
Hydrogen Peroxide and Amyotrophic Lateral Sclerosis: From Biochemistry to Pathophysiology



Abstract
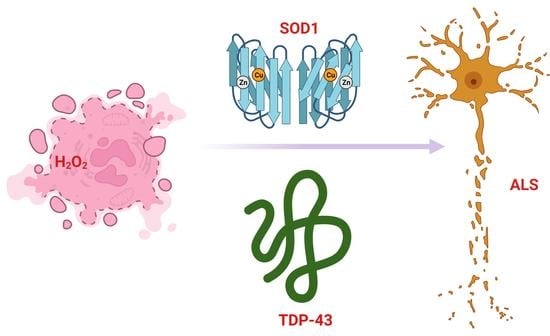
{kind=link}
{kind=link}
{kind=link}
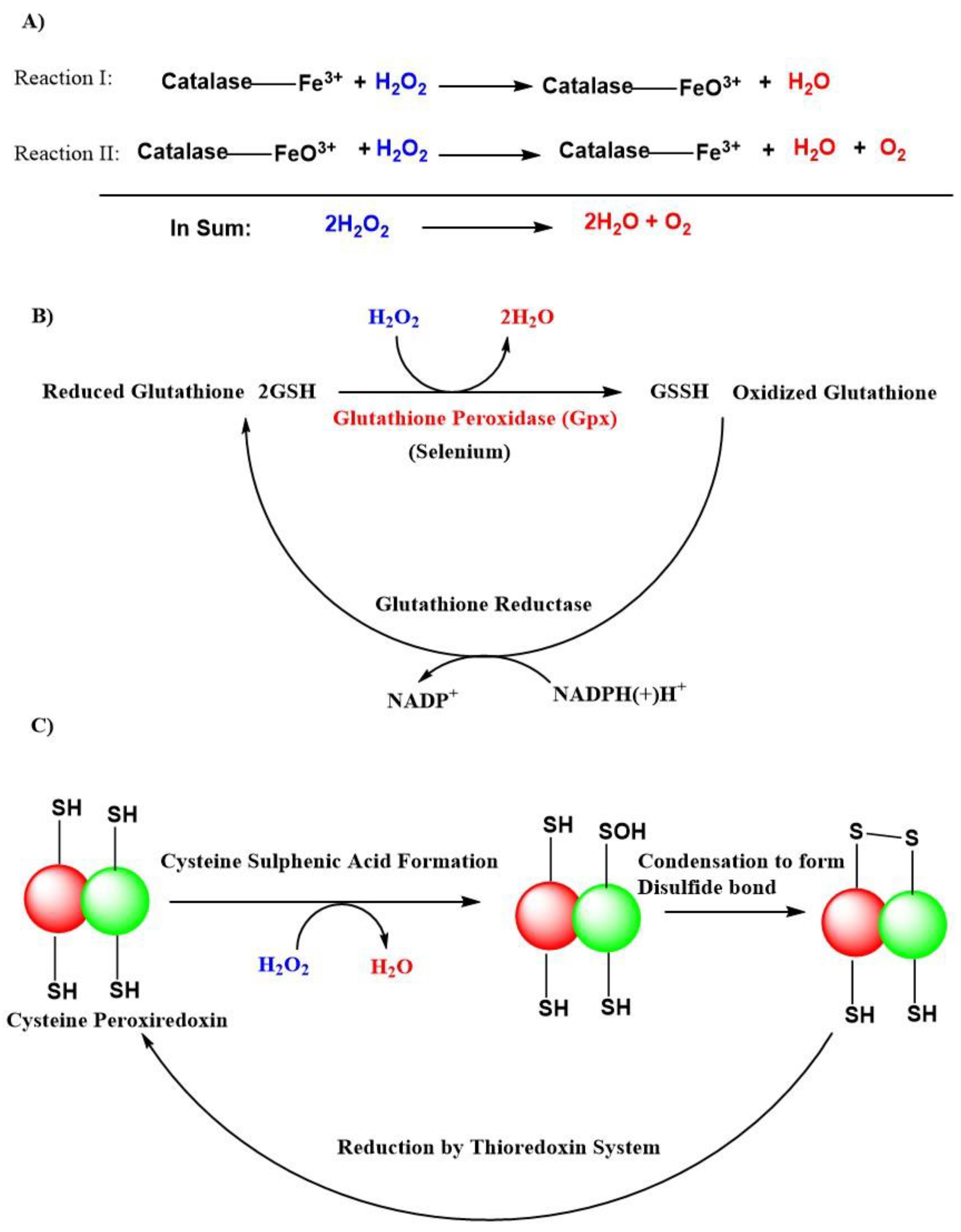{kind=link}
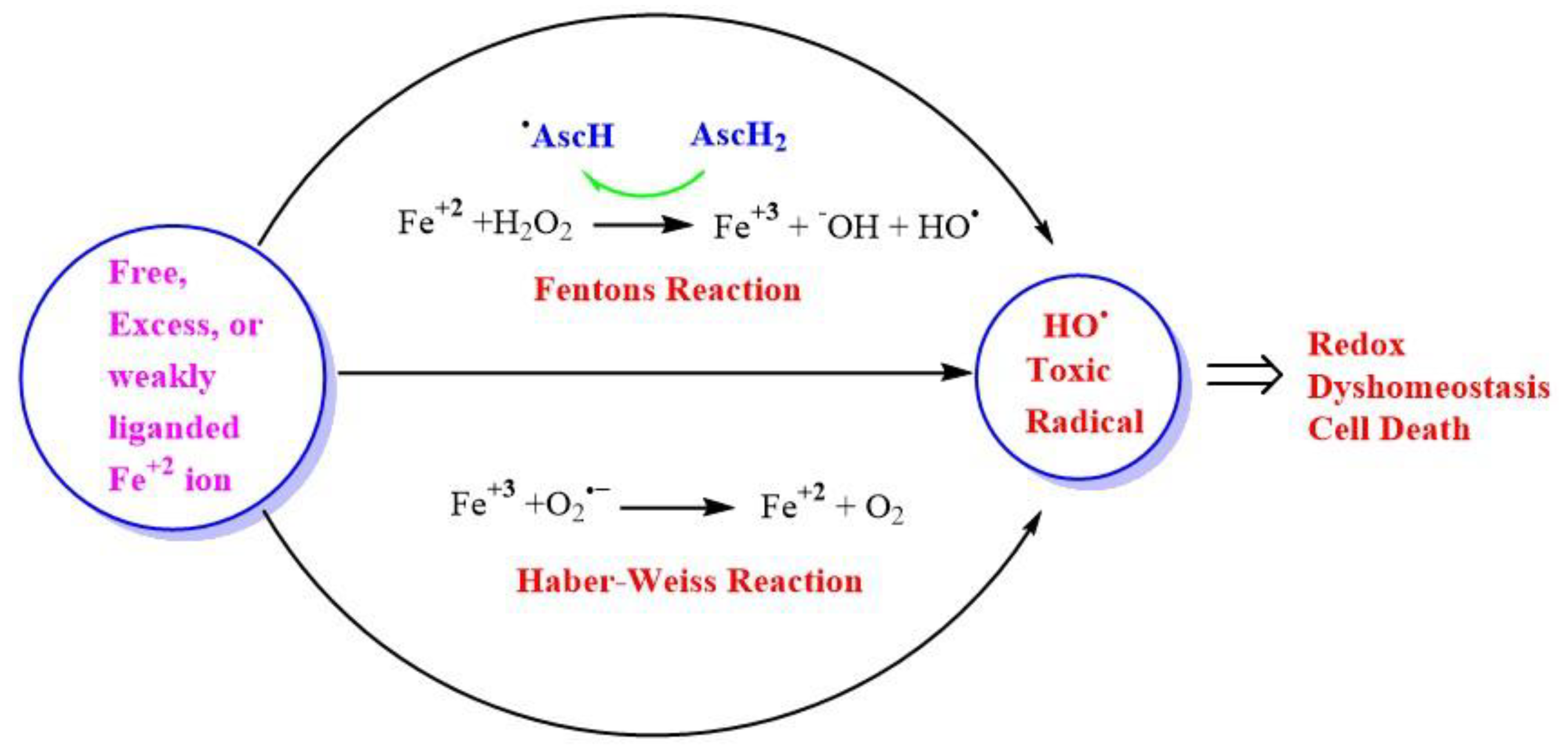{kind=link}
{kind=link}
{kind=link}
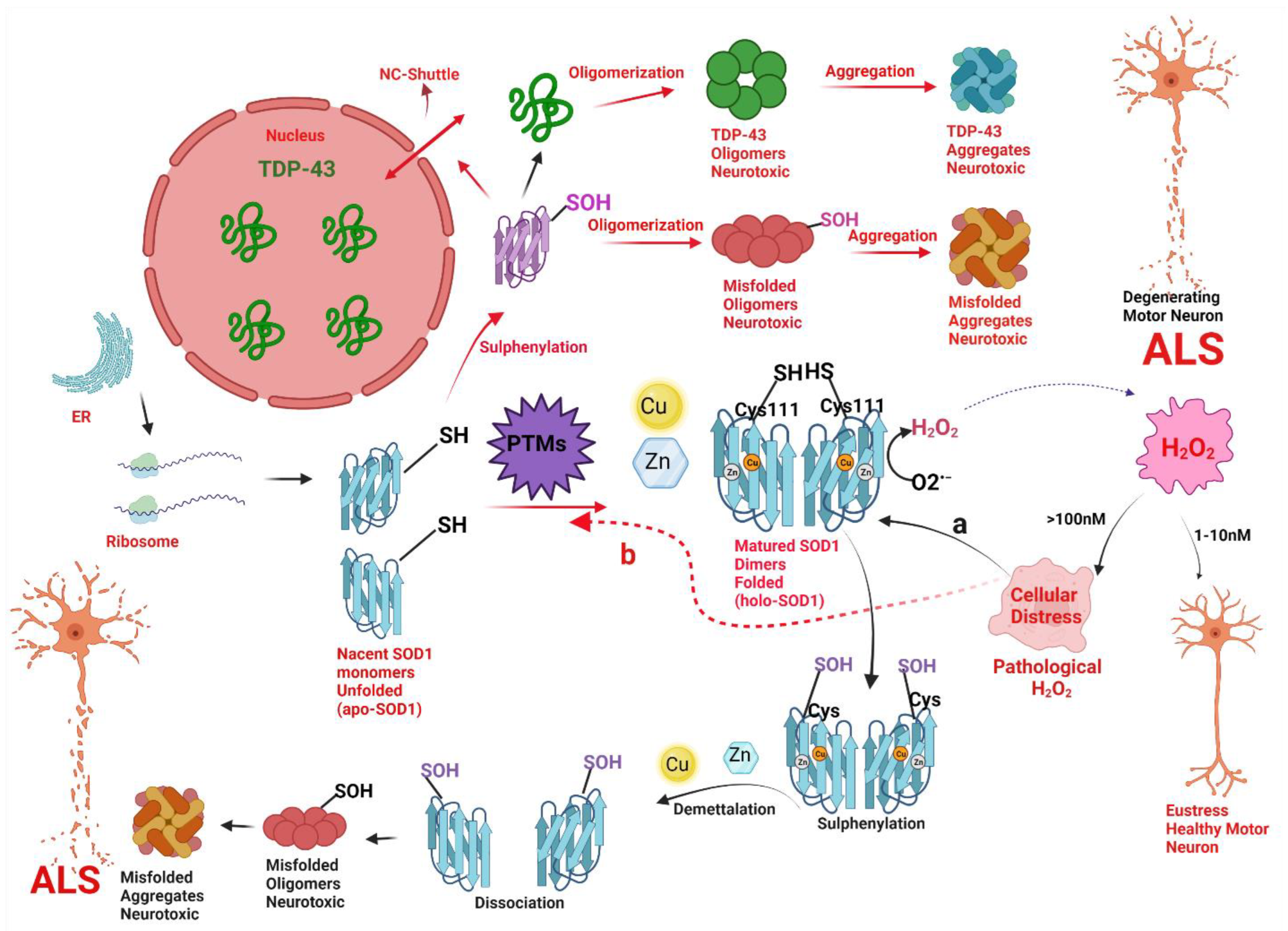{kind=link}
1. Introduction
2. Bio-Reactive Oxygen Species in Living Cells
3. The Chemistry of Hydrogen Peroxide
3.1. Hydrogen Peroxide as Double Edge Sword in Living Cells
3.2. Metabolic Sources and Sinks of Hydrogen Peroxide
3.3. Generation of Highly Unstable and Reactive Free Radical Species, the HO• from H2O2
4. Amyotrophic Lateral Sclerosis
5. Cu, Zn Superoxide Dismutase Enzyme
6. Role of H2O2 in Misfolding of Cu, Zn SOD1
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef]
- Barber, S.C.; Mead, R.J.; Shaw, P.J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 2006, 1762, 1051–1067. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef]
- Olanow, C.W. An introduction to the free radical hypothesis in Parkinson’s disease. Ann. Neurol. 1992, 32, S2–S9. [Google Scholar] [CrossRef] [PubMed]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. Adv. Exp. Med. Biol. 2018, 1049, 59–83. [Google Scholar] [PubMed]
- Simpson, C.L.; Al-Chalabi, A. Amyotrophic lateral sclerosis as a complex genetic disease. Biochim. Biophys. Acta 2006, 1762, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Paré, B.; Lehmann, M.; Beaudin, M.; Nordström, U.; Saikali, S.; Julien, J.P.; Gilthorpe, J.D.; Marklund, S.L.; Cashman, N.R.; Andersen, P.M.; et al. Misfolded SOD1 pathology in sporadic Amyotrophic Lateral Sclerosis. Sci. Rep. 2018, 8, 14223. [Google Scholar] [CrossRef] [PubMed]
- Bunton-Stasyshyn, R.K.; Saccon, R.A.; Fratta, P.; Fisher, E.M. SOD1 Function and Its Implications for Amyotrophic Lateral Sclerosis Pathology: New and Renascent Themes. Neuroscientist 2015, 21, 519–529. [Google Scholar] [CrossRef]
- Saccon, R.A.; Bunton-Stasyshyn, R.K.; Fisher, E.M.; Fratta, P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 2013, 136 Pt 8, 2342–2358. [Google Scholar] [CrossRef] [PubMed]
- Rakhit, R.; Chakrabartty, A. Structure, folding, and misfolding of Cu,Zn superoxide dismutase in amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 1025–1037. [Google Scholar] [CrossRef]
- Rakhit, R.; Cunningham, P.; Furtos-Matei, A.; Dahan, S.; Qi, X.F.; Crow, J.P.; Cashman, N.R.; Kondejewski, L.H.; Chakrabartty, A. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J. Biol. Chem. 2002, 277, 47551–47556. [Google Scholar] [CrossRef] [PubMed]
- Carrì, M.T.; Valle, C.; Bozzo, F.; Cozzolino, M. Oxidative stress and mitochondrial damage: Importance in non-SOD1 ALS. Front. Cell. Neurosci. 2015, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Martins, D.; English, A.M. SOD1 oxidation and formation of soluble aggregates in yeast: Relevance to sporadic ALS development. Redox Biol. 2014, 2, 632–639. [Google Scholar] [CrossRef]
- Furukawa, Y. Pathological roles of wild-type cu, zn-superoxide dismutase in amyotrophic lateral sclerosis. Neurol. Res. Int. 2012, 2012, 323261. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.; Daura, X.; Zagrovic, B. Effect of Oxidative Damage on the Stability and Dimerization of Superoxide Dismutase 1. Biophys. J. 2016, 110, 1499–1509. [Google Scholar] [CrossRef]
- Gill, C.; Phelan, J.P.; Hatzipetros, T.; Kidd, J.D.; Tassinari, V.R.; Levine, B.; Wang, M.Z.; Moreno, A.; Thompson, K.; Maier, M.; et al. SOD1-positive aggregate accumulation in the CNS predicts slower disease progression and increased longevity in a mutant SOD1 mouse model of ALS. Sci. Rep. 2019, 9, 6724. [Google Scholar] [CrossRef]
- Benkler, C.; O’Neil, A.L.; Slepian, S.; Qian, F.; Weinreb, P.H.; Rubin, L.L. Aggregated SOD1 causes selective death of cultured human motor neurons. Sci. Rep. 2018, 8, 16393. [Google Scholar] [CrossRef]
- Bourassa, M.W.; Brown, H.H.; Borchelt, D.R.; Vogt, S.; Miller, L.M. Metal-deficient aggregates and diminished copper found in cells expressing SOD1 mutations that cause ALS. Front. Aging Neurosci. 2014, 6, 110. [Google Scholar] [CrossRef]
- Brasil, A.A.; de Carvalho, M.D.C.; Gerhardt, E.; Queiroz, D.D.; Pereira, M.D.; Outeiro, T.F.; Eleutherio, E.C.A. Characterization of the activity, aggregation, and toxicity of heterodimers of WT and ALS-associated mutant Sod1. Proc. Natl. Acad. Sci. USA 2019, 116, 25991–26000. [Google Scholar] [CrossRef]
- Xu, W.C.; Liang, J.Z.; Li, C.; He, Z.X.; Yuan, H.Y.; Huang, B.Y.; Liu, X.-L.; Tang, B.; Pang, D.-W.; Du, H.-N.; et al. Pathological hydrogen peroxide triggers the fibrillization of wild-type SOD1 via sulfenic acid modification of Cys-111. Cell Death Dis. 2018, 9, 67. [Google Scholar] [CrossRef]
- Mulligan, V.K.; Kerman, A.; Laister, R.C.; Sharda, P.R.; Arslan, P.E.; Chakrabartty, A. Early steps in oxidation-induced SOD1 misfolding: Implications for non-amyloid protein aggregation in familial ALS. J. Mol. Biol. 2012, 421, 631–652. [Google Scholar] [CrossRef]
- Yim, M.B.; Chock, P.B.; Stadtman, E.R. Copper, zinc superoxide dismutase catalyzes hydroxyl radical production from hydrogen peroxide. Proc. Natl. Acad. Sci. USA 1990, 87, 5006–5010. [Google Scholar] [CrossRef]
- Goto, J.J.; Gralla, E.B.; Valentine, J.S.; Cabelli, D.E. Reactions of hydrogen peroxide with familial amyotrophic lateral sclerosis mutant human copper-zinc superoxide dismutases studied by pulse radiolysis. J. Biol. Chem. 1998, 273, 30104–30109. [Google Scholar] [CrossRef]
- Valentine, J.S.; Hart, P.J. Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 3617–3622. [Google Scholar] [CrossRef]
- Jeon, G.S.; Shim, Y.M.; Lee, D.Y.; Kim, J.S.; Kang, M.; Ahn, S.H.; Shin, J.-Y.; Geum, D.; Hong, Y.-H.; Sung, J.-J. Pathological Modification of TDP-43 in Amyotrophic Lateral Sclerosis with SOD1 Mutations. Mol. Neurobiol. 2019, 56, 2007–2021. [Google Scholar] [CrossRef] [PubMed]
- Suk, T.R.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 45. [Google Scholar] [CrossRef] [PubMed]
- Volkening, K.; Leystra-Lantz, C.; Yang, W.; Jaffee, H.; Strong, M.J. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res. 2009, 1305, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Marusawa, H.; Ichikawa, K.; Narita, N.; Murakami, H.; Ito, K.; Tezuka, T. Hydroxyl radical as a strong electrophilic species. Bioorganic Med. Chem. 2002, 10, 2283–2290. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef] [PubMed]
- Southorn, P.A.; Powis, G. Free radicals in medicine. I. Chemical nature and biologic reactions. Mayo Clin. Proc. 1988, 63, 381–389. [Google Scholar] [CrossRef]
- Shields, H.J.; Traa, A.; Van Raamsdonk, J.M. Beneficial and Detrimental Effects of Reactive Oxygen Species on Lifespan: A Comprehensive Review of Comparative and Experimental Studies. Front. Cell Dev. Biol. 2021, 9, 628157. [Google Scholar] [CrossRef]
- Borden, W.T.; Hoffmann, R.; Stuyver, T.; Chen, B. Dioxygen: What Makes This Triplet Diradical Kinetically Persistent? J. Am. Chem. Soc. 2017, 139, 9010–9018. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M. The evolution of free radicals and oxidative stress. Am. J. Med. 2000, 108, 652–659. [Google Scholar] [CrossRef]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Linnane, A.W.; Kios, M.; Vitetta, L. Coenzyme Q(10)–its role as a prooxidant in the formation of superoxide anion/hydrogen peroxide and the regulation of the metabolome. Mitochondrion 2007, 7, S51–S61. [Google Scholar] [CrossRef]
- Beyer, R.E. An analysis of the role of coenzyme Q in free radical generation and as an antioxidant. Biochem. Cell Biol. 1992, 70, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, E.; Ivanova, D.; Zhelev, Z.; Bakalova, R.; Gulubova, M.; Aoki, I. Mitochondrial Dysfunction and Redox Imbalance as a Diagnostic Marker of “Free Radical Diseases”. Anticancer Res. 2017, 37, 5373–5381. [Google Scholar]
- Ježek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Reis, A.; Spickett, C.M. Chemistry of phospholipid oxidation. Biochim. Biophys. Acta 2012, 1818, 2374–2387. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Baba, S.P.; Bhatnagar, A. Role of Thiols in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 133–139. [Google Scholar] [CrossRef]
- Chung, H.S.; Wang, S.B.; Venkatraman, V.; Murray, C.I.; Van Eyk, J.E. Cysteine oxidative posttranslational modifications: Emerging regulation in the cardiovascular system. Circ. Res. 2013, 112, 382–392. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Carroll, K.S. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef]
- Arnold, W.P.; Mittal, C.K.; Katsuki, S.; Murad, F. Nitric oxide activates guanylate cyclase and increases guanosine 3’:5’-cyclic monophosphate levels in various tissue preparations. Proc. Natl. Acad. Sci. USA 1977, 74, 3203–3207. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Moreno, J.J.; Pryor, W.A.; Ischiropoulos, H.; Beckman, J.S. Peroxynitrite, a cloaked oxidant formed by nitric oxide and superoxide. Chem. Res. Toxicol. 1992, 5, 834–842. [Google Scholar] [CrossRef]
- Bandookwala, M.; Sengupta, P. 3-Nitrotyrosine: A versatile oxidative stress biomarker for major neurodegenerative diseases. Int. J. Neurosci. 2020, 130, 1047–1062. [Google Scholar] [CrossRef]
- Beal, M.F.; Ferrante, R.J.; Browne, S.E.; Matthews, R.T.; Kowall, N.W.; Brown, R.H., Jr. Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann. Neurol. 1997, 42, 644–654. [Google Scholar] [CrossRef]
- Fernández-Espejo, E.; Rodríguez de Fonseca, F.; Suárez, J.; Tolosa, E.; Vilas, D.; Aldecoa, I.; Berenguer, J.; Damas-Hermoso, F. Native α-Synuclein, 3-Nitrotyrosine Proteins, and Patterns of Nitro-α-Synuclein-Immunoreactive Inclusions in Saliva and Submandibulary Gland in Parkinson’s Disease. Antioxidants 2021, 10, 715. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Role of metabolic H2O2 generation: Redox signaling and oxidative stress. J. Biol. Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Teong, S.P.; Li, X.; Zhang, Y. Hydrogen peroxide as an oxidant in biomass-to-chemical processes of industrial interest. Green Chem. 2019, 21, 5753–5780. [Google Scholar] [CrossRef]
- Boateng, M.K.; Price, S.L.; Huddersman, K.D.; Walsh, S.E. Antimicrobial activities of hydrogen peroxide and its activation by a novel heterogeneous Fenton’s-like modified PAN catalyst. J. Appl. Microbiol. 2011, 111, 1533–1543. [Google Scholar] [CrossRef]
- Goti, A.; Cardona, F. (Eds.) Hydrogen Peroxide in Green Oxidation Reactions: Recent Catalytic Processes; Springer: Dordrecht, The Netherlands, 2008. [Google Scholar]
- Elango, M.; Parthasarathi, R.; Subramanian, V.; Ramachandran, C.N.; Sathyamurthy, N. Hydrogen peroxide clusters: The role of open book motif in cage and helical structures. J. Phys. Chem. A 2006, 110, 6294–6300. [Google Scholar] [CrossRef]
- Engdahl, A.; Nelander, B.; Karlström, G. A matrix isolation and ab initio study of the hydrogen peroxide dimer. J. Phys. Chem. A 2001, 105, 8393–8398. [Google Scholar] [CrossRef]
- Winterbourn, C.C. The biological chemistry of hydrogen peroxide. Methods Enzymol. 2013, 528, 3–25. [Google Scholar]
- Halliwell, B. Free radicals and antioxidants: Updating a personal view. Nutr. Rev. 2012, 70, 257–265. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Comparative reactivities of various biological compounds with myeloperoxidase-hydrogen peroxide-chloride, and similarity of the oxidant to hypochlorite. Biochim. Biophys. Acta 1985, 840, 204–210. [Google Scholar] [CrossRef]
- Prütz, W.A. Hypochlorous acid interactions with thiols, nucleotides, DNA, and other biological substrates. Arch. Biochem. Biophys. 1996, 332, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef]
- Ischiropoulos, H.; Zhu, L.; Chen, J.; Tsai, M.; Martin, J.C.; Smith, C.D.; Beckman, J.S. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch. Biochem. Biophys. 1992, 298, 431–437. [Google Scholar] [CrossRef]
- Douki, T.; Cadet, J. Peroxynitrite mediated oxidation of purine bases of nucleosides and isolated DNA. Free Radic. Res. 1996, 24, 369–380. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef]
- Schöpfer, F.; Riobó, N.; Carreras, M.C.; Alvarez, B.; Radi, R.; Boveris, A.; Radi, R.; Boveris, A.; Cadenas, E.; Poderoso, J.J. Oxidation of ubiquinol by peroxynitrite: Implications for protection of mitochondria against nitrosative damage. Biochem. J. 2000, 349 Pt 1, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, B. Hydroxyl radical and its scavengers in health and disease. Oxid. Med. Cell. Longev. 2011, 2011, 809696. [Google Scholar] [CrossRef]
- Pinto, E.; Sigaud-kutner, T.C.; Leitao, M.A.; Okamoto, O.K.; Morse, D.; Colepicolo, P. Heavy metal–induced oxidative stress in algae 1. J. Phycol. 2003, 39, 1008–1018. [Google Scholar] [CrossRef]
- Rojanasakul, Y.; Wang, L.; Hoffman, A.H.; Shi, X.; Dalal, N.S.; Banks, D.E.; Ma, J.K.H. Mechanisms of hydroxyl free radical-induced cellular injury and calcium overloading in alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 1993, 8, 377–383. [Google Scholar] [CrossRef]
- Bach, R.D.; Ayala, P.Y.; Schlegel, H. A reassessment of the bond dissociation energies of peroxides. An ab initio study. J. Am. Chem. Soc. 1996, 118, 12758–12765. [Google Scholar] [CrossRef]
- Behrman, E.J.; John, O.; Edwards, A. Half-Century of Peroxide Chemistry: A translation from R. Curci, L. D’Accolti, & C. Fusco. Chim. E L’Industria 2020, 88, 32–39. [Google Scholar]
- Chu, J.W.; Trout, B.L. On the mechanisms of oxidation of organic sulfides by H2O2 in aqueous solutions. J. Am. Chem. Soc. 2004, 126, 900–908. [Google Scholar] [CrossRef]
- Cardey, B.; Enescu, M. Selenocysteine versus cysteine reactivity: A theoretical study of their oxidation by hydrogen peroxide. J. Phys. Chem. A 2007, 111, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Carroll, K.S. Profiling the Reactivity of Cyclic C-Nucleophiles towards Electrophilic Sulfur in Cysteine Sulfenic Acid. Chem. Sci. 2016, 7, 400–415. [Google Scholar] [CrossRef]
- Zeida, A.; Babbush, R.; Lebrero, M.C.; Trujillo, M.; Radi, R.; Estrin, D.A. Molecular basis of the mechanism of thiol oxidation by hydrogen peroxide in aqueous solution: Challenging the SN2 paradigm. Chem. Res. Toxicol. 2012, 25, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Roos, G.; Foloppe, N.; Messens, J. Understanding the pK(a) of redox cysteines: The key role of hydrogen bonding. Antioxid. Redox Signal. 2013, 18, 94–127. [Google Scholar] [CrossRef]
- Pearson, R.G.; Edgington, D.N. Nucleophilic reactivity of the hydrogen peroxide anion: Distinction between SN2 and SN1 CB mechanisms. J. Am. Chem. Soc. 1962, 84, 4607–4608. [Google Scholar] [CrossRef]
- Brown, H.; Rao, B. Communications-selective conversion of olefins into organoboranes through competitive hydroboration, isomerization and displacement reactions. J. Org. Chem. 1957, 22, 1137–1138. [Google Scholar] [CrossRef]
- Yim, M.B.; Yim, H.S.; Boon Chock, P.; Stadtman, E.R. Pro-oxidant activity of Cu,Zn-superoxide dismutase. Age 1998, 21, 91–93. [Google Scholar] [CrossRef][Green Version]
- Krols, M.; Bultynck, G.; Janssens, S. ER-Mitochondria contact sites: A new regulator of cellular calcium flux comes into play. J. Cell Biol. 2016, 214, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.M.; Gladyshev, V.N. Cysteine function governs its conservation and degeneration and restricts its utilization on protein surfaces. J. Mol. Biol. 2010, 404, 902–916. [Google Scholar] [CrossRef]
- Gülden, M.; Jess, A.; Kammann, J.; Maser, E.; Seibert, H. Cytotoxic potency of H2O2 in cell cultures: Impact of cell concentration and exposure time. Free Radic. Biol. Med. 2010, 49, 1298–1305. [Google Scholar] [CrossRef] [PubMed]
- Juarez, J.C.; Manuia, M.; Burnett, M.E.; Betancourt, O.; Boivin, B.; Shaw, D.E.; Tonks, N.K.; Mazar, A.P.; Donate, F. Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 7147–7152. [Google Scholar] [CrossRef] [PubMed]
- Ezzi, S.A.; Urushitani, M.; Julien, J.P. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J. Neurochem. 2007, 102, 170–178. [Google Scholar] [CrossRef]
- Clément, M.V.; Ponton, A.; Pervaiz, S. Apoptosis induced by hydrogen peroxide is mediated by decreased superoxide anion concentration and reduction of intracellular milieu. FEBS Lett. 1998, 440, 13–18. [Google Scholar] [CrossRef]
- Gutiérrez-Venegas, G.; Guadarrama-Solís, A.; Muñoz-Seca, C.; Arreguín-Cano, J.A. Hydrogen peroxide-induced apoptosis in human gingival fibroblasts. Int. J. Clin. Exp. Pathol. 2015, 8, 15563–15572. [Google Scholar]
- Zhao, L.; Lin, H.; Chen, S.; Chen, S.; Cui, M.; Shi, D.; Wang, B.; Ma, K.; Shao, Z. Hydrogen peroxide induces programmed necrosis in rat nucleus pulposus cells through the RIP1/RIP3-PARP-AIF pathway. J. Orthop. Res. 2018, 36, 1269–1282. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Saito, Y.; Nishio, K.; Ogawa, Y.; Kimata, J.; Kinumi, T.; Yoshida, Y.; Noguchi, N.; Niki, E. Turning point in apoptosis/necrosis induced by hydrogen peroxide. Free Radic. Res. 2006, 40, 619–630. [Google Scholar] [CrossRef]
- Teramoto, S.; Tomita, T.; Matsui, H.; Ohga, E.; Matsuse, T.; Ouchi, Y. Hydrogen peroxide-induced apoptosis and necrosis in human lung fibroblasts: Protective roles of glutathione. Jpn. J. Pharmacol. 1999, 79, 33–40. [Google Scholar] [PubMed]
- Troyano, A.; Sancho, P.; Fernández, C.; de Blas, E.; Bernardi, P.; Aller, P. The selection between apoptosis and necrosis is differentially regulated in hydrogen peroxide-treated and glutathione-depleted human promonocytic cells. Cell Death Differ. 2003, 10, 889–898. [Google Scholar] [CrossRef]
- Cookson, M.R.; Ince, P.G.; Shaw, P.J. Peroxynitrite and hydrogen peroxide induced cell death in the NSC34 neuroblastoma x spinal cord cell line: Role of poly (ADP-ribose) polymerase. J. Neurochem. 1998, 70, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.Y.; Luan, P.; Huang, S.X.; Xiao, S.H.; Zhao, J.; Zhang, B.; Gu, B.-B.; Pi, R.-B.; Liu, J. Edaravone protects HT22 neurons from H2O2-induced apoptosis by inhibiting the MAPK signaling pathway. CNS Neurosci. Ther. 2013, 19, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.Y.; Guo, J.; Majeed, H.; Zhu, K.X.; Guo, X.N.; Peng, W.; Zhou, H.-M. Ferulic acid renders protection to HEK293 cells against oxidative damage and apoptosis induced by hydrogen peroxide. Vitr. Cell. Dev. Biol. Anim. 2015, 51, 722–729. [Google Scholar] [CrossRef]
- Xiang, J.; Wan, C.; Guo, R.; Guo, D. Is Hydrogen Peroxide a Suitable Apoptosis Inducer for All Cell Types? Biomed. Res. Int. 2016, 2016, 7343965. [Google Scholar] [CrossRef]
- Ali, M.A.; Kandasamy, A.D.; Fan, X.; Schulz, R. Hydrogen peroxide-induced necrotic cell death in cardiomyocytes is independent of matrix metalloproteinase-2. Toxicol. In Vitro 2013, 27, 1686–1692. [Google Scholar] [CrossRef]
- Ohguro, N.; Fukuda, M.; Sasabe, T.; Tano, Y. Concentration dependent effects of hydrogen peroxide on lens epithelial cells. Br. J. Ophthalmol. 1999, 83, 1064–1068. [Google Scholar] [CrossRef]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. Hydrogen peroxide-mediated neuronal cell death induced by an endogenous neurotoxin, 3-hydroxykynurenine. Proc. Natl. Acad. Sci. USA 1996, 93, 12553–12558. [Google Scholar] [CrossRef]
- Huang, B.K.; Sikes, H.D. Quantifying intracellular hydrogen peroxide perturbations in terms of concentration. Redox Biol. 2014, 2, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide-production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 39. [Google Scholar] [CrossRef]
- Go, Y.M.; Chandler, J.D.; Jones, D.P. The cysteine proteome. Free Radic. Biol. Med. 2015, 84, 227–245. [Google Scholar] [CrossRef]
- Bleier, L.; Wittig, I.; Heide, H.; Steger, M.; Brandt, U.; Dröse, S. Generator-specific targets of mitochondrial reactive oxygen species. Free Radic. Biol. Med. 2015, 78, 1–10. [Google Scholar] [CrossRef]
- Chang, C.; Worley, B.L.; Phaëton, R.; Hempel, N. Extracellular Glutathione Peroxidase GPx3 and Its Role in Cancer. Cancers 2020, 12, 2197. [Google Scholar] [CrossRef]
- West, J.D.; Roston, T.J.; David, J.B.; Allan, K.M.; Loberg, M.A. Piecing Together How Peroxiredoxins Maintain Genomic Stability. Antioxidants 2018, 7, 177. [Google Scholar] [CrossRef]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef]
- Day, B.J. Catalase and glutathione peroxidase mimics. Biochem. Pharmacol. 2009, 77, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase A₂ activities. Antioxid. Redox Signal. 2011, 15, 831–844. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L.; Niu, Y.; Sitia, R.; Wang, C.C. Glutathione peroxidase 7 utilizes hydrogen peroxide generated by Ero1α to promote oxidative protein folding. Antioxid. Redox Signal. 2014, 20, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.; Ward, D.M. The essential nature of iron usage and regulation. Curr. Biol. 2013, 23, 2325. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wen, J.; Liu, J.; Li, L. The roles of free radicals in amyotrophic lateral sclerosis: Reactive oxygen species and elevated oxidation of protein, DNA, and membrane phospholipids. FASEB J. 1999, 13, 2318–2328. [Google Scholar] [CrossRef]
- Bu, X.L.; Xiang, Y.; Guo, Y. The Role of Iron in Amyotrophic Lateral Sclerosis. Adv. Exp. Med. Biol. 2019, 1173, 145–152. [Google Scholar] [PubMed]
- Piñero, D.J.; Connor, J.R. Iron in the brain: An important contributor in normal and diseased states. Neuroscientist 2000, 6, 435–453. [Google Scholar] [CrossRef]
- McCann, S.; Perapoch Amadó, M.; Moore, S.E. The Role of Iron in Brain Development: A Systematic Review. Nutrients 2020, 12, 2001. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Ndayisaba, A.; Kaindlstorfer, C.; Wenning, G.K. Iron in Neurodegeneration-Cause or Consequence? Front. Neurosci. 2019, 13, 180. [Google Scholar] [CrossRef]
- Ince, P.G.; Shaw, P.J.; Candy, J.M.; Mantle, D.; Tandon, L.; Ehmann, W.D.; Markesbery, W. Iron, selenium and glutathione peroxidase activity are elevated in sporadic motor neuron disease. Neurosci. Lett. 1994, 182, 87–90. [Google Scholar] [CrossRef]
- Ignjatović, A.; Stević, Z.; Lavrnić, S.; Daković, M.; Bačić, G. Brain iron MRI: A biomarker for amyotrophic lateral sclerosis. J. Magn. Reson. Imaging 2013, 38, 1472–1479. [Google Scholar] [CrossRef]
- Yasui, M.; Ota, K.; Garruto, R.M. Concentrations of zinc and iron in the brains of Guamanian patients with amyotrophic lateral sclerosis and parkinsonism-dementia. Neurotoxicology 1993, 14, 445–450. [Google Scholar]
- Veyrat-Durebex, C.; Corcia, P.; Mucha, A.; Benzimra, S.; Mallet, C.; Gendrot, C.; Moreau, C.; Devos, D.; Piver, E.; Pages, J.-C.; et al. Iron metabolism disturbance in a French cohort of ALS patients. Biomed. Res. Int. 2014, 2014, 485723. [Google Scholar] [CrossRef]
- Zheng, Y.; Gao, L.; Wang, D.; Zang, D. Elevated levels of ferritin in the cerebrospinal fluid of amyotrophic lateral sclerosis patients. Acta Neurol. Scand. 2017, 136, 145–150. [Google Scholar] [CrossRef]
- Popović-Bijelić, A.; Mojović, M.; Stamenković, S.; Jovanović, M.; Selaković, V.; Andjus, P.; Bačić, G. Iron-sulfur cluster damage by the superoxide radical in neural tissues of the SOD1(G93A) ALS rat model. Free Radic. Biol. Med. 2016, 96, 313–322. [Google Scholar] [CrossRef]
- Sheykhansari, S.; Kozielski, K.; Bill, J.; Sitti, M.; Gemmati, D.; Zamboni, P.; Singh, A.V. Redox metals homeostasis in multiple sclerosis and amyotrophic lateral sclerosis: A review. Cell Death Dis. 2018, 9, 348. [Google Scholar] [CrossRef]
- Bozzo, F.; Mirra, A.; Carrì, M.T. Oxidative stress and mitochondrial damage in the pathogenesis of ALS: New perspectives. Neurosci. Lett. 2017, 636, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef]
- Cadet, J.; Delatour, T.; Douki, T.; Gasparutto, D.; Pouget, J.P.; Ravanat, J.L.; Sauvaigo, S. Hydroxyl radicals and DNA base damage. Mutat. Res. 1999, 424, 9–21. [Google Scholar] [CrossRef]
- Uchida, K.; Kawakishi, S. 2-Oxo-histidine as a novel biological marker for oxidatively modified proteins. FEBS Lett. 1993, 332, 208–210. [Google Scholar] [CrossRef]
- Schöneich, C.; Pogocki, D.; Hug, G.L.; Bobrowski, K. Free radical reactions of methionine in peptides: Mechanisms relevant to beta-amyloid oxidation and Alzheimer’s disease. J. Am. Chem. Soc. 2003, 125, 13700–13713. [Google Scholar] [CrossRef]
- Masuda, T.; Shinohara, H.; Eda, M.; Kondo, M. Reactivity of nucleotides and polynucleotides toward hydroxyl radical in aqueous solution. J. Radiat. Res. 1980, 21, 173–179. [Google Scholar] [CrossRef]
- Gardner, H.W. Oxygen radical chemistry of polyunsaturated fatty acids. Free Radic. Biol. Med. 1989, 7, 65–86. [Google Scholar] [CrossRef]
- Stadtman, E.R. Protein oxidation in aging and age-related diseases. Ann. N. Y. Acad. Sci. 2001, 928, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Ramana, K.V.; Srivastava, S.; Singhal, S.S. Lipid peroxidation products in human health and disease 2014. Oxid. Med. Cell. Longev. 2014, 2014, 162414. [Google Scholar] [CrossRef]
- Kehrer, J.P. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef]
- Shen, J.; Griffiths, P.T.; Campbell, S.J.; Utinger, B.; Kalberer, M.; Paulson, S.E. Ascorbate oxidation by iron, copper and reactive oxygen species: Review, model development, and derivation of key rate constants. Sci. Rep. 2021, 11, 7417. [Google Scholar] [CrossRef]
- Timoshnikov, V.A.; Kobzeva, T.V.; Polyakov, N.E.; Kontoghiorghes, G.J. Redox Interactions of Vitamin C and Iron: Inhibition of the Pro-Oxidant Activity by Deferiprone. Int. J. Mol. Sci. 2020, 21, 3967. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.F.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Safer, A.; Wöhrle, J.C.; Palm, F.; Nix, W.A.; Maschke, M.; Grau, A.J. Factors predicting one-year mortality in amyotrophic lateral sclerosis patients--data from a population-based registry. BMC Neurol. 2014, 14, 197. [Google Scholar] [CrossRef]
- Román, G.C. Neuroepidemiology of amyotrophic lateral sclerosis: Clues to aetiology and pathogenesis. J. Neurol. Neurosurg. Psychiatry 1996, 61, 131–137. [Google Scholar] [CrossRef]
- Longinetti, E.; Regodón Wallin, A.; Samuelsson, K.; Press, R.; Zachau, A.; Ronnevi, L.O.; Kierkegaard, M.; Andersen, P.M.; Hillert, J.; Fang, F.; et al. The Swedish motor neuron disease quality registry. Amyotroph Lateral Scler. Front. Degener. 2018, 19, 528–537. [Google Scholar] [CrossRef]
- Palese, F.; Sartori, A.; Verriello, L.; Ros, S.; Passadore, P.; Manganotti, P.; Barbone, F.; Pisa, F.E. Epidemiology of amyotrophic lateral sclerosis in Friuli-Venezia Giulia, North-Eastern Italy, 2002-2014: A retrospective population-based study. Amyotroph Lateral Scler. Front. Degener. 2019, 20, 90–99. [Google Scholar] [CrossRef]
- Rose, L.; McKim, D.; Leasa, D.; Nonoyama, M.; Tandon, A.; Bai, Y.Q.; Amin, R.; Katz, S.; Goldstein, R.; Gershon, A. Trends in incidence, prevalence, and mortality of neuromuscular disease in Ontario, Canada: A population-based retrospective cohort study (2003–2014). PLoS ONE 2019, 14, e0210574. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zhou, Y.; Qian, S.; Chang, W.; Wang, L.; Fan, D. Amyotrophic lateral sclerosis in Beijing: Epidemiologic features and prognosis from 2010 to 2015. Brain Behav. 2018, 8, e01131. [Google Scholar] [CrossRef] [PubMed]
- Nelson, L.M.; Topol, B.; Kaye, W.; Williamson, D.; Horton, D.K.; Mehta, P.; Wagner, T. Estimation of the Prevalence of Amyotrophic Lateral Sclerosis in the United States Using National Administrative Healthcare Data from 2002 to 2004 and Capture-Recapture Methodology. Neuroepidemiology 2018, 51, 149–157. [Google Scholar] [CrossRef]
- Nakken, O.; Lindstrøm, J.C.; Tysnes, O.B.; Holmøy, T. Assessing amyotrophic lateral sclerosis prevalence in Norway from 2009 to 2015 from compulsory nationwide health registers. Amyotroph Lateral Scler. Front. Degener. 2018, 19, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Kaye, W.; Raymond, J.; Punjani, R.; Larson, T.; Cohen, J.; Muravov, O.; Horton, K. Prevalence of Amyotrophic Lateral Sclerosis-United States, 2015. MMWR Morb. Mortal Wkly. Rep. 2018, 67, 1285–1289. [Google Scholar] [CrossRef]
- Longinetti, E.; Fang, F. Epidemiology of amyotrophic lateral sclerosis: An update of recent literature. Curr. Opin. Neurol. 2019, 32, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.M.; Masson, G.L. Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Rouleau, G.A. Oxidized/misfolded superoxide dismutase-1: The cause of all amyotrophic lateral sclerosis? Ann. Neurol. 2007, 62, 553–559. [Google Scholar] [CrossRef]
- Bosco, D.A.; Landers, J.E. Genetic determinants of amyotrophic lateral sclerosis as therapeutic targets. CNS Neurol. Disord. Drug Targets 2010, 9, 779–790. [Google Scholar] [CrossRef]
- Orrell, R.W. Amyotrophic lateral sclerosis: Copper/zinc superoxide dismutase (SOD1) gene mutations. Neuromuscul. Disord. 2000, 10, 63–68. [Google Scholar] [CrossRef]
- Andersen, P.M. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr. Neurol. Neurosci. Rep. 2006, 6, 37–46. [Google Scholar] [CrossRef]
- Rotunno, M.S.; Bosco, D.A. An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front. Cell. Neurosci. 2013, 7, 253. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Birsa, N.; Bentham, M.P.; Fratta, P. Cytoplasmic functions of TDP-43 and FUS and their role in ALS. Semin. Cell Dev. Biol. 2020, 99, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Forsberg, K.; Andersen, P.M.; Marklund, S.L.; Brännström, T. Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2011, 121, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.; Price, D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef]
- Wiedau-Pazos, M.; Goto, J.J.; Rabizadeh, S.; Gralla, E.B.; Roe, J.A.; Lee, M.K.; Valentine, J.S.; Bredesen, D.E. Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science 1996, 271, 515–518. [Google Scholar] [CrossRef]
- Wang, J.; Xu, G.; Li, H.; Gonzales, V.; Fromholt, D.; Karch, C.; Valentine, J.S.; Bredesen, D.E. Somatodendritic accumulation of misfolded SOD1-L126Z in motor neurons mediates degeneration: αB-crystallin modulates aggregation. Hum. Mol. Genet. 2005, 14, 2335–2347. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, P.A.; Graffmo, K.S.; Andersen, P.M.; Brännström, T.; Lindberg, M.; Oliveberg, M.; Marklund, S.L. Disulphide-reduced superoxide dismutase-1 in CNS of transgenic amyotrophic lateral sclerosis models. Brain 2006, 129 Pt 2, 451–464. [Google Scholar] [CrossRef]
- Julien, J.P.; Kriz, J. Transgenic mouse models of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 1013–1024. [Google Scholar] [CrossRef]
- De Giorgio, F.; Maduro, C.; Fisher, E.M.C.; Acevedo-Arozena, A. Transgenic and physiological mouse models give insights into different aspects of amyotrophic lateral sclerosis. Dis. Model Mech. 2019, 12, dmm037424. [Google Scholar] [CrossRef]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- Pardo, C.A.; Xu, Z.; Borchelt, D.R.; Price, D.L.; Sisodia, S.S.; Cleveland, D.W. Superoxide dismutase is an abundant component in cell bodies, dendrites, and axons of motor neurons and in a subset of other neurons. Proc. Natl. Acad. Sci. USA 1995, 92, 954–958. [Google Scholar] [CrossRef]
- Furukawa, Y.; Torres, A.S.; O’Halloran, T.V. Oxygen-induced maturation of SOD1: A key role for disulfide formation by the copper chaperone CCS. EMBO J. 2004, 23, 2872–2881. [Google Scholar] [CrossRef]
- Lepock, J.R.; Arnold, L.D.; Torrie, B.H.; Andrews, B.; Kruuv, J. Structural analyses of various Cu2+, Zn2+-superoxide dismutases by differential scanning calorimetry and Raman spectroscopy. Arch. Biochem. Biophys. 1985, 241, 243–251. [Google Scholar] [CrossRef]
- Tainer, J.A.; Getzoff, E.D.; Richardson, J.S.; Richardson, D.C. Structure and mechanism of copper, zinc superoxide dismutase. Nature 1983, 306, 284–287. [Google Scholar] [CrossRef]
- Tainer, J.A.; Getzoff, E.D.; Beem, K.M.; Richardson, J.S.; Richardson, D.C. Determination and analysis of the 2 A-structure of copper, zinc superoxide dismutase. J. Mol. Biol. 1982, 160, 181–217. [Google Scholar] [CrossRef]
- Kayatekin, C.; Zitzewitz, J.A.; Matthews, C.R. Zinc binding modulates the entire folding free energy surface of human Cu,Zn superoxide dismutase. J. Mol. Biol. 2008, 384, 540–555. [Google Scholar] [CrossRef]
- Dafforn, T.R.; Smith, C.J. Natively unfolded domains in endocytosis: Hooks, lines and linkers. EMBO Rep. 2004, 5, 1046–1052. [Google Scholar] [CrossRef]
- Anzai, I.; Tokuda, E.; Handa, S.; Misawa, H.; Akiyama, S.; Furukawa, Y. Oxidative misfolding of Cu/Zn-superoxide dismutase triggered by non-canonical intramolecular disulfide formation. Free Radic. Biol. Med. 2020, 147, 187–199. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Liu, J. Oxidation versus aggregation-how do SOD1 mutants cause ALS? Nat. Med. 2000, 6, 1320–1321. [Google Scholar] [CrossRef]
- Jewett, S.L.; Rocklin, A.M.; Ghanevati, M.; Abel, J.M.; Marach, J.A. A new look at a time-worn system: Oxidation of CuZn-SOD by H2O2. Free Radic. Biol. Med. 1999, 26, 905–918. [Google Scholar] [CrossRef]
- Sampson, J.B.; Beckman, J.S. Hydrogen peroxide damages the zinc-binding site of zinc-deficient Cu,Zn superoxide dismutase. Arch. Biochem. Biophys. 2001, 392, 8–13. [Google Scholar] [CrossRef]
- Seetharaman, S.V.; Winkler, D.D.; Taylor, A.B.; Cao, X.; Whitson, L.J.; Doucette, P.A.; Valentine, J.S.; Schirf, V.; Demeler, B.; Carroll, M.C.; et al. Disrupted zinc-binding sites in structures of pathogenic SOD1 variants D124V and H80R. Biochemistry 2010, 49, 5714–5725. [Google Scholar] [CrossRef]
- Li, H.T.; Jiao, M.; Chen, J.; Liang, Y. Roles of zinc and copper in modulating the oxidative refolding of bovine copper, zinc superoxide dismutase. Acta Biochim. Biophys. Sin. 2010, 42, 183–194. [Google Scholar] [CrossRef]
- Boyd, S.D.; Ullrich, M.S.; Calvo, J.S.; Behnia, F.; Meloni, G.; Winkler, D.D. Mutations in Superoxide Dismutase 1 (Sod1) Linked to Familial Amyotrophic Lateral Sclerosis Can Disrupt High-Affinity Zinc-Binding Promoted by the Copper Chaperone for Sod1 (Ccs). Molecules 2020, 25, 1086. [Google Scholar] [CrossRef]
- Mendonça, D.M.; Chimelli, L.; Martinez, A.M. Quantitative evidence for neurofilament heavy subunit aggregation in motor neurons of spinal cords of patients with amyotrophic lateral sclerosis. Braz. J. Med. Biol. Res. 2005, 38, 925–933. [Google Scholar] [CrossRef][Green Version]
- Zucchi, E.; Bonetto, V.; Sorarù, G.; Martinelli, I.; Parchi, P.; Liguori, R.; Mandrioli, J. Neurofilaments in motor neuron disorders: Towards promising diagnostic and prognostic biomarkers. Mol. Neurodegener. 2020, 15, 58. [Google Scholar] [CrossRef]
- Falzone, Y.M.; Russo, T.; Domi, T.; Pozzi, L.; Quattrini, A.; Filippi, M. Current application of neurofilaments in amyotrophic lateral sclerosis and future perspectives. Neural Regen. Res. 2021, 16, 1985–1991. [Google Scholar]
- Pierson, K.B.; Evenson, M.A. 200 Kd neurofilament protein binds Al, Cu and Zn. Biochem. Biophys. Res. Commun. 1988, 152, 598–604. [Google Scholar] [CrossRef]
- Crow, J.P.; Sampson, J.B.; Zhuang, Y.; Thompson, J.A.; Beckman, J.S. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J. Neurochem. 1997, 69, 1936–1944. [Google Scholar] [CrossRef]
- Crow, J.P.; Ye, Y.Z.; Strong, M.; Kirk, M.; Barnes, S.; Beckman, J.S. Superoxide dismutase catalyzes nitration of tyrosines by peroxynitrite in the rod and head domains of neurofilament-L. J. Neurochem. 1997, 69, 1945–1953. [Google Scholar] [CrossRef]
- Gaggelli, E.; Kozlowski, H.; Valensin, D.; Valensin, G. Copper homeostasis and neurodegenerative disorders (Alzheimer’s, prion, and Parkinson’s diseases and amyotrophic lateral sclerosis). Chem. Rev. 2006, 106, 1995–2044. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.R.; Lim, N.K.; McAllum, E.J.; Donnelly, P.S.; Hare, D.J.; Doble, P.A.; Turner, B.J.; Price, K.A.; Lim, S.C.; Paterson, B.M.; et al. Oral treatment with Cu(II)(atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 8021–8031. [Google Scholar] [CrossRef] [PubMed]
- Nikseresht, S.; Hilton, J.B.W.; Kysenius, K.; Liddell, J.R.; Crouch, P.J. Copper-ATSM as a Treatment for ALS: Support from Mutant SOD1 Models and Beyond. Life 2020, 10, 271. [Google Scholar] [CrossRef]
- Kurahashi, T.; Miyazaki, A.; Suwan, S.; Isobe, M. Extensive investigations on oxidized amino acid residues in H(2)O(2)-treated Cu,Zn-SOd protein with LC-ESI-Q-TOF-MS, MS/MS for the determination of the copper-binding site. J. Am. Chem. Soc. 2001, 123, 9268–9278. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, E.K.; Fridovich, I. The interaction of bovine erythrocyte superoxide dismutase with hydrogen peroxide: Inactivation of the enzyme. Biochemistry 1975, 14, 5294–5299. [Google Scholar] [CrossRef]
- Hough, M.A.; Grossmann, J.G.; Antonyuk, S.V.; Strange, R.W.; Doucette, P.A.; Rodriguez, J.A.; Whitson, L.J.; Hart, P.J.; Hayward, L.J.; Valentine, J.S.; et al. Dimer destabilization in superoxide dismutase may result in disease-causing properties: Structures of motor neuron disease mutants. Proc. Natl. Acad. Sci. USA 2004, 101, 5976–5981. [Google Scholar] [CrossRef]
- Arnesano, F.; Banci, L.; Bertini, I.; Martinelli, M.; Furukawa, Y.; O’Halloran, T.V. The unusually stable quaternary structure of human Cu,Zn-superoxide dismutase 1 is controlled by both metal occupancy and disulfide status. J. Biol. Chem. 2004, 279, 47998–48003. [Google Scholar] [CrossRef]
- Lynch, S.M.; Boswell, S.A.; Colón, W. Kinetic stability of Cu/Zn superoxide dismutase is dependent on its metal ligands: Implications for ALS. Biochemistry 2004, 43, 16525–16531. [Google Scholar] [CrossRef]
- Julien, J.P. Amyotrophic lateral sclerosis. unfolding the toxicity of the misfolded. Cell 2001, 104, 581–591. [Google Scholar] [CrossRef]
- Coelho, F.R.; Iqbal, A.; Linares, E.; Silva, D.F.; Lima, F.S.; Cuccovia, I.M.; Augusto, O. Oxidation of the tryptophan 32 residue of human superoxide dismutase 1 caused by its bicarbonate-dependent peroxidase activity triggers the non-amyloid aggregation of the enzyme. J. Biol. Chem. 2014, 289, 30690–30701. [Google Scholar] [CrossRef]
- Graffmo, K.S.; Forsberg, K.; Bergh, J.; Birve, A.; Zetterström, P.; Andersen, P.M.; Marklund, S.L.; Brännström, T. Expression of wild-type human superoxide dismutase-1 in mice causes amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Guareschi, S.; Cova, E.; Cereda, C.; Ceroni, M.; Donetti, E.; Bosco, D.A.; Trotti, D.; Pasinelli, P. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc. Natl. Acad. Sci. USA 2012, 109, 5074–5079. [Google Scholar] [CrossRef]
- Bakavayev, S.; Chetrit, N.; Zvagelsky, T.; Mansour, R.; Vyazmensky, M.; Barak, Z.; Israelson, A.; Engel, S. Cu/Zn-superoxide dismutase and wild-type like fALS SOD1 mutants produce cytotoxic quantities of H(2)O(2) via cysteine-dependent redox short-circuit. Sci. Rep. 2019, 9, 10826. [Google Scholar] [CrossRef]
- Fujiwara, N.; Nakano, M.; Kato, S.; Yoshihara, D.; Ookawara, T.; Eguchi, H.; Taniguchi, N.; Suzuki, K. Oxidative modification to cysteine sulfonic acid of Cys111 in human copper-zinc superoxide dismutase. J. Biol. Chem. 2007, 282, 35933–35944. [Google Scholar] [CrossRef] [PubMed]
- De Beus, M.D.; Chung, J.; Colón, W. Modification of cysteine 111 in Cu/Zn superoxide dismutase results in altered spectroscopic and biophysical properties. Protein Sci. 2004, 13, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shang, H.; Qiu, X.; Fujiwara, N.; Cui, L.; Li, X.M.; Gao, T.-M.; Kong, J. Oxidative modification of cysteine 111 promotes disulfide bond-independent aggregation of SOD1. Neurochem. Res. 2012, 37, 835–845. [Google Scholar] [CrossRef]
- Shafiq, K.; Sanghai, N.; Guo, Y.; Kong, J. Implication of post-translationally modified SOD1 in pathological aging. Geroscience 2021, 43, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Bosco, D.A.; Morfini, G.; Karabacak, N.M.; Song, Y.; Gros-Louis, F.; Pasinelli, P.; Goolsby, H.; A Fontaine, B.; Lemay, N.; McKenna-Yasek, D.; et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 2010, 13, 1396–1403. [Google Scholar] [CrossRef]
- Strange, R.W.; Antonyuk, S.V.; Hough, M.A.; Doucette, P.A.; Valentine, J.S.; Hasnain, S.S. Variable metallation of human superoxide dismutase: Atomic resolution crystal structures of Cu-Zn, Zn-Zn and as-isolated wild-type enzymes. J. Mol. Biol. 2006, 356, 1152–1162. [Google Scholar] [CrossRef]
- Cohen, T.J.; Hwang, A.W.; Unger, T.; Trojanowski, J.Q.; Lee, V.M. Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. EMBO J. 2012, 31, 1241–1252. [Google Scholar] [CrossRef]
- Pokrishevsky, E.; Grad, L.I.; Yousefi, M.; Wang, J.; Mackenzie, I.R.; Cashman, N.R. Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e35050. [Google Scholar] [CrossRef]
- Jiang, L.L.; Che, M.X.; Zhao, J.; Zhou, C.J.; Xie, M.Y.; Li, H.Y.; He, J.-H.; Hu, H.-Y. Structural transformation of the amyloidogenic core region of TDP-43 protein initiates its aggregation and cytoplasmic inclusion. J. Biol. Chem. 2013, 288, 19614–19624. [Google Scholar] [CrossRef]
- Robinson, J.L.; Geser, F.; Stieber, A.; Umoh, M.; Kwong, L.K.; Van Deerlin, V.M.; Lee, V.M.-Y.; Trojanowski, J.Q. TDP-43 skeins show properties of amyloid in a subset of ALS cases. Acta Neuropathol. 2013, 125, 121–131. [Google Scholar] [CrossRef]
- Chang, C.K.; Chiang, M.H.; Toh, E.K.; Chang, C.F.; Huang, T.H. Molecular mechanism of oxidation-induced TDP-43 RRM1 aggregation and loss of function. FEBS Lett. 2013, 587, 575–582. [Google Scholar] [CrossRef]
- Lin, Y.; Zhou, X.; Kato, M.; Liu, D.; Ghaemmaghami, S.; Tu, B.P.; McKnight, S.L. Redox-mediated regulation of an evolutionarily conserved cross-β structure formed by the TDP43 low complexity domain. Proc. Natl. Acad. Sci. USA 2020, 117, 28727–28734. [Google Scholar] [CrossRef]
- Wood, A.; Gurfinkel, Y.; Polain, N.; Lamont, W.; Lyn Rea, S. Molecular Mechanisms Underlying TDP-43 Pathology in Cellular and Animal Models of ALS and FTLD. Int. J. Mol. Sci. 2021, 22, 4705. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanghai, N.; Tranmer, G.K. Hydrogen Peroxide and Amyotrophic Lateral Sclerosis: From Biochemistry to Pathophysiology. Antioxidants 2022, 11, 52. https://doi.org/10.3390/antiox11010052
Sanghai N, Tranmer GK. Hydrogen Peroxide and Amyotrophic Lateral Sclerosis: From Biochemistry to Pathophysiology. Antioxidants. 2022; 11(1):52. https://doi.org/10.3390/antiox11010052
Chicago/Turabian StyleSanghai, Nitesh, and Geoffrey K. Tranmer. 2022. "Hydrogen Peroxide and Amyotrophic Lateral Sclerosis: From Biochemistry to Pathophysiology" Antioxidants 11, no. 1: 52. https://doi.org/10.3390/antiox11010052
APA StyleSanghai, N., & Tranmer, G. K. (2022). Hydrogen Peroxide and Amyotrophic Lateral Sclerosis: From Biochemistry to Pathophysiology. Antioxidants, 11(1), 52. https://doi.org/10.3390/antiox11010052