. [Google Scholar] [CrossRef]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.-Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef]
- Finck, B.N.; Kelly, D.P. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J. Clin. Investig. 2006, 116, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Irrcher, I.; Ljubicic, V.; Hood, D.A. Interactions between ROS and AMP kinase activity in the regulation of PGC-1α transcription in skeletal muscle cells. Am. J. Physiol. Physiol. 2009, 296, C116–C123. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Bartsakoulia, M.; Pyle, A.; Troncoso-Chandía, D.; Vial-Brizzi, J.; Paz-Fiblas, M.V.; Duff, J.; Griffin, H.; Boczonadi, V.; Lochmüller, H.; Kleinle, S.; et al. A novel mechanism causing imbalance of mitochondrial fusion and fission in human myopathies. Hum. Mol. Genet. 2018, 27, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Irving, B.A.; Lanza, I.; Henderson, G.C.; Rao, R.R.; Spiegelman, B.M.; Nair, K.S. Combined Training Enhances Skeletal Muscle Mitochondrial Oxidative Capacity Independent of Age. J. Clin. Endocrinol. Metab. 2015, 100, 1654–1663. [Google Scholar] [CrossRef]
- Kang, T.-C. Nuclear Factor-Erythroid 2-Related Factor 2 (Nrf2) and Mitochondrial Dynamics/Mitophagy in Neurological Diseases. Antioxidants 2020, 9, 617. [Google Scholar] [CrossRef]
- Mottillo, E.P.; Bloch, A.E.; Leff, T.; Granneman, J.G. Lipolytic Products Activate Peroxisome Proliferator-activated Receptor (PPAR) α and δ in Brown Adipocytes to Match Fatty Acid Oxidation with Supply. J. Biol. Chem. 2012, 287, 25038–25048. [Google Scholar] [CrossRef] [PubMed]
- Monsalve, F.A.; Pyarasani, R.D.; Delgado-Lopez, F.; Moore-Carrasco, R. Peroxisome Proliferator-Activated Receptor Targets for the Treatment of Metabolic Diseases. Mediat. Inflamm. 2013, 2013, 1–18. [Google Scholar] [CrossRef]
- Muthusamy, V.R.; Kannan, S.; Sadhaasivam, K. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic. Biol. Med. 2012, 52, 366–376. [Google Scholar] [CrossRef]
- Spanidis, Y.; Veskoukis, A.S.; Papanikolaou, C.; Stagos, D.; Priftis, A.; Deli, C.K.; Jamurtas, A.Z.; Kouretas, D. Exercise-Induced Reductive Stress Is a Protective Mechanism against Oxidative Stress in Peripheral Blood Mononuclear Cells. Oxidative Med. Cell. Longev. 2018, 2018, 1–9. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.-C.; Borrás, C.; Pallardó, F.V.; Sastre, J.; Ji, L.L.; Viña, J. Decreasing xanthine oxidase-mediated oxidative stress prevents useful cellular adaptations to exercise in rats. J. Physiol. 2005, 567, 113–120. [Google Scholar] [CrossRef]
- Zacarías-Flores, M.; Sánchez-Rodríguez, M.A.; García-Anaya, O.D.; Correa-Muñoz, E.; Mendoza-Núñez, V.M. Relación entre el estrés oxidativo y la pérdida de masa muscular en la posmenopausia temprana: Estudio exploratorio. Endocrinol. Diabetes Nutr. 2018, 65, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K.; Oberbach, A.; Klöting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Blüher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.M.; Bagatini, M.D.; Roth, M.A. Acute effects of resistance exercise and intermittent intense aerobic exercise on blood cell count and oxidative stress. Braz. J. Med. Biol. Res. 2012, 45, 1172–1182. [Google Scholar] [CrossRef] [PubMed]
- Mounier, R.; Théret, M.; Lantier, L.; Foretz, M.; Viollet, B. Expanding roles for AMPK in skeletal muscle plasticity. Trends Endocrinol. Metab. 2015, 26, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Vercauteren, K.; Pasko, R.A.; Gleyzer, N.; Marino, V.M.; Scarpulla, R.C. PGC-1-Related Coactivator: Immediate Early Expression and Characterization of a CREB/NRF-1 Binding Domain Associated with Cytochrome c Promoter Occupancy and Respiratory Growth. Mol. Cell. Biol. 2006, 26, 7409–7419. [Google Scholar] [CrossRef]
- Handschin, C.; Spiegelman, B.M. The role of exercise and PGC1α in inflammation and chronic disease. Nature 2008, 454, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Romanello, V.; Sandri, M. Mitochondrial Biogenesis and Fragmentation as Regulators of Muscle Protein Degradation. Curr. Hypertens. Rep. 2010, 12, 433–439. [Google Scholar] [CrossRef]
- Park, J.-H.; Lee, Y.-E. Effects of exercise on glycemic control in type 2 diabetes mellitus in Koreans: The fifth Korea National Health and Nutrition Examination Survey (KNHANES V). J. Phys. Ther. Sci. 2015, 27, 3559–3564. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, S.; Houle, F.; Kotanides, H.; Witte, L.; Waltenberger, J.; Landry, J.; Huot, J. Vascular Endothelial Growth Factor (VEGF)-driven Actin-based Motility Is Mediated by VEGFR2 and Requires Concerted Activation of Stress-activated Protein Kinase 2 (SAPK2/p38) and Geldanamycin-sensitive Phosphorylation of Focal Adhesion Kinase. J. Biol. Chem. 2000, 275, 10661–10672. [Google Scholar] [CrossRef] [PubMed]
- Morales-Ruiz, M.; Fulton, D.; Sowa, G. Vascular endothelial growth factor-stimulated actin reorganization and migration of endothelial cells is regu- lated via the serine/threonine kinase Akt. Circ Res. 2000, 86, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Gee, S.L.; Hargreaves, M. Exercise and skeletal muscle glucose transporter 4 expression: Molecular mechanism. Clin. Exp. Pharmacol. Physiol. 2006, 33, 395–399. [Google Scholar]
- Hutchison, S.K.; Teede, H.J.; Rachoń, D.; Harrison, C.L.; Strauss, B.J.; Stepto, N.K. Effect of exercise training on insulin sensitivity, mitochondria and computed tomography muscle attenuation in overweight women with and without polycystic ovary syndrome. Diabetology 2012, 55, 1424–1434. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, J.M.; Moreli, M.; Tewari, S.; Benite-Ribeiro, S.A. The effect of exercise on skeletal muscle glucose uptake in type 2 diabetes: An epigenetic perspective. Metabolism 2015, 64, 1619–1628. [Google Scholar] [CrossRef]
- Steinbacher, P.; Eckl, P. Impact of Oxidative Stress on Exercising Skeletal Muscle. Biomolecules 2015, 5, 356–377. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Duarte, J.A.; Kavazis, A.N.; Talbert, E.E. Reactive oxygen species are signalling molecules for skeletal muscle adaptation. Exp. Physiol. 2009, 95, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, D.M.; West, D.W.D.; Churchward-Venne, T.A.; Breen, L.; Baker, S.K.; Phillips, S.M. Influence of aerobic exercise intensity on myofibrillar and mitochondrial protein synthesis in young men during early and late postexercise recovery. Am. J. Physiol. Metab. 2014, 306, E1025–E1032. [Google Scholar] [CrossRef]
- Kang, C.; O’Moore, K.M.; Dickman, J.R.; Ji, L.L. Exercise activation of muscle peroxisome proliferator-activated receptor-γ coactivator-1α signaling is redox sensitive. Free Radic. Biol. Med. 2009, 47, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Dumke, C.L.; Davis, J.M.; Murphy, E.A.; Nieman, D.C.; Carmichael, M.D.; Quindry, J.C.; Triplett, N.T.; Utter, A.C.; Gowin, S.J.G.; Henson, D.A.; et al. Successive bouts of cycling stimulates genes associated with mitochondrial biogenesis. Graefe’s Arch. Clin. Exp. Ophthalmol. 2009, 107, 419–427. [Google Scholar] [CrossRef]
- Margaritis, I.; Tessier, F.; Prou, E.; Marconnet, P.; Marini, J.-F. Effects of Endurance Training on Skeletal Muscle Oxidative Capacities with and without Selenium Supplementation. J. Trace Elements Med. Biol. 1997, 11, 37–43. [Google Scholar] [CrossRef]
- Stepto, N.K.; Benziane, B.; Wadley, G.D.; Chibalin, A.V.; Canny, B.J.; Eynon, N.; McConell, G.K. Short-Term Intensified Cycle Training Alters Acute and Chronic Responses of PGC1α and Cytochrome C Oxidase IV to Exercise in Human Skeletal Muscle. PLoS ONE 2012, 7, e53080. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J. Keap1-dependent Proteasomal Degradation of Transcription Factor Nrf2 Contributes to the Negative Regulation of Antioxidant Response Element-driven Gene Expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef]
- Kobayashi, A.; Ohta, T.; Yamamoto, M. Unique Function of the Nrf2—Keap1 Pathway in the Inducible Expression of Antioxidant and Detoxifying Enzymes. Methods Enzymol. 2004, 378, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, E.; Giammarioli, A.M.; Chiandotto, S. Exercise-Induced Skeletal Muscle Remodeling and Metabolic Adaptation: Redox Signaling and Role of Autophagy. Antioxid. Redox Signal. 2014, 21, 154–176. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription Factor Nrf2 Coordinately Regulates a Group of Oxidative Stress-inducible Genes in Macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T. Coordinate Regulation of Glutathione Biosynthesis and Release by Nrf2-Expressing Glia Potently Protects Neurons from Oxidative Stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Torres, I.; Guarner-Lans, V.; Rubio-Ruiz, M.E. Reductive Stress in Inflammation-Associated Diseases and the Pro-Oxidant Effect of Antioxidant Agents. Int. J. Mol. Sci. 2017, 18, 2098. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K. How increased Hormesis in Aging and Neurodegeneration—A Prodigy Awaiting Dissection mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y.; Li, F.; Wei, Y. Conserved roles of glucose in suppressing reactive oxygen species-induced cell death and animal survival. Aging 2019, 11, 5726–5743. [Google Scholar] [CrossRef] [PubMed]
- Zgheib, E. Bioinformatic and Modelling Approaches for a System-Level Understanding of Oxidative Stress Toxicity; Université de Technologie de Compiègne: Compiègne, France, 2018. [Google Scholar]
- Andrews, Z.B.; Liu, Z.-W.; Wallingford, N.; Erion, D.M.; Borok, E.; Friedman, J.M.; Tschöp, M.H.; Shanabrough, M.; Cline, G.; Shulman, G.; et al. UCP2 mediates ghrelin’s action on NPY/Ag, RP neurons by lowering free radicals. Nature 2008, 454, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Kroemer, G.; Kepp, O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2020, 8, 200. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, L.R.; Daniel De Magalhaes Filho, C.; McQuary, P.R.; Chu, C.-C.; Visvikis, O.; Chang, J.T.; Gelino, S.; Ong, B.; Davis, A.E.; Irazoqui, J.E.; et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat. Commun. 2013, 4, 2267. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.-O.; Yoo, S.-M.; Ahn, H.-H.; Nah, J.; Hong, S.-H.; Kam, T.-I.; Jung, S.; Jung, Y.-K. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.; Cumming, R.C.; Brech, A.; Isakson, P.; Schubert, D.R.; Finley, K.D. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008, 4, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Talalay, P.; Sharkey, J.; Zhang, Y.; Holtzclaw, W.D.; Wang, X.J.; David, E.; Schiavoni, K.H.; Finlayson, S.; Mierke, D.F.; et al. An Exceptionally Potent Inducer of Cytoprotective Enzymes: Elucidation of the structural features that determine inducer potency and reactivity with Keap1. J. Biol. Chem. 2010, 285, 33747–33755. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural Regen. Res. 2012, 7, 376–385. [Google Scholar] [PubMed]
- Lévy, E.; El Banna, N.; Baïlle, D.; Heneman-Masurel, A.; Truchet, S.; Rezaei, H.; Huang, M.-E.; Béringue, V.; Martin, D.; Vernis, L. Causative Links between Protein Aggregation and Oxidative Stress: A Review. Int. J. Mol. Sci. 2019, 20, 3896. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-H.; Lee, M.J.; Park, S.; Oh, D.-C.; Elsasser, S.; Chen, P.-C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP. Nature 2010, 467, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Obrador, E.; Salvador, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 901. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Mhyre, T.R.; Boyd, J.T.; Hamill, R.W.; Maguire-Zeiss, K.A. Parkinson’s Disease. Subcell Biochem. 2012, 65, 389–455. [Google Scholar] [CrossRef] [PubMed]
- Krüger, R.; Klucken, J.; Weiss, D.; Tönges, L.; Kolber, P.; Unterecker, S.; Lorrain, M.; Baas, H.; Müller, T.; Riederer, P. Classification of advanced stages of Parkinson’s disease: Translation into stratified treatments. J. Neural Transm. 2017, 124, 1015–1027. [Google Scholar] [CrossRef] [PubMed]
- Jîtcă, G.; Ősz, B.; Tero-Vescan, A.; Vari, C. Psychoactive Drugs—From Chemical Structure to Oxidative Stress Related to Dopaminergic Neurotransmission. A Review. Antioxidants 2021, 10, 381. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxidative Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Larson, E.B.; Wang, L.; Bowen, J.D.; McCormick, W.C.; Teri, L.; Crane, P.; Kukull, W. Exercise Is Associated with Reduced Risk for Incident Dementia among Persons 65 Years of Age and Older. Ann. Intern. Med. 2006, 144, 73–81. [Google Scholar] [CrossRef] [PubMed]
- van Gelder, B.M.; Tijhuis, M.A.; Kalmijn, S.; Giampaoli, S.; Nissinen, A.; Kromhout, D. Physical activity in relation to cognitive decline in elderly men. Neurology 2004, 63, 2316–2321. [Google Scholar] [CrossRef]
- Paillard, T.; Rolland, Y.; Barreto, P. Protective Effects of Physical Exercise in Alzheimer’s Disease and Parkinson’s Disease: A Narrative Review. J. Clin. Neurol. 2015, 11, 212–219. [Google Scholar] [CrossRef] [PubMed]
- E Lucas, S.J.; Cotter, J.D.; Brassard, P.; Bailey, D.M. High-Intensity Interval Exercise and Cerebrovascular Health: Curiosity, Cause, and Consequence. J. Cereb. Blood Flow Metab. 2015, 35, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: Overview. Ann. N. Y. Acad. Sci. 2006, 928, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.J.; Kirkwood, A.; Pizzorusso, T.; Porciatti, V.; Morales, B.; Bear, M.F.; Maffei, L.; Tonegawa, S. BDNF Regulates the Maturation of Inhibition and the Critical Period of Plasticity in Mouse Visual Cortex. Cell 1999, 98, 739–755. [Google Scholar] [CrossRef]
- Araya, A.V.; Orellana, X.; Espinoza, J. Evaluation of the effect of caloric restriction on serum BDNF in overweight and obese subjects: Preliminary evidences. Endocrine 2008, 33, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Gredilla, R.; Barja, G. Minireview: The Role of Oxidative Stress in Relation to Caloric Restriction and Longevity. Endocrinology 2005, 146, 3713–3717. [Google Scholar] [CrossRef]
- Radak, Z.; Chung, H.Y.; Goto, S. Exercise and hormesis: Oxidative stress-related adaptation for successful aging. Biogerontology 2005, 6, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Pickering, A.M.; Vojtovich, L.; Tower, J.; Davies, K.J.A. Oxidative stress adaptation with acute, chronic, and repeated stress. Free Radic. Biol. Med. 2012, 55, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.; Liu, S.; So, K.-F. Dietary restriction and brain health. Neurosci. Bull. 2010, 26, 55–65. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Van Cauwenberghe, C.; Vandendriessche, C.; Libert, C.; Vandenbroucke, R. Caloric restriction: Beneficial effects on brain aging and Alzheimer’s disease. Mamm. Genome 2016, 27, 300–319. [Google Scholar] [CrossRef]
- Radak, Z.; Zhao, Z.; Koltai, E.; Ohno, H.; Atalay, M. Oxygen Consumption and Usage during Physical Exercise: The Balance Between Oxidative Stress and ROS-Dependent Adaptive Signaling. Antioxidants Redox Signal. 2013, 18, 1208–1246. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Naito, H.; Kaneko, T.; Tahara, S.; Nakamoto, H.; Takahashi, R.; Cardozo-Pelaez, F.; Goto, S. Exercise training decreases DNA damage and increases DNA repair and resistance against oxidative stress of proteins in aged rat skeletal muscle. Pflügers Arch. 2002, 445, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Kaneko, T.; Tahara, S.; Nakamoto, H.; Pucsok, J.; Sasvári, M.; Nyakas, C.; Goto, S. Regular exercise improves cognitive function and decreases oxidative damage in rat brain. Neurochem. Int. 2001, 38, 17–23. [Google Scholar] [CrossRef]
- Ji, L.L. Antioxidants and Oxidative Stress in Exercise. Proc. Soc. Exp. Boil. Med. 1999, 222, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Bouzid, M.A.; Filaire, E.; Matran, R.; Robin, S.; Fabre, C. Lifelong Voluntary Exercise Modulates Age-Related Changes in Oxidative Stress. Int. J. Sports Med. 2018, 39, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Vezzoli, A.; Mrakic-Sposta, S.; Montorsi, M.; Porcelli, S.; Vago, P.; Cereda, F.; Longo, S.; Maggio, M.; Narici, M.; Sposta, M.-; et al. Moderate Intensity Resistive Training Reduces Oxidative Stress and Improves Muscle Mass and Function in Older Individuals. Antioxidants 2019, 8, 431. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Jentoft, A.J.; Baeyens, J.P.; Bauer, J.M.; Boirie, Y.; Cederholm, T.; Landi, F.; Martin, F.C.; Michel, J.-P.; Rolland, Y.; Schneider, S.M.; et al. Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Ageing 2010, 39, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Rygiel, K.A.; Picard, M.; Turnbull, D. The ageing neuromuscular system and sarcopenia: A mitochondrial perspective. J. Physiol. 2016, 594, 4499–4512. [Google Scholar] [CrossRef] [PubMed]
- Chabi, B.; Ljubicic, V.; Menzies, K.J.; Huang, J.H.; Saleem, A.; Hood, D.A. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 2008, 7, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.L.; Robinson, M.M.; Nair, K.S. Skeletal muscle aging and the mitochondrion. Trends Endocrinol. Metab. 2013, 24, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.J.; Candau, R.; Bernardi, H. FoxO transcription factors: Their roles in the maintenance of skeletal muscle homeostasis. Cell. Mol. Life Sci. 2013, 71, 1657–1671. [Google Scholar] [CrossRef] [PubMed]
- Meydani, M.; Das, S.; Band, M.; Epstein, S.; Roberts, S. The effect of caloric restriction and glycemic load on measures of oxidative stress and antioxidants in humans: Results from the calerie trial of human caloric restriction. J. Nutr. Health Aging 2011, 15, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-S.; Lin, S.-C. AMPK Promotes Autophagy by Facilitating Mitochondrial Fission. Cell Metab. 2016, 23, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Cunha, T.F.; Moreira, J.B.N.; Paixão, N.A.; Campos, J.C.; Monteiro, A.W.A.; Bacurau, A.V.N.; Bueno, C.R.; Ferreira, J.C.B.; Brum, P.C. Aerobic exercise training upregulates skeletal muscle calpain and ubiquitin-proteasome systems in healthy mice. J. Appl. Physiol. 2012, 112, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Weindruch, R. Oxidative Stress, Caloric Restriction, and Aging. Science 1996, 273, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Mut-Salud, N.; Alvarez, P.; Garrido, J.M.; Carrasco, E.; Aránega, A.; Rodríguez-Serrano, F. Antioxidant Intake and Antitumor Therapy: Toward Nutritional Recommendations for Optimal Results. Oxidative Med. Cell. Longev. 2016, 2016, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef]
- Salehi, B.; Martorell, M.; Arbiser, J.; Sureda, A.; Martins, N.; Maurya, P.; Sharifi-Rad, M.; Kumar, P.; Sharifi-Rad, J. Antioxidants: Positive or Negative Actors? Biomolecules 2018, 8, 124. [Google Scholar] [CrossRef]
- Halagappa, V.K.M.; Guo, Z.; Pearson, M.; Matsuoka, Y.; Cutler, R.G.; LaFerla, F.M.; Mattson, M.P. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 2007, 26, 212–220. [Google Scholar] [CrossRef]
- Mao, L.; Franke, J. Hormesis in Aging and Neurodegeneration—A Prodigy Awaiting Dissection. Int. J. Mol. Sci. 2013, 14, 13109–13128. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-T.; Shih, Y.-R.V.; Kuo, T.K.; Lee, O.K.; Wei, Y.-H. Coordinated Changes of Mitochondrial Biogenesis and Antioxidant Enzymes during Osteogenic Differentiation of Human Mesenchymal Stem Cells. Stem Cells 2008, 26, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.M.; Esain, V.; Frechette, G.M.; Harris, L.J.; Cox, A.G.; Cortes, M.; Garnaas, M.K.; Carroll, K.J.; Cutting, C.C.; Khan, T.; et al. Glucose metabolism impacts the spatiotemporal onset and magnitude of HSC induction in vivo. Blood 2013, 121, 2483–2493. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Marsboom, G.; Toth, P.T.; Rehman, J. Mitochondrial Respiration Regulates Adipogenic Differentiation of Human Mesenchymal Stem Cells. PLoS ONE 2013, 8, e77077. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Fang, Y.; Cai, J.; Li, X.; Xu, F.; Yuan, N.; Zhang, S.; Wang, J. ROS functions as an upstream trigger for autophagy to drive hematopoietic stem cell differentiation. Hematology 2016, 21, 613–618. [Google Scholar] [CrossRef]
- Wagatsuma, A.; Sakuma, K. Mitochondria as a Potential Regulator of Myogenesis. Sci. World J. 2013, 2013, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Vieira, H.L.; Alves, P.; Vercelli, A. Modulation of neuronal stem cell differentiation by hypoxia and reactive oxygen species. Prog. Neurobiol. 2011, 93, 444–455. [Google Scholar] [CrossRef]
- Tormos, K.V.; Anso, E.; Hamanaka, R.B.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.; Chandel, N.S. Mitochondrial Complex III ROS Regulate Adipocyte Differentiation. Cell Metab. 2011, 14, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Tak, E.; Lee, J.; Rashid, M.; Murphy, M.P.; Ha, J.; Kim, S.S. Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation. Cell Res. 2011, 21, 817–834. [Google Scholar] [CrossRef]
- Menon, S.G.; Sarsour, E.H.; Spitz, D.R.; Higashikubo, R.; Sturm, M.; Zhang, H.; Goswami, P.C. Redox regulation of the G1 to S phase transition in the mouse embryo fibroblast cell cycle. Cancer Res. 2003, 63, 2109. [Google Scholar] [PubMed]
- Pashkovskaia, N.; Gey, U.; Rödel, G. Mitochondrial ROS direct the differentiation of murine pluripotent P19 cells. Stem Cell Res. 2018, 30, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Miyata, M.; Kasai, H.; Kawai, K.; Yamada, N.; Tokudome, M.; Ichikawa, H.; Goto, C.; Tokudome, Y.; Kuriki, K.; Hoshino, H.; et al. Changes of Urinary 8-Hydroxydeoxyguanosine Levels during a Two-Day Ultramarathon Race Period in Japanese Non-Professional Runners. Int. J. Sports Med. 2008, 29, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Kiffin, R.; Bandyopadhyay, U.; Cuervo, A.M. Oxidative stress and autophagy. Antioxid. Redox Signal. 2006, 8, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.-J. Positive oxidative stress in aging and aging-related disease tolerance. Redox Biol. 2014, 2, 165–169. [Google Scholar] [CrossRef]
- Zuo, L.; Zhou, T.; Pannell, B.K.; Ziegler, A.C.; Best, T.M. Biological and physiological role of reactive oxygen species—The good, the bad and the ugly. Acta Physiol. 2015, 214, 329–348. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Hormesis Defined. Ageing Res. Rev. 2008, 7, 1–7. [Google Scholar] [CrossRef]
- Gomes, A.P.; Price, N.L.; Ling, A.J.Y.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD+ Induces a Pseudohypoxic State Disrupting Nuclear-Mitochondrial Communication during Aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef]
- Nunn, A.V.; Guy, G.W.; Brodie, J.S.; Bell, J.D. Inflammatory modulation of exercise salience: Using hormesis to return to a healthy lifestyle. Nutr. Metab. 2010, 7, 87. [Google Scholar] [CrossRef]
- Woods, J.A.; Vieira-Potter, V.; Keylock, K.T. Exercise, Inflammation, and Innate Immunity. Immunol. Allergy Clin. N. Am. 2009, 29, 381–393. [Google Scholar] [CrossRef]
- Bastos Maia, S.; Rolland Souza, A.S.; Costa Caminha, M.d.F.; Lins da Silva, S.; Callou Cruz, R.d.S.B.L.; Carvalho dos Santos, C.; Batista Filho, M. Vitamin A and Pregnancy: A Narrative Review. Nutrients 2019, 11, 681. [Google Scholar] [CrossRef]
- McGuire, S. WHO Guideline: Vitamin A Supplementation in Pregnant Women. Geneva: WHO, 2011; WHO Guideline: Vitamin A Supplementation in Postpartum Women. Geneva: WHO. Adv. Nutr. Int. Rev. J. 2012, 3, 215–216. [Google Scholar] [CrossRef] [PubMed]
- Ambalavanan, N.; Tyson, J.E.; Kennedy, K.A.; Hansen, N.I.; Vohr, B.R.; Wright, L.L.; Carlo, W.A. National Institute of Child Health and Human Development Neonatal Research Network. Vitamin A supplementation for extremely low birth weight infants: Outcome at 18 to 22 months. Pediatrics 2005, 115, e249-54. [Google Scholar] [CrossRef] [PubMed]
- Farhangi, M.A.; Keshavarz, S.A.; Eshraghian, M.; Ostadrahimi, A.; Saboor-Yaraghi, A.A. Vitamin a supplementation, serum lipids, liver enzymes and C-reactive protein concentrations in obese women of reproductive age. Ann. Clin. Biochem. Int. J. Lab. Med. 2013, 50, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, S.M.; Sheril, A.; Vajreswari, A. Chronic vitamin A-enriched diet feeding induces body weight gain and adiposity in lean and glucose-intolerant obese rats of WNIN/GR-Ob strain. Exp. Physiol. 2015, 100, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Crandall, C. Vitamin A Intake and Osteoporosis: A Clinical Review. J. Women’s Health 2004, 13, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Feskanich, D.; Singh, V.; Willett, W.C.; Colditz, G. Vitamin A Intake and Hip Fractures Among Postmenopausal Women. JAMA 2002, 287, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.S.; Harnack, L.J.; Lazovich, D.; Folsom, A.R. Vitamin A intake and the risk of hip fracture in postmenopausal women: The Iowa Women?s Health Study. Osteoporos. Int. 2004, 15, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. The Effect of Vitamin E and Beta Carotene on the Incidence of Lung Cancer and Other Cancers in Male Smokers. N. Engl. J. Med. 1994, 330, 1029–1035. [Google Scholar] [CrossRef]
- Ferraro, P.M.; Curhan, G.C.; Gambaro, G.; Taylor, E.N. Total, Dietary, and Supplemental Vitamin C Intake and Risk of Incident Kidney Stones. Am. J. Kidney Dis. 2016, 67, 400–407. [Google Scholar] [CrossRef]
- Jiang, K.; Tang, K.; Liu, H.; Xu, H.; Ye, Z.; Chen, Z. Ascorbic Acid Supplements and Kidney Stones Incidence Among Men and Women: A systematic review and meta-analysis. Urol. J. 2018, 16, 115–120. [Google Scholar]
- Martín-Calvo, N.; Martínez-González, M. Ángel Vitamin C Intake is Inversely Associated with Cardiovascular Mortality in a Cohort of Spanish Graduates: The SUN Project. Nutrients 2017, 9, 954. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.B.; Gambardella, J.; Castellanos, V.; Trimarco, V.; Santulli, G. Vitamin C and Cardiovascular Disease: An Update. Antioxidants 2020, 9, 1227. [Google Scholar] [CrossRef]
- Lee, D.-H.; Folsom, A.R.; Harnack, L.; Halliwell, B.; Jacobs, D.R. Does supplemental vitamin C increase cardiovascular disease risk in women with diabetes? Am. J. Clin. Nutr. 2004, 80, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.M.; Wu, C.B.; Joung, S.J.; Tsai, W.P.; Su, K.Y. Multi-model approach on growth estimation and association with life history trait for elasmobranchs. Front. Mar. Sci. 2021, 8, 591692. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.M., Jr.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the Risk of Prostate Cancer: The selenium and vitamin e cancer prevention trial (SELECT). The selenium and vitamin e cancer prevention trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Kositamongkol, S.; Suthutvoravut, U.; Chongviriyaphan, N.; Feungpean, B.; Nuntnarumit, P. Vitamin A and E status in very low birth weight infants. J. Perinatol. 2011, 31, 471–476. [Google Scholar] [CrossRef]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants Accelerate Lung Cancer Progression in Mice. Sci. Transl. Med. 2014, 6, 221ra15. [Google Scholar] [CrossRef] [PubMed]
- Lippman, S.M.; Klein, E.A.; Goodman, P.J.; Lucia, M.S.; Thompson, I.M.; Ford, L.G.; Parnes, H.L.; Minasian, L.M.; Gaziano, J.M.; Hartline, J.A.; et al. Effect of Selenium and Vitamin E on Risk of Prostate Cancer and Other Cancers: The selenium and vitamin E cancer prevention trial (SELECT). JAMA 2009, 301, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, R.; Gasnier, B.C.H.; Dietrich, J.W.; Krebs, B.; Angstwurm, M.W.A. Selenium Supplementation in Patients with Autoimmune Thyroiditis Decreases Thyroid Peroxidase Antibodies Concentrations. J. Clin. Endocrinol. Metab. 2002, 87, 1687–1691. [Google Scholar] [CrossRef] [PubMed]
- Ventura, M.; Melo, M.; Carrilho, F. Selenium and Thyroid Disease: From Pathophysiology to Treatment. Int. J. Endocrinol. 2017, 2017, 1–9. [Google Scholar] [CrossRef]
- Roozbeh, J.; Hedayati, P.; Sagheb, M.M.; Sharifian, M.; Jahromi, A.H.; Shaabani, S.; Jalaeian, H.; Raeisjalali, G.A.; Behzadi, S. Effect of Zinc Supplementation on Triglyceride, Cholesterol, LDL, and HDL Levels in Zinc-Deficient Hemodialysis Patients. Ren. Fail. 2009, 31, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-S.; Marbán, E. Physiological Levels of Reactive Oxygen Species Are Required to Maintain Genomic Stability in Stem Cells. Stem Cells 2010, 28, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Owusu-Ansah, E.; Banerjee, U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature 2009, 461, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.P.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Atalay, M.; Oksala, N.K.J.; Laaksonen, D.E.; Khanna, S.; Nakao, C.; Lappalainen, J.; Roy, S.; Hänninen, O.; Sen, C.K. Exercise training modulates heat shock protein response in diabetic rats. J. Appl. Physiol. 2004, 97, 605–611. [Google Scholar] [CrossRef]
- Davidov-Pardo, G.; McClements, D.J. Nutraceutical delivery systems: Resveratrol encapsulation in grape seed oil nanoemulsions formed by spontaneous emulsification. Food Chem. 2015, 167, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Gliemann, L.; Nyberg, M.; Hellsten, Y. Effects of exercise training and resveratrol on vascular health in aging. Free Radic. Biol. Med. 2016, 98, 165–176. [Google Scholar] [CrossRef]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A Double-Edged Sword in Health Benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.-C.; Lee, C.-H.; Song, T.-Y. Evaluation of Resveratrol Oxidationin Vitroand the Crucial Role of Bicarbonate Ions. Biosci. Biotechnol. Biochem. 2010, 74, 63–68. [Google Scholar] [CrossRef]
- Rüweler, M.; Gülden, M.; Maser, E.; Murias, M.; Seibert, H. Cytotoxic, cytoprotective and antioxidant activities of resveratrol and analogues in C6 astroglioma cells in vitro. Chem. Interact. 2009, 182, 128–135. [Google Scholar] [CrossRef]
- Gehm, B.D.; McAndrews, J.M.; Chien, P.-Y.; Jameson, J.L. Resveratrol, a polyphenolic compound found in grapes and wine, is an agonist for the estrogen receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 14138–14143. [Google Scholar] [CrossRef] [PubMed]
- Ferry-Dumazet, H.; Garnier, O.; Mamani-Matsuda, M.; Vercauteren, J.; Belloc, F.; Billiard, C.; Dupouy, M.; Thiolat, D.; Kolb, J.P.; Marit, G.; et al. Resveratrol inhibits the growth and induces the apoptosis of both normal and leukemic hematopoietic cells. Carcinogenesis 2002, 23, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Baron, S.; Bedarida, T.; Cottart, C.-H.; Vibert, F.; Vessieres, E.; Ayer, A.; Henrion, D.; Hommeril, B.; Paul, J.-L.; Renault, G.; et al. Dual Effects of Resveratrol on Arterial Damage Induced by Insulin Resistance in Aged Mice. J. Gerontol. Ser. A 2013, 69A, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Moini, H.; Packer, L.; Saris, N.-E.L. Antioxidant and Prooxidant Activities of α-Lipoic Acid and Dihydrolipoic Acid. Toxicol. Appl. Pharmacol. 2002, 182, 84–90. [Google Scholar] [CrossRef]
- Han, D.; Handelman, G.; Marcocci, L.; Sen, C.K.; Roy, S.; Kobuchi, H.; Tritschler, H.J.; Flohé, L.; Packer, L. Lipoic acid increasesde novosynthesis of cellular glutathione by improving cystine utilization. BioFactors 1997, 6, 321–338. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Yoshizawa, K.; Kaido, Y.; Takenouchi, A.; Terao, K.; Yasui, H.; Yoshikawa, Y. Exercise Performance Upregulatory Effect of R-α-Lipoic Acid with γ-Cyclodextrin. Nutriens 2021, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Marangon, K.; Devaraj, S.; Tirosh, O.; Packer, L.; Jialal, I. Comparison of the effect of α-lipoic acid and α-tocopherol supplementation on measures of oxidative stress. Free Radic. Biol. Med. 1999, 27, 1114–1121. [Google Scholar] [CrossRef]
- Suh, J.H.; Shigeno, E.T.; Morrow, J.D.; Cox, J.B.; Rocha, A.E.; Frei, B.; Hagen, T.M. Oxidative stress in the aging rat heart is reversed by dietary supplementation with (R)-α-lipoic acid. FASEB J. 2001, 15, 700–706. [Google Scholar] [CrossRef]
- Çakatay, U.; Kayaliı, R. Plasma protein oxidation in aging rats after alpha-lipoic acid administration. Biogerontology 2005, 6, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.-R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria Are Required for Antigen-Specific T Cell Activation through Reactive Oxygen Species Signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef]
- Kamiński, M.M.; Röth, D.; Krammer, P.H.; Gülow, K. Mitochondria as Oxidative Signaling Organelles in T-cell Activation: Physiological Role and Pathological Implications. Arch. Immunol. Ther. Exp. 2013, 61, 367–384. [Google Scholar] [CrossRef] [PubMed]
- Chávez, M.D.; Tse, H.M. Targeting Mitochondrial-Derived Reactive Oxygen Species in T Cell-Mediated Autoimmune Diseases. Front. Immunol. 2021, 12, 703972. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Liu, H.; Ge, B. Innate immunity in tuberculosis: Host defense vs pathogen evasion. Cell. Mol. Immunol. 2017, 14, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Garaude, J.; Acín-Pérez, R.; Martínez-Cano, S.; Enamorado, M.; Ugolini, M.; Nistal-Villán, E.; Hervás-Stubbs, S.; Pelegrín, P.; Sander, L.E.; Enríquez, J.A.; et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat. Immunol. 2016, 17, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.-Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef]
- Roca, F.J.; Whitworth, L.J.; Redmond, S.; Jones, A.A.; Ramakrishnan, L. TNF Induces Pathogenic Programmed Macrophage Necrosis in Tuberculosis through a Mitochondrial-Lysosomal-Endoplasmic Reticulum Circuit. Cell 2019, 178, 1344–1361.e11. [Google Scholar] [CrossRef] [PubMed]
- Kamiński, M.M.; Kiessling, M.; Süss, D.; Krammer, P.H.; Gülow, K. Novel Role for Mitochondria: Protein Kinase C-Dependent Oxidative Signaling Organelles in Activation-Induced T-Cell Death. Mol. Cell. Biol. 2007, 27, 3625–3639. [Google Scholar] [CrossRef]
- Hohn, D.C.; I Lehrer, R. NADPH oxidase deficiency in X-linked chronic granulomatous disease. J. Clin. Investig. 1975, 55, 707–713. [Google Scholar] [CrossRef]
- Ben-Ari, J.; Wolach, O.; Gavrieli, R.; Wolach, B. Infections associated with chronic granulomatous disease: Linking genetics to phenotypic expression. Expert Rev. Anti-Infective Ther. 2012, 10, 881–894. [Google Scholar] [CrossRef]
- Chin, W.-Y.; He, C.-Y.; Chow, T.W.; Yu, Q.-Y.; Lai, L.-C.; Miaw, S.-C. Adenylate Kinase 4 Promotes Inflammatory Gene Expression via Hif1α and AMPK in Macrophages. Front. Immunol. 2021, 12, 630318. [Google Scholar] [CrossRef]
- Bylund, J.; Brown, K.L.; Movitz, C.; Dahlgren, C.; Karlsson, A. Intracellular generation of superoxide by the phagocyte NADPH oxidase: How, where, and what for? Free. Radic. Biol. Med. 2010, 49, 1834–1845. [Google Scholar] [CrossRef] [PubMed]
- Dupré-Crochet, S.; Erard, M.; Nüβe, O. ROS production in phagocytes: Why, when, and where? J. Leukoc. Biol. 2013, 94, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Angelini, G.; Gardella, S.; Ardy, M.; Ciriolo, M.R.; Filomeni, G.; Di Trapani, G.; Clarke, F.; Sitia, R.; Rubartelli, A. Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc. Natl. Acad. Sci. USA 2002, 99, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Mak, T.W.; Grusdat, M.; Duncan, G.S.; Dostert, C.; Nonnenmacher, Y.; Cox, M.; Binsfeld, C.; Hao, Z.; Bruestle, A.; Itsumi, M.; et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunology 2017, 46, 675–689. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Pérez, P.S.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Louzada, R.A.N.; Bouviere, J.; Matta, L.P.; de Castro, J.P.S.W.; Dupuy, C.; de Carvalho, D.P.; Fortunato, R.S. Redox Signaling in Widespread Health Benefits of Exercise. Antioxid. Redox Signal. 2020, 33, 745–760. [Google Scholar] [CrossRef] [PubMed]
- Wan, T.; Zhou, X.; Chen, G.; An, H.; Chen, T.; Zhang, W.; Liu, S.; Jiang, Y.; Yang, F.; Wu, Y.; et al. Novel heat shock protein Hsp70L1 activates dendritic cells and acts as a Th1 polarizing adjuvant. Blood 2004, 103, 1747–1754. [Google Scholar] [CrossRef] [PubMed]
- Kurashova, N.A.; Madaeva, I.M.; Kolesnikova, L.I. Expression of HSP70 Heat-Shock Proteins under Oxidative Stress. Adv. Gerontol. 2020, 10, 20–25. [Google Scholar] [CrossRef]
- Taherkhani, S.; Suzuki, K.; Castell, L. A Short Overview of Changes in Inflammatory Cytokines and Oxidative Stress in Response to Physical Activity and Antioxidant Supplementation. Antioxidants 2020, 9, 886. [Google Scholar] [CrossRef]
- Reshi, L.; Su, Y.-C.; Hong, J.-R. RNA Viruses: ROS-Mediated Cell Death. Int. J. Cell Biol. 2014, 2014, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Cook, N.R.; Albert, C.; Zaharris, E.; Gaziano, J.M.; Van Denburgh, M.; Buring, J.E.; Manson, J.E. Vitamins C and E and Beta Carotene Supplementation and Cancer Risk: A Randomized Controlled Trial. J. Natl. Cancer Inst. 2008, 101, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxidative Med. Cell. Longev. 2019, 2019, 5381692. [Google Scholar] [CrossRef] [PubMed]
- Van Loenhout, J.; Peeters, M.; Bogaerts, A.; Smits, E.; Deben, C. Oxidative Stress-Inducing Anticancer Therapies: Taking a Closer Look at Their Immunomodulating Effects. Antioxidants 2020, 9, 1188. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, Y.; Watanabe, S.; Kizaki, T.; Sakurai, T.; Takemasa, T.; Haga, S.; Ookawara, T.; Suzuki, K.; Ohno, H. Acute exercise increases expression of extracellular superoxide dismutase in skeletal muscle and the aorta. Redox Rep. 2008, 13, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Dodd, S.L.; Bs, B.J.G.; Senf, S.M.; Hain, B.; Judge, S. Ros-mediated activation of NF-κB and Foxo during muscle disuse. Muscle Nerve 2009, 41, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Derbre, F.; Ferrando, B.; Gomez-Cabrera, M.C.; Sanchis-Gomar, F.; Martinez-Bello, V.E.; Olaso-Gonzalez, G.; Diaz, A.; Gratas-Delamarche, A.; Cerda, M.; Viña, J. Inhibition of Xanthine Oxidase by Allopurinol Prevents Skeletal Muscle Atrophy: Role of p38 MAPKinase and E3 Ubiquitin Ligases. PLoS ONE 2012, 7, e46668. [Google Scholar] [CrossRef] [PubMed]
- Kröller-Schön, S.; Jansen, T.; Hauptmann, F.; Schüler, A.; Heeren, T.; Hausding, M.; Oelze, M.; Viollet, B.; KeaneyJr, J.F.; Wenzel, P.; et al. α1AMP-Activated Protein Kinase Mediates Vascular Protective Effects of Exercise. Arter. Thromb. Vasc. Biol. 2012, 32, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, A.; Leiva, A.; Peña, M.; Müller, M.; Debandi, A.; Hidalgo, C.; Carrasco, M.A.; Jaimovich, E. Myotube depolarization generates reactive oxygen species through NAD(P)H oxidase; ROS-elicited Ca2+ stimulates ERK, CREB, early genes. J. Cell. Physiol. 2006, 209, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, C.; Sánchez, G.; Barrientos, G.; Aracena-Parks, P. A Transverse Tubule NADPH Oxidase Activity Stimulates Calcium Release from Isolated Triads via Ryanodine Receptor Type 1 S -Glutathionylation. J. Biol. Chem. 2006, 281, 26473–26482. [Google Scholar] [CrossRef] [PubMed]
- Tsatmali, M.; Walcott, E.C.; Crossin, K.L. Newborn neurons acquire high levels of reactive oxygen species and increased mitochondrial proteins upon differentiation from progenitors. Brain Res. 2005, 1040, 137–150. [Google Scholar] [CrossRef]
- Tsatmali, M.; Walcott, E.C.; Makarenkova, H.; Crossin, K.L. Reactive oxygen species modulate the differentiation of neurons in clonal cortical cultures. Mol. Cell. Neurosci. 2006, 33, 345–357. [Google Scholar] [CrossRef]
- Hu, D.; Cao, P.; Thiels, E.; Chu, C.T.; Wu, G.-Y.; Oury, T.D.; Klann, E. Hippocampal long-term potentiation, memory, and longevity in mice that overexpress mitochondrial superoxide dismutase. Neurobiol. Learn. Mem. 2007, 87, 372–384. [Google Scholar] [CrossRef]
- Kishida, K.T.; Hoeffer, C.A.; Hu, D.; Pao, M.; Holland, S.M.; Klann, E. Synaptic Plasticity Deficits and Mild Memory Impairments in Mouse Models of Chronic Granulomatous Disease. Mol. Cell. Biol. 2006, 26, 5908–5920. [Google Scholar] [CrossRef] [PubMed]
- Massaad, C.A.; Washington, T.M.; Pautler, R.G.; Klann, E. Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 13576–13581. [Google Scholar] [CrossRef] [PubMed]
- Mursaleen, L.; Noble, B.; Chan, S.; Somavarapu, S.; Zariwala, M. N-Acetylcysteine Nanocarriers Protect against Oxidative Stress in a Cellular Model of Parkinson’s Disease. Antioxidants 2020, 9, 600. [Google Scholar] [CrossRef] [PubMed]
- Percario, S.; da Silva Barbosa, A.; Varela, E.L.P.; Gomes, A.R.Q.; Ferreira, M.E.S.; de Nazare Araujo Moreira, T.; Dolabela, M.F. Oxidative Stress in Parkinson’s Disease: Potential Benefits of Antioxidant Supplementation. Oxidative Med. Cell. Longev. 2020, 2020, 1–23. [Google Scholar] [CrossRef]
- Chang, K.-H.; Chen, C.-M. The Role of Oxidative Stress in Parkinson’s Disease. Antioxidants 2020, 9, 597. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Li, Z.; Xu, X.; Leng, X.; He, M.; Wang, J.; Cheng, S.; Wu, H. Roles of reactive oxygen species in cell signaling pathways and immune responses to viral infections. Arch. Virol. 2016, 162, 603–610. [Google Scholar] [CrossRef]
- D’Aiuto, F.; Nibali, L.; Parkar, M.; Patel, K.; Suvan, J.; Donos, N. Oxidative Stress, Systemic Inflammation, and Severe Periodontitis. J. Dent. Res. 2010, 89, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Tucker, P.S.; Scanlan, A.; Dalbo, V.J. Chronic Kidney Disease Influences Multiple Systems: Describing the Relationship between Oxidative Stress, Inflammation, Kidney Damage, and Concomitant Disease. Oxidative Med. Cell. Longev. 2015, 2015, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Piechota-Polanczyk, A.; Fichna, J. Review article: The role of oxidative stress in pathogenesis and treatment of inflammatory bowel diseases. Naunyn-Schmiedebergs Arch. Exp. Pathol. Pharmakol. 2014, 387, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-J.; Yoo, S.-A.; Kim, W.-U. Role of Endoplasmic Reticulum Stress in Rheumatoid Arthritis Pathogenesis. J. Korean Med. Sci. 2014, 29, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, M.; Werner, S. Oxidative stress in normal and impaired wound repair. Pharmacol. Res. 2008, 58, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Jaishankar, G.B.; Saleh, H.; Jithpratuck, W.; Sahni, R.; Krishnaswamy, G. Chronic granulomatous disease: A review of the infectious and inflammatory complications. Clin. Mol. Allergy 2011, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Siti, H.N.; Kamisah, Y.; Kamsiah, J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vasc. Pharmacol. 2015, 71, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Lacy, F.; Kailasam, M.T.; O’Connor, D.T.; Schmid-Schönbein, G.W.; Parmer, R.J. Plasma Hydrogen Peroxide Production in Human Essential Hypertension. Hypertension 2000, 36, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Tanito, M.; Nakamura, H.; Kwon, Y.-W.; Teratani, A.; Masutani, H.; Shioji, K.; Kishimoto, C.; Ohira, A.; Horie, R.; Yodoi, J. Enhanced Oxidative Stress and Impaired Thioredoxin Expression in Spontaneously Hypertensive Rats. Antioxid. Redox Signal. 2004, 6, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Z. Increased Oxidative Stress as a Selective Anticancer Therapy. Oxidative Med. Cell. Longev. 2015, 2015, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.-W.; Chen, Y.-A.; Ho, S.-Y.; Wang, Y.-J. Arsenic Trioxide Enhances the Radiation Sensitivity of Androgen-Dependent and -Independent Human Prostate Cancer Cells. PLoS ONE 2012, 7, e31579. [Google Scholar] [CrossRef] [PubMed]
- Sobhakumari, A.; Love-Homan, L.; Fletcher, E.V.M.; Martin, S.M.; Parsons, A.D.; Spitz, D.R.; Knudson, C.M.; Simons, A.L. Susceptibility of Human Head and Neck Cancer Cells to Combined Inhibition of Glutathione and Thioredoxin Metabolism. PLoS ONE 2012, 7, e48175. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jiang, H.; Van De Gucht, M.; De Ridder, M. Hypoxic Radioresistance: Can ROS Be the Key to Overcome It? Cancers 2019, 11, 112. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Kamendulis, L.M.; Hocevar, B.A. Oxidative Stress and Oxidative Damage in Carcinogenesis. Toxicol. Pathol. 2010, 38, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Salomon, D.S.; Brandt, R.; Ciardiello, F.; Normanno, N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit. Rev. Oncol. 1995, 19, 183–232. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Rhee, D.B.; Ghosh, A.; Lu, J.; Bohr, V.A.; Liu, Y. Factors that influence telomeric oxidative base damage and repair by DNA glycosylase OGG. DNA Repair 2011, 10, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Aeby, E.; Ahmed, W.; Redon, S.; Simanis, V.; Lingner, J. Peroxiredoxin 1 Protects Telomeres from Oxidative Damage and Preserves Telomeric DNA for Extension by Telomerase. Cell Rep. 2016, 17, 3107–3114. [Google Scholar] [CrossRef]
- Kurz, D.J.; Decary, S.; Hong, Y.; Trivier, E.; Akhmedov, A.; Erusalimsky, J.D. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J. Cell Sci. 2004, 117, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Fouquerel, E.; Barnes, R.; Uttam, S.; Watkins, S.; Bruchez, M.P.; Opresko, P.L. Targeted and Persistent 8-Oxoguanine Base Damage at Telomeres Promotes Telomere Loss and Crisis. Mol. Cell 2019, 75, 117–130.e6. [Google Scholar] [CrossRef] [PubMed]
- Gavia-García, G.; Rosado-Pérez, J.; Arista-Ugalde, T.; Aguiñiga-Sánchez, I.; Santiago-Osorio, E.; Mendoza-Núñez, V. Telomere Length and Oxidative Stress and Its Relation with Metabolic Syndrome Components in the Aging. Biology 2021, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Barnes, R.P.; Fouquerel, E.; Opresko, P.L. The impact of oxidative DNA damage and stress on telomere homeostasis. Mech. Ageing Dev. 2019, 177, 37–45. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).