Abstract
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are severe neurodegenerative disorders that belong to a common disease spectrum. The molecular and cellular aetiology of the spectrum is a highly complex encompassing dysfunction in many processes, including mitochondrial dysfunction and oxidative stress. There is a paucity of treatment options aside from therapies with subtle effects on the post diagnostic lifespan and symptom management. This presents great interest and necessity for the discovery and development of new compounds and therapies with beneficial effects on the disease. Polyphenols are secondary metabolites found in plant-based foods and are well known for their antioxidant activity. Recent research suggests that they also have a diverse array of neuroprotective functions that could lead to better treatments for neurodegenerative diseases. We present an overview of the effects of various polyphenols in cell line and animal models of ALS/FTD. Furthermore, possible mechanisms behind actions of the most researched compounds (resveratrol, curcumin and green tea catechins) are discussed.
1. Introduction
With the ageing population, the treatment and management of neurodegenerative diseases is a major and increasing challenge for health care systems and societies around the world [1]. Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that affects motor neurons, resulting in deterioration of motor function, and frontotemporal dementia (FTD) is a neurodegenerative disorder characterised by changes in personality, behaviour, and language. The development of both diseases is a progressive and ultimately fatal multistep process with a complex genetic and molecular background. Despite extensive research efforts, only two treatment options with limited effects on survival and motor function are currently approved for ALS. The vast majority of compounds researched as possible ALS therapies until today were found to be ineffective in clinical trials, highlighting the need for further research [2]. Currently, only symptomatic treatments with limited effects are available for FTD [3].
Polyphenols are natural compounds whose neuroprotective effects have been demonstrated in various models of neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease. These compounds are being explored for possible dietary intervention and supplementation as preventive measures against neurodegenerative diseases, and also as possible candidates for therapies to slow disease progression and alleviate symptoms [4]. Due to the lack of disease-changing treatments for ALS/FTD and the growing interest in natural compounds as therapeutic agents, this article reviews an intriguing topic of potential use of polyphenols in the development of treatments for ALS/FTD symptoms.
1.1. Amyotrophic Lateral Sclerosis and Frontotemporal Dementia
ALS is a neurodegenerative disease characterised by progressive loss of both upper and lower motor neurons. Initial signs of the disease may include weakness of the limbs (in spinal-onset ALS) or difficulties with speech and swallowing (in bulbar-onset ALS) [5]. Disease progression eventually leads to paralysis and death from respiratory failure, on average 24 to 50 months after onset [6,7,8,9,10]. The worldwide incidence of ALS is 1.75 with a reported mean age at diagnosis between 51 and 69 years [11,12]. ALS cases can be divided into the familial form of the disease (fALS, 5–15% of patients), where there is a clear family history, and the predominant sporadic form (sALS) [13]. Frontotemporal dementia (FTD) is a type of dementia primarily associated with alterations in the frontal and temporal lobes. Symptoms manifest as changes in behaviour, personality, language, and motor skills [14,15]. The incidence of FTD is 1.6 and the mean age of onset is 65 years [16]. FTD can be divided into one behavioural (bvFTD) and two language variants (or primary progressive aphasias (PPA)) [14]. Mean survival time for most forms of FTD is approximately 8 years [17]. Up to 40% of FTD patients have a family history of the disease [18,19].
Clinical, genetic, pathological and biochemical data show that there is an overlap between ALS and FTD. First observations that ALS and FTD might be connected were made in the early 1990s [20,21]. Data show that about half of ALS patients have cognitive impairment and 15% meet the criteria for FTD [22,23]. Similarly, about 30% of patients with FTD develop signs of motor dysfunction and 10–15% have ALS [24,25]. The discovery of common genetic causes and biological mechanisms further confirmed that ALS and FTD are closely associated (Figure 1) [5,26].

Figure 1.
Genes involved in pathologies along the ALS-FTD spectrum. The most common genetic causes of the disease are highlighted in bold.
ALS and FTD pathologies are multistep processes that affect many aspects of cellular activity. The most prominent pathological hallmark of both ALS and FTD are changes in protein homeostasis, including protein misfolding and aggregation, altered localisation, and defects in autophagic and proteasomal degradation. The combination of these mechanisms leads to the formation of toxic cytoplasmic inclusions in motor neurons and surrounding cells. Proteins that predominantly form these structures are two RNA-binding proteins, TAR DNA binding protein (TDP-43, protein product of TARDBP), and fused in sarcoma (FUS), microtubule-associated protein tau (gene MAPT), and superoxide dismutase 1 (SOD1) [27,28]. The correlation between pathology and genetics is complex [29,30,31]. Pathologically, 97% of ALS cases have pathognomonic TDP-43 aggregates, while only 1% of those are associated with mutations in TDP-43 and in the rest TDP-43 is not mutated. A total of 1% of ALS shows FUS aggregates, all of which are associated with mutations in FUS. Mutations in FUS or TDP-43 are extremely rare in FTD; however, 50% of FTD have TDP-43 aggregates and 10% of FTD have FUS aggregates. A total of 40% of FTD is tau aggregates. Impairments in protein turnover and clearance are also observed. Mutations in genes associated with different stages of autophagy are also causative for ALS/FTD, from autophagy regulating activities of C9ORF72 to impaired functions of autophagic receptors SQSTM1 and optineurin [32,33,34,35,36,37].
In healthy cells, TDP-43 and FUS are predominantly nuclear RNA/DNA-binding proteins with functions in RNA splicing, transcription, microRNA biogenesis, and mRNA transport [38,39,40,41,42,43,44,45,46,47]. Both play important parts in ribonucleoprotein coacervates that form membrane-less organelles such as stress granules in the cytoplasm and paraspeckles in the nucleus [48,49]. In ALS/FTD, FUS or TDP-43 mislocalise to the cytoplasm and form aggregates that are most likely toxic, although loss of function from the nucleus may also be the key disease-causing factor. This mislocalisation is instigated by a number of disruptions, including dysfunctions in proteostasis, nucleocytoplasmic shuttling, and the cellular stress response [50,51,52]. Upon stress, TDP-43, FUS, and some other ALS-associated RNA-binding proteins separate into stress granules, which may be the first step in the formation of insoluble aggregates [53,54]. Another common factor in the disruption of RNA metabolism is G4C2 repeat expansions in the C9ORF72 gene, which are the most common cause of familiar forms of ALS/FTD [55,56,57]. The repeats form stable nucleic acid secondary structures known as G-quadruplexes, hairpin loops, and i-motifs, that sequester RNA-binding proteins and form nuclear foci similar to paraspeckles, or can be translated into toxic dipeptide repeats via repeat-associated non-ATG translation [58,59,60,61,62,63,64,65].
Mitochondria play a central role in neurons, primarily fulfilling high needs for energy. ALS/FTD-associated changes include defects in oxidative phosphorylation and calcium homeostasis, elevated production of ROS, structural impairments, and reduced clearance of damaged mitochondria [66]. Changes in mitochondrial morphology are observed in cells overexpressing mutant SOD1, FUS, or TDP-43 [67,68,69,70]. The increased localisation of mutant SOD1 in the mitochondrial intermembrane space causes mitochondrial dysfunction and toxicity to neurons [71,72,73]. Overall, mitochondrial changes result in decreased electron transport chain activity and reduced ATP production [66]. Moreover, oxidative stress has been proposed to be crucial in ALS pathogenesis and has been well documented in patient samples [74,75,76].
1.2. Currently Used Therapies for ALS/FTD
Treatments currently in clinical trials for ALS/FTD were comprehensively reviewed by Liscic et al. [26]. Therapeutic targets include a reduction in glutamate excitotoxicity and protein aggregation, upregulation of certain heat shock proteins, and activation of troponin in skeletal muscle. Interesting novel strategies for ALS/FTD treatment may also come from stem cell therapy, non-invasive brain stimulation, and the growing knowledge of the influence of the gut microbiota on the development of neurological diseases [26]. Currently, only two drugs are approved for the treatment of ALS. Riluzole was approved for clinical use in 1995 and trials observed reduced one year mortality and slower deterioration of muscle function [26,77,78]. The mechanisms behind the beneficial effects of riluzole are not entirely clear. Different neuroprotective actions have been proposed, such as inhibition of glutamate excitotoxicity, blockade of Ca2+- or Na+-ion channels, and modulation of GABA pathways [79]. In recent years, some countries have also approved the use of edaravone (also known as MCI-186 or Radicava) for the treatment of ALS [26]. Its actions could benefit a subgroup of patients with early onset and rapidly progressive disease [80]. Edaravone is thought to act as an antioxidant and free radical scavenger, but the mechanisms are not well understood [81]. There are currently no approved direct treatments for FTD, other than symptom management [82].
2. Therapeutic Potentials of Polyphenols in ALS/FTD
Many potential therapeutic compounds have antioxidant and anti-inflammatory properties. Polyphenols (Figure 2) are a diverse group of naturally occurring compounds with a characteristic chemical structure that has one or more phenolic rings. They are found in plant foods such as fruits, vegetables, and whole grains [83,84]. In plants, polyphenols are categorised as secondary metabolites and have functions in normal growth as well as in the plant defense system [85]. They are synthesised in the shikimate and phenylpropanoid pathways [86]. Many different polyphenols have been described to have neuroprotective effects in mammalian cell and animal models of ALS/FTD [87]. In this review, the focus will be on resveratrol, epigallocatechin gallate (EGCG), and curcumin (Figure 2). We will also explore the effects of some other flavonoids and phenolic acids in the context of ALS/FTD.
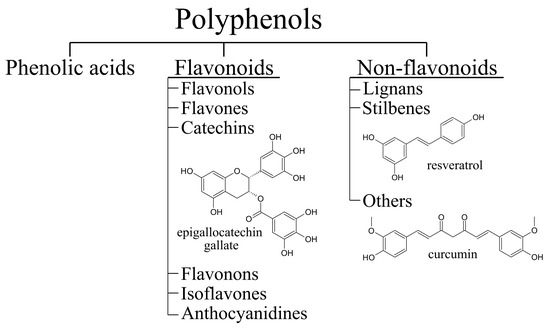
Figure 2.
Classification of polyphenols with structural formulas of epigallocatechin gallate (EGCG), resveratrol and curcumin.
2.1. Resveratrol
Resveratrol (3,5,4′-trihydroxystilbene) is a polyphenol found in grapes, red wine, berries, and peanuts [88]. Both cis- and trans- isomers occur naturally, with trans-form being the focus in terms of potential neuroprotective activity [89]. Effects of resveratrol in ALS were first demonstrated in neuronal cell lines expressing the SOD1G93A mutant [90,91,92]. Resveratrol treatment halved the cell death observed as a consequence of SOD1-mediated toxicity [90]. Treatments of mouse motor neuron cells NSC34 expressing SOD1G93A showed a minor dose-dependent improvement in cell viability and a simultaneous reduction in the concentration of cytosolic ROS [91]. Administration of resveratrol protected rat cortical motor neurons from the toxic effects of cerebrospinal fluid (CSF) from ALS patients [93]. Further studies in mice ALS models expressing mutant SOD1G93A showed conflicting results, which are probably a consequence of different protocols on dosing and route of administration. Chronic oral administration of resveratrol at 25 mg/kg/day did not improve motor abilities and life span of ALS mice [94]. On the other hand, intraperitoneal injections of 20 mg/kg/twice a week improved survival and delayed the onset of ALS [95]. A similar positive effect on survival and motor function was observed with a higher dose (160 mg/kg/day) administered orally [96]. The neuroprotective effects of resveratrol in ALS mice have been further demonstrated in coadministration with other potential therapeutics [97,98]. Resveratrol has also been researched in models of tauopathies, a hallmark of FTD, but the overall effects on tau aggregation are inconclusive [99].
The predominant mechanism behind the neuroprotective effect of resveratrol is the activation of SIRT1, a NAD+-dependent protein deacetylase [90,92,95,96,100]. Structural studies suggested a mechanism in which resveratrol acts as an adaptor for the interaction between the peptide substrate and SIRT1 [101]. Many downstream mechanisms of SIRT1 targets have been proposed as possible mediators of the beneficial effects. SIRT1 deacetylates p53 [90,96], a known tumor suppressor protein involved in mechanisms of motor neuron cell death [102]. Resveratrol treatment upregulates factors involved in mitochondrial biogenesis, which could improve altered energy metabolism observed in ALS [92,96]. SIRT1 also targets HSF1 (heat shock factor 1) that activates several heat shock proteins. Their activity as chaperones possibly mitigates formation of toxic protein aggregates [95]. Normalisation of autophagic flux was also observed in resveratrol-treated ALS mice, but it is not clear whether SIRT1 is involved [96]. Independent of SIRT1, resveratrol can also activate AMPK (AMP-activated protein kinase) [96,103] that has downstream targets involved in neuroprotective mechanisms [104]. Moreover, a molecular mechanistic study on SOD1G93A showed a stabilising effect of resveratrol that could impede the aggregation of mutant protein [105]. A similar inhibitory effect was observed in aggregation studies of wt SOD1 [106].
2.2. Curcumin
Curcumin (diferuloylmethane) is the predominant curcuminoid found in turmeric (Curcuma longa), which is widely used in traditional Indian medicine. The potential benefits of curcumin are being explored in many neurodegenerative diseases. In models of Alzheimer’s and Parkinson’s disease, curcumin can reduce oxidative stress, affect toxic protein aggregation, and protect against apoptosis [107,108].
Regarding ALS, curcumin was shown to impede aggregation of reduced wt SOD1 in vitro by binding its aggregation prone regions. Curcumin-bound SOD1 aggregates were smaller, unstructured, and less cytotoxic [109]. A similar effect of inhibiting beta-sheet formation and aggregation was observed with tau, a protein involved in FTD [110]. In contrast, the binding of curcumin to tau aggregates was not observed in post-mortem brain tissue sections from FTD patients [111].
Curcumin presents a challenge for in vivo use due to its poor absorption, fast metabolism, and rapid elimination. Several strategies can be utilised to overcome the low oral bioavailability of curcumin [112]. The protective effect of an analogue, dimethoxy curcumin, was demonstrated in a neuronal cell line expressing TDP-43 mutants Q331K or M337V. Dimethoxy curcumin restored mitochondrial damage by improving transmembrane potential, increasing electron transfer chain complex I activity, and upregulating UCP2 (uncoupling protein 2) [113]. The same compound also improved abnormally high excitability of cells expressing mutant TDP-43 [114]. Furthermore, an improved curcumin analogue, monocarbonyl dimethoxycurcumin C, prevented aggregation of mutant TDP-43 and reduced oxidative stress, possibly due to increased expression of heme oxygenase-1 [115].
Another approach to improve the bioavailability of curcumin is delivery using nanoparticles. The potential for ALS treatment was demonstrated with curcumin-loaded inulin-d-alfa-tocopherol succinate micelles, which were effectively delivered into mesenchymal stromal cells [116]. Furthermore, the efficiency of a turmeric supplement in nanomicelles was tested in a clinical trial involving 54 ALS patients treated primarily with riluzole. Nanocurcumin improved the survival probability of the patients, but did not significantly improve their motor function [117].
2.3. Catechins
Green tea, produced from the leaves and buds of Camellia sinensis, is rich in polyphenols catechins, predominantly (−)epigallocatechin gallate (EGCG), but also (−)-epigallocatechin (EGC), (−)-epicatechin gallate (ECG), (−)-epicatechin (EC), and (+)-catechin [118]. In ALS models, EGCG has been shown to protect motor neuron cells from oxidative stress and mitochondrial damage [119]. Presymptomatic oral supplementation of EGCG at doses of at least 2.9 mg EGCG/kg body weight in SOD1G93A mice significantly delayed symptom onset, improved motor function, and increased lifespan [120,121].
EGCG likely acts by upregulating a prosurvival signaling pathway PI3K/Akt. Among other pathways, PI3K/Akt regulates the activity of GSK-3. Increased GSK-3 levels are associated with the formation of neurofibrillary tangles and neuronal death. In addition, GSK-3 induces apoptosis through downstream signaling, including mitochondrial damage and caspase-3 activation. It was shown that Akt phosphorylates GSK-3, resulting in less mitochondrial damage [119]. Observations in ALS mice further confirm an increase in PI3K/Akt and a decrease in death signals such as caspase-3, cytosolic cytochrome c, and cleaved PARP (poly (ADP-ribose) polymerase) [120]. EGCG also has antioxidant and anti-inflammatory effects on microglia and astrocytes [121]. In addition, it can decrease lipid peroxidation, but has no effect on iron metabolism despite its presumed chelating abilities [122]. A molecular docking study showed the potential of EGCG to reduce mutant SOD1 aggregates [123]. In vitro studies confirmed an inhibitory effect on apo-SOD1 aggregation [124]. It has also been shown that the addition of EGCG induces oligomerisation of TDP-43 and inhibits its degradation into toxic aggregation-prone fragments [125]. In FTD, inhibition of tau filament formation was observed for ECG, but not for EC [126].
2.4. Other Flavonoids
In addition to green tea catechins, several other flavonoids have been tested in ALS/FTD models. Presymptomatic administration of 2 mg/kg body weight of an anthocyanin-enriched strawberry extract with callistephin (pelargonidin 3-glucoside) as the predominant component delayed ALS onset, preserved grip strength, and prolonged survival in SOD1G93A mice [127]. Oral supplementation of fisetin (3,3,4,7-tetrahydroxyflavone) improved motor functions, delayed disease onset, and increased survival in SOD1G93A mice (at a dosage of 9 mg/kg) and SOD1G85R Drosophila melanogaster. The predominant mechanism behind the activity of fisetin in motor neuron cell lines expressing SOD1G93A appears to be the activation of the ERK pathway involved in the regulation of cell survival. Moreover, fisetin decreased both wt and mutant SOD1 levels in cells, possibly by activating autophagy [128].
A computational study confirmed the binding of kaempferol (3,4′,5,7-tetrahydroxyflavone) and kaempferide to mutant SOD1G85R [129]. Both compounds were experimentally shown to have antioxidant properties and could reduce the formation of SOD1G85R aggregates in N2a mouse neuroblastoma cells. Kaempferol could act via increased phosphorylation of AMPK and downstream induction of autophagy [130]. The antioxidant effect of quercetin (3,3′,4′,5,7-pentahyroxyflavone) was first observed in lymphoblast cell lines from ALS patients [131]. In vitro tests showed that quercetin glycosides, namely quercitrin and quercetin 3-beta-d-glucoside, inhibit misfolding and aggregation of SOD1A4V mutant [132]. A similar effect on aggregation was observed with quercetin and baicalein [133]. Furthermore, preventive administration of quercetin in rats reduced oxidative stress, defective mitochondria, and brain cell death caused by aluminium exposure [134].
SOD1G93A mice treated with 5 mg/kg 7,8-dihydroxyflavone exhibited significantly improved motor performance and increased numbers of spinal motor neurons compared with untreated animals [135]. Interestingly, it was observed that treatment with 16 mg/kg genistein (4′,5,7-trihydroxyisoflavone) had a protective effect on disease progression in male SOD1G93A mice [136]. In contrast, in further studies, a delay in symptoms and higher survival of motor neurons was observed in both sexes, possibly due to anti-inflammatory effects and restored autophagy [137]. Twice-daily administration of 700 mg luteolin (3′,4′,5,7-tetrahydroxyflavone) in combination with palmitoylethanolamide showed some improvement of symptoms in patients with FTD [138].
2.5. Phenolic Acids and Derivatives
Phenolic acids are found in fruits, coffee, tea, and grains. Their diverse neuroprotective effects make them interesting candidates for better ALS therapies. It has been reported that protocatechuic acid administration at 100 mg/kg in SOD1G93A mice prolongs survival, improves motor function, and reduces gliosis [139]. Caffeic acid phenethyl ester (CAPE) showed a dose-dependent improvement in survival and a simultaneous reduction in cytosolic ROS in the NCS34 cell line expressing SOD1G93A. CAPE decreased the activation of the oxidative stress-associated transcription factor NF-κB and activated the antioxidant response element (ARE) [91]. Further studies in SOD1G93A mice confirmed that daily administration of 10 mg/kg CAPE after disease onset slowed symptom progression and prolonged survival. A reduction in glial activation and phospho-p38 levels was observed as a result [140]. Gallic acid and wedelolactone improved locomotor function and motor learning abilities in an aluminium or quinolinic acid-induced rat model of sALS. The effects may be due to a reduction in inflammatory cytokines, normalisation of L-glutamate levels, and decreased activation of caspase-3 [141,142]. Rosmarinic acid, the main compound in rosemary (Rosmarinus officinalis) extract, reduced weight loss, improved motor performance, and prolonged survival of SOD1G93A mice [143,144]. The effects of treatment with higher doses were compared with the established ALS therapeutic agent riluzole, but were not found to be more effective [144].
2.6. Overview of Potential Therapeutic Effects of Polyphenols in ALS and FTD
We have summarised the therapeutic implications of polyphenols, including their proposed mechanisms in animal and cell line models of ALS and FTD (Table 1). The predominant mechanism behind the neuroprotective role of resveratrol is the activation of SIRT1. Its downstream targets may impact processes such as neuronal survival, mitochondrial biogenesis, and prevention of protein aggregate formation, all of which contribute to the observed delay in symptoms and increased viability in ALS models [90,92,95,96]. Curcumin derivatives show neuroprotective value through several mechanisms, such as restoring mitochondrial functions, normalising cell excitability, and preventing the formation of toxic protein aggregates [113,114,115]. Green tea catechin EGCG has been observed to upregulate a prosurvival signaling pathway PI3K/Akt and decrease signals leading to cell death, such as activation of caspase-3, which is associated with apoptosis [119,120]. Both resulted in the delayed onset of ALS and increased survival in mice models treated with EGCG [120,121]. Fisetin acts by activating the ERK pathway, which modulates cell survival and upregulates HO-1, both of which contribute to the cellular response against oxidative stress [128]. Another mechanism exerted by some polyphenols is the downregulation of the NF-κB pathway that, overall, has an anti-inflammatory effect [91].

Table 1.
Therapeutic implications of different polyphenols in ALS and FTD models.
The importance of the gut–brain axis in ALS/FTD has been recognised. On the one hand, polyphenols may serve as prebiotics and alter the gut microbiota, affecting disease pathogenesis [146], (for a detailed review, see [147]). On the other hand, certain polyphenols such as EGCG are degraded by some gut microbiota, which reduces their bioavailability [148,149]. However, some metabolites do target the brain and have beneficial effects on neurons [118,150].
3. Conclusions
Polyphenols offer new possibilities for the development of therapies for ALS/FTD. However, more research is needed in this field, including strategies for effective targeting and delivery to the site of action. When evaluating the therapeutic potential of polyphenols, we must also consider their uptake in the gut, degradation by the microbiota, and the delivery to the brain. Therefore, it is important whether polyphenols are consumed or administered intravenously and how well they can cross the blood–brain barrier [151,152]. Another hurdle for potential ALS/FTD medication is translating findings from animal models into successful clinical trials. Additional aspect of potential variability in successful treatment lies in the use of purified polyphenols or plant extracts that may act synergistically. Most of the findings reviewed here come from various successful preclinical stages and have yet to be tested in humans. Nevertheless, polyphenols have the potential to improve the treatment of ALS/FTD, either through the development of new drugs or as dietary supplements.
Funding
This work was funded by Slovenian Research Agency grants (P4-0127, P1-0207, J3-9263, J3-8201, J7-9399 and N3-0141) and CRP-ICGEB research grant (CRP/SVN19-03).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shah, H.; Albanese, E.; Duggan, C.; Rudan, I.; Langa, K.M.; Carrillo, M.C.; Chan, K.Y.; Joanette, Y.; Prince, M.; Rossor, M.; et al. Research priorities to reduce the global burden of dementia by 2025. Lancet Neurol. 2016, 15, 1285–1294. [Google Scholar] [CrossRef]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment? Front. Aging Neurosci. 2017, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Panza, F.; Lozupone, M.; Seripa, D.; Daniele, A.; Watling, M.; Giannelli, G.; Imbimbo, B.P. Development of disease-modifying drugs for frontotemporal dementia spectrum disorders. Nat. Rev. Neurol. 2020, 16, 213–228. [Google Scholar] [CrossRef]
- Silva, R.F.M.; Pogačnik, L. Polyphenols from food and natural products: Neuroprotection and safety. Antioxidants 2020, 9, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Longinetti, E.; Regodón Wallin, A.; Samuelsson, K.; Press, R.; Zachau, A.; Ronnevi, L.O.; Kierkegaard, M.; Andersen, P.M.; Hillert, J.; Fang, F.; et al. The Swedish motor neuron disease quality registry. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 528–537. [Google Scholar] [CrossRef]
- Jun, K.Y.; Park, J.; Oh, K.W.; Kim, E.M.; Bae, J.S.; Kim, I.; Kim, S.H. Epidemiology of ALS in Korea using nationwide big data. J. Neurol. Neurosurg. Psychiatry 2019, 90, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Benjaminsen, E.; Alstadhaug, K.B.; Gulsvik, M.; Baloch, F.K.; Odeh, F. Amyotrophic lateral sclerosis in Nordland county, Norway, 2000–2015: Prevalence, incidence, and clinical features. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 522–527. [Google Scholar] [CrossRef]
- Ryan, M.; Zaldívar Vaillant, T.; McLaughlin, R.L.; Doherty, M.A.; Rooney, J.; Heverin, M.; Gutierrez, J.; Lara-Fernández, G.E.; Pita Rodríguez, M.; Hackembruch, J.; et al. Comparison of the clinical and genetic features of amyotrophic lateral sclerosis across Cuban, Uruguayan and Irish clinic-based populations. J. Neurol. Neurosurg. Psychiatry 2019, 90, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Qadri, S.; Langefeld, C.D.; Milligan, C.; Caress, J.B.; Cartwright, M.S. Racial differences in intervention rates in individuals with ALS: A case-control study. Neurology 2019, 92, E1969–E1974. [Google Scholar] [CrossRef]
- Marin, B.; Boumé diene, F.; Logroscino, G.; Couratier, P.; Babron, M.C.; Leutenegger, A.L.; Copetti, M.; Preux, P.M.; Beghi, E. Variation in world wide incidence of amyotrophic lateral sclerosis: A meta-analysis. Int. J. Epidemiol. 2017, 46, 57–74. [Google Scholar] [CrossRef] [Green Version]
- Longinetti, E.; Fang, F. Epidemiology of amyotrophic lateral sclerosis: An update of recent literature. Curr. Opin. Neurol. 2019, 32, 771–776. [Google Scholar] [CrossRef]
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Greaves, C.V.; Rohrer, J.D. An update on genetic frontotemporal dementia. J. Neurol. 2019, 266, 2075–2086. [Google Scholar] [CrossRef] [Green Version]
- Couratier, P.; Corcia, P.; Lautrette, G.; Nicol, M.; Marin, B. ALS and frontotemporal dementia belong to a common disease spectrum. Rev. Neurol. (Paris) 2017, 173, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Coyle-Gilchrist, I.T.S.; Dick, K.M.; Patterson, K.; Rodríquez, P.V.; Wehmann, E.; Wilcox, A.; Lansdall, C.J.; Dawson, K.E.; Wiggins, J.; Mead, S.; et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology 2016, 86, 1736–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kansal, K.; Mareddy, M.; Sloane, K.L.; Minc, A.A.; Rabins, P.V.; McGready, J.B.; Onyike, C.U. Survival in Frontotemporal Dementia Phenotypes: A Meta-Analysis. Dement. Geriatr. Cogn. Disord. 2016, 41, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.S.; Farmer, J.M.; Wood, E.M.; Johnson, J.K.; Boxer, A.; Neuhaus, J.; Lomen-Hoerth, C.; Wilhelmsen, K.C.; Lee, V.M.Y.; Grossman, M.; et al. Comparison of family histories in FTLD subtypes and related tauopathies. Neurology 2005, 65, 1817–1819. [Google Scholar] [CrossRef]
- Rohrer, J.D.; Guerreiro, R.; Vandrovcova, J.; Uphill, J.; Reiman, D.; Beck, J.; Isaacs, A.M.; Authier, A.; Ferrari, R.; Fox, N.C.; et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology 2009, 73, 1451–1456. [Google Scholar] [CrossRef] [Green Version]
- Neary, D.; Snowden, J.S.; Mann, D.M.A.; Northern, B.; Goulding, P.J.; Macdermott, N. Frontal lobe dementia and motor neuron disease. J. Neurol. Neurosurg. Psychiatry 1990, 53, 23–32. [Google Scholar] [CrossRef]
- Mitsuyama, Y. Presenile dementia with motor neuron disease. Dement. Geriatr. Cogn. Disord. 1993, 4, 137–142. [Google Scholar] [CrossRef]
- Ringholz, G.M.; Appel, S.H.; Bradshaw, M.; Cooke, N.A.; Mosnik, D.M.; Schulz, P.E. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 2005, 65, 586–590. [Google Scholar] [CrossRef]
- Wheaton, M.W.; Salamone, A.R.; Mosnik, D.M.; McDonald, R.O.; Appel, S.H.; Schmolck, H.I.; Ringholz, G.M.; Schulz, P.E. Cognitive impairment in familial ALS. Neurology 2007, 69, 1411–1417. [Google Scholar] [CrossRef] [PubMed]
- Burrell, J.R.; Kiernan, M.C.; Vucic, S.; Hodges, J.R. Motor Neuron dysfunction in frontotemporal dementia. Brain 2011, 134, 2582–2594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Langenhove, T.; Piguet, O.; Burrell, J.R.; Leyton, C.; Foxe, D.; Abela, M.; Bartley, L.; Kim, W.S.; Jary, E.; Huang, Y.; et al. Predicting development of amyotrophic lateral sclerosis in frontotemporal dementia. J. Alzheimer Dis. 2017, 58, 163–170. [Google Scholar] [CrossRef]
- Liscic, R.M.; Alberici, A.; Cairns, N.J.; Romano, M.; Buratti, E. From basic research to the clinic: Innovative therapies for ALS and FTD in the pipeline. Mol. Neurodegener. 2020, 15, 1–17. [Google Scholar] [CrossRef]
- Gao, F.; Almeida, S.; Lopez-Gonzalez, R. Dysregulated molecular pathways in amyotrophic lateral sclerosis–frontotemporal dementia spectrum disorder. EMBO J. 2017, 36, 2931–2950. [Google Scholar] [CrossRef]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bampton, A.; Gittings, L.M.; Fratta, P.; Lashley, T.; Gatt, A. The role of hnRNPs in frontotemporal dementia and amyotrophic lateral sclerosis. Acta Neuropathol. 2020, 140, 599–623. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.A.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK 1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef]
- Goode, A.; Butler, K.; Long, J.; Cavey, J.; Scott, D.; Shaw, B.; Sollenberger, J.; Gell, C.; Johansen, T.; Oldham, N.J.; et al. Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy 2016, 12, 1094–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, Y.C.; Holzbaur, E.L.F. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [Green Version]
- Hortobágyi, T.; Troakes, C.; Nishimura, A.L.; Vance, C.; van Swieten, J.C.; Seelaar, H.; King, A.; Al-Sarraj, S.; Rogelj, B.; Shaw, C.E. Optineurin inclusions occur in a minority of TDP-43 positive ALS and FTLD-TDP cases and are rarely observed in other neurodegenerative disorders. Acta Neuropathol. 2011, 121, 519–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markovinovic, A.; Cimbro, R.; Ljutic, T.; Kriz, J.; Rogelj, B.; Munitic, I. Optineurin in amyotrophic lateral sclerosis: Multifunctional adaptor protein at the crossroads of different neuroprotective mechanisms. Prog. Neurobiol. 2017, 154, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Chua, J.P.; De Calbiac, H.; Kabashi, E.; Barmada, S.J. Autophagy and ALS: Mechanistic insights and therapeutic implications. Autophagy 2021, 1–29. [Google Scholar] [CrossRef]
- Buratti, E.; Baralle, F.E. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J. Biol. Chem. 2001, 276, 36337–36343. [Google Scholar] [CrossRef] [Green Version]
- Fallini, C.; Bassell, G.J.; Rossoll, W. The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum. Mol. Genet. 2012, 21, 3703–3718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butti, Z.; Patten, S.A. RNA dysregulation in amyotrophic lateral sclerosis. Front. Genet. 2019, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Zupunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Rogelj, B.; Easton, L.E.; Bogu, G.K.; Stanton, L.W.; Rot, G.; Curk, T.; Zupan, B.; Sugimoto, Y.; Modic, M.; Haberman, N.; et al. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci. Rep. 2012, 2, 603. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Arai, S.; Song, X.; Reichart, D.; Du, K.; Pascual, G.; Tempst, P.; Rosenfeld, M.G.; Glass, C.K.; Kurokawa, R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008, 454, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.Y.; Riley, T.R.; Coady, T.; Bussemaker, H.J.; Manley, J.L. TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proc. Natl. Acad. Sci. USA 2012, 109, 6030–6035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoell, J.I.; Larsson, E.; Runge, S.; Nusbaum, J.D.; Duggimpudi, S.; Farazi, T.A.; Hafner, M.; Borkhardt, A.; Sander, C.; Tuschl, T. RNA targets of wild-type and mutant FET family proteins. Nat. Struct. Mol. Biol. 2011, 18, 1428–1431. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, S.; Masuda, A.; Fujioka, Y.; Iguchi, Y.; Katsuno, M.; Shibata, A.; Urano, F.; Sobue, G.; Ohno, K. Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci. Rep. 2012, 2, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; MacKenzie, I.R.A.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef]
- Webber, C.J.; Lei, S.E.; Wolozin, B. The pathophysiology of neurodegenerative disease: Disturbing the balance between phase separation and irreversible aggregation. Prog. Mol. Biol. Transl. Sci. 2020, 174, 187–223. [Google Scholar] [CrossRef]
- Modic, M.; Grosch, M.; Rot, G.; Schirge, S.; Lepko, T.; Yamazaki, T.; Lee, F.C.Y.; Rusha, E.; Shaposhnikov, D.; Palo, M.; et al. Cross-Regulation between TDP-43 and Paraspeckles Promotes Pluripotency-Differentiation Transition. Mol. Cell 2019, 74, 951–965.e13. [Google Scholar] [CrossRef] [Green Version]
- Prpar Mihevc, S.; Darovic, S.; Kovanda, A.; Bajc Česnik, A.; Župunski, V.; Rogelj, B. Nuclear trafficking in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Brain 2017, 140, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Pasha, T.; Zatorska, A.; Sharipov, D.; Rogelj, B.; Hortobágyi, T.; Hirth, F. Karyopherin abnormalities in neurodegenerative proteinopathies. Brain 2021. [Google Scholar] [CrossRef] [PubMed]
- Cicardi, M.E.; Marrone, L.; Azzouz, M.; Trotti, D. Proteostatic imbalance and protein spreading in amyotrophic lateral sclerosis. EMBO J. 2021, 40, e106389. [Google Scholar] [CrossRef]
- Vance, C.; Scotter, E.L.; Nishimura, A.L.; Troakes, C.; Mitchell, J.C.; Kathe, C.; Urwin, H.; Manser, C.; Miller, C.C.; Hortobágyi, T.; et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum. Mol. Genet. 2013, 22, 2676–2688. [Google Scholar] [CrossRef] [PubMed]
- Portz, B.; Lee, B.L.; Shorter, J. FUS and TDP-43 Phases in Health and Disease. Trends Biochem. Sci. 2021, 46, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.N.; Newhouse, S.; Shatunov, A.; Vance, C.; Topp, S.; Johnson, L.; Miller, J.; Lee, Y.; Troakes, C.; Scott, K.M.; et al. The C9ORF72 expansion mutation is a common cause of ALS+/−FTD in Europe and has a single founder. Eur. J. Hum. Genet. 2013, 21, 102–108. [Google Scholar] [CrossRef]
- Malnar, M.; Rogelj, B. SFPQ regulates the accumulation of RNA foci and dipeptide repeat proteins from the expanded repeat mutation in C9orf72. J. Cell Sci. 2021, 134. [Google Scholar] [CrossRef]
- Božič, T.; Zalar, M.; Rogelj, B.; Plavec, J.; Šket, P. Structural Diversity of Sense and Antisense RNA Hexanucleotide Repeats Associated with ALS and FTLD. Molecules 2020, 25, 525. [Google Scholar] [CrossRef] [Green Version]
- Bajc Česnik, A.; Darovic, S.; Prpar Mihevc, S.; Štalekar, M.; Malnar, M.; Motaln, H.; Lee, Y.-B.; Mazej, J.; Pohleven, J.; Grosch, M.; et al. Nuclear RNA foci from C9ORF72 expansion mutation form paraspeckle-like bodies. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Solomon, D.A.; Stepto, A.; Au, W.H.; Adachi, Y.; Diaper, D.C.; Hall, R.; Rekhi, A.; Boudi, A.; Tziortzouda, P.; Lee, Y.-B.; et al. A feedback loop between dipeptide-repeat protein, TDP-43 and karyopherin-α mediates C9orf72-related neurodegeneration. Brain 2018, 141, 2908–2924. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-B.; Baskaran, P.; Gomez-Deza, J.; Chen, H.-J.; Nishimura, A.L.; Smith, B.N.; Troakes, C.; Adachi, Y.; Stepto, A.; Petrucelli, L.; et al. C9orf72 poly GA RAN-translated protein plays a key role in amyotrophic lateral sclerosis via aggregation and toxicity. Hum. Mol. Genet. 2017, 26, 4765–4777. [Google Scholar] [CrossRef] [Green Version]
- Kovanda, A.; Zalar, M.; Šket, P.; Plavec, J.; Rogelj, B. Anti-sense DNA d(GGCCCC)n expansions in C9ORF72 form i-motifs and protonated hairpins. Sci. Rep. 2015, 5, 17944. [Google Scholar] [CrossRef]
- Šket, P.; Pohleven, J.; Kovanda, A.; Štalekar, M.; Župunski, V.; Zalar, M.; Plavec, J.; Rogelj, B. Characterization of DNA G-quadruplex species forming from C9ORF72 G4C2-expanded repeats associated with amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Neurobiol. Aging 2015, 36, 1091–1096. [Google Scholar] [CrossRef]
- Vatovec, S.; Kovanda, A.; Rogelj, B. Unconventional features of C9ORF72 expanded repeat in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Neurobiol. Aging 2014, 35, 2421.e1–2421.e12. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magrané, J.; Cortez, C.; Gan, W.-B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgins, C.M.J.; Jung, C.; Xu, Z. ALS-associated mutant SODIG93A causes mitochondrial vacuolation by expansion of the intermembrane space by involvement of SODI aggregation and peroxisomes. BMC Neurosci. 2003, 4, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Yang, M.; Chen, Y.; Chen, X.; Liu, J.; Sun, S.; Cheng, H.; Li, Y.; Bigio, E.H.; Mesulam, M.; et al. FUS interacts with HSP60 to promote mitochondrial damage. PLoS Genet. 2015, 11, e1005357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magrané, J.; Hervias, I.; Henning, M.S.; Damiano, M.; Kawamata, H.; Manfredi, G. Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamics abnormalities. Hum. Mol. Genet. 2009, 18, 4552–4564. [Google Scholar] [CrossRef] [Green Version]
- Cozzolino, M.; Pesaresi, M.G.; Amori, I.; Crosio, C.; Ferri, A.; Nencini, M.; Carrì, M.T. Oligomerization of mutant SOD1 in mitochondria of motoneuronal cells drives mitochondrial damage and cell toxicity. Antioxid. Redox Signal. 2009, 11, 1547–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igoudjil, A.; Magrané, J.; Fischer, L.R.; Kim, H.J.; Hervias, I.; Dumont, M.; Cortez, C.; Glass, J.D.; Starkov, A.A.; Manfredi, G. In vivo pathogenic role of mutant SOD1 localized in the mitochondrial intermembrane space. J. Neurosci. 2011, 31, 15826–15837. [Google Scholar] [CrossRef] [Green Version]
- Bogdanov, M.; Brown, R.H.; Matson, W.; Smart, R.; Hayden, D.; O’Donnell, H.; Flint Beal, M.; Cudkowicz, M. Increased oxidative damage to DNA in ALS patients. Free Radic. Biol. Med. 2000, 29, 652–658. [Google Scholar] [CrossRef]
- Simpson, E.P.; Henry, Y.K.; Henkel, J.S.; Smith, R.G.; Appel, S.H. Increased lipid peroxidation in sera of ALS patients: A potential biomarker of disease burden. Neurology 2004, 62, 1758–1765. [Google Scholar] [CrossRef]
- Mitsumoto, H.; Santella, R.; Liu, X.; Bogdanov, M.; Zipprich, J.; Wu, H.C.; Mahata, J.; Kilty, M.; Bednarz, K.; Bell, D.; et al. Oxidative stress biomarkers in sporadic ALS. Amyotroph. Lateral Scler. 2008, 9, 177–183. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Lacomblez, L.; Bensimon, G.; Leigh, P.N.; Guillet, P.; Meininger, V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Lancet 1996, 347, 1425–1431. [Google Scholar] [CrossRef]
- Cheah, B.C.; Vucic, S.; Krishnan, A.; Kiernan, M.C. Riluzole, neuroprotection and amyotrophic lateral sclerosis. Curr. Med. Chem. 2010, 17, 1942–1959. [Google Scholar] [CrossRef]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K.; et al. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef]
- Hodges, J.R.; Piguet, O. Progress and challenges in frontotemporal dementia research: A 20-year review. J. Alzheimer Dis. 2018, 62, 1467–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swallah, M.S.; Sun, H.; Affoh, R.; Fu, H.; Yu, H. Antioxidant Potential Overviews of Secondary Metabolites (Polyphenols) in Fruits. Int. J. Food Sci. 2020, 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rambaran, T.F. Nanopolyphenols: A review of their encapsulation and anti-diabetic effects. SN Appl. Sci. 2020, 2, 1–26. [Google Scholar] [CrossRef]
- Yang, B.; Dong, Y.; Wang, F.; Zhang, Y. Nanoformulations to enhance the bioavailability and physiological functions of polyphenols. Molecules 2020, 25, 4613. [Google Scholar] [CrossRef] [PubMed]
- Tsao, R. Chemistry and biochemistry of dietary polyphenols. Nutrients 2010, 2, 1231–1246. [Google Scholar] [CrossRef]
- Solanki, I.; Parihar, P.; Mansuri, M.L.; Parihar, M.S. Flavonoid-based therapies in the early management of neurodegenerative diseases. Adv. Nutr. 2015, 6, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Tian, B.; Liu, J. Resveratrol: A review of plant sources, synthesis, stability, modification and food application. J. Sci. Food Agric. 2020, 100, 1392–1404. [Google Scholar] [CrossRef]
- Tellone, E.; Galtieri, A.; Russo, A.; Giardina, B.; Ficarra, S. Resveratrol: A focus on several neurodegenerative diseases. Oxid. Med. Cell. Longev. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Nguyen, M.D.; Dobbin, M.M.; Fischer, A.; Sananbenesi, F.; Rodgers, J.T.; Delalle, I.; Baur, J.A.; Sui, G.; Armour, S.M.; et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007, 26, 3169–3179. [Google Scholar] [CrossRef]
- Barber, S.C.; Higginbottom, A.; Mead, R.J.; Barber, S.; Shaw, P.J. An in vitro screening cascade to identify neuroprotective antioxidants in ALS. Free Radic. Biol. Med. 2009, 46, 1127–1138. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, Y.; Tang, L.; Zhang, N.; Fan, D. Protective effects of resveratrol through the up-regulation of SIRT1 expression in the mutant hSOD1-G93A-bearing motor neuron-like cell culture model of amyotrophic lateral sclerosis. Neurosci. Lett. 2011, 503, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Yáñez, M.; Galán, L.; Matías-Guiu, J.; Vela, A.; Guerrero, A.; García, A.G. CSF from amyotrophic lateral sclerosis patients produces glutamate independent death of rat motor brain cortical neurons: Protection by resveratrol but not riluzole. Brain Res. 2011, 1423, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Markert, C.D.; Kim, E.; Gifondorwa, D.J.; Childers, M.K.; Milligan, C.E. A single-dose resveratrol treatment in a mouse model of amyotrophic lateral sclerosis. J. Med. Food 2010, 13, 1081–1085. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Choi, J.R.; Soon Shin, K.; Kang, S.J. Resveratrol upregulated heat shock proteins and extended the survival of G93A-SOD1 mice. Brain Res. 2012, 1483, 112–117. [Google Scholar] [CrossRef]
- Mancuso, R.; del Valle, J.; Modol, L.; Martinez, A.; Granado-Serrano, A.B.; Ramirez-Núñez, O.; Pallás, M.; Portero-Otin, M.; Osta, R.; Navarro, X. Resveratrol improves motoneuron function and extends survival in SOD1G93A ALS Mice. Neurotherapeutics 2014, 11, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, R.; Del Valle, J.; Morell, M.; Pallás, M.; Osta, R.; Navarro, X. Lack of synergistic effect of resveratrol and sigma-1 receptor agonist (PRE-084) in SOD1G93A ALS mice: Overlapping effects or limited therapeutic opportunity? Orphanet J. Rare Dis. 2014, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Schiaffino, L.; Bonafede, R.; Scambi, I.; Parrella, E.; Pizzi, M.; Mariotti, R. Acetylation state of RelA modulated by epigenetic drugs prolongs survival and induces a neuroprotective effect on ALS murine model. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.C.; Kwan, P.; Cheung, S.K.K.; Ho, A.; Baum, L. Effects of resveratrol and morin on insoluble tau in tau transgenic mice. Transl. Neurosci. 2018, 9, 54–60. [Google Scholar] [CrossRef]
- Laudati, G.; Mascolo, L.; Guida, N.; Sirabella, R.; Pizzorusso, V.; Bruzzaniti, S.; Serani, A.; Di Renzo, G.; Canzoniero, L.M.T.; Formisano, L. Resveratrol treatment reduces the vulnerability of SH-SY5Y cells and cortical neurons overexpressing SOD1-G93A to Thimerosal toxicity through SIRT1/DREAM/PDYN pathway. Neurotoxicology 2019, 71, 6–15. [Google Scholar] [CrossRef]
- Cao, D.; Wang, M.; Qiu, X.; Liu, D.; Jiang, H.; Yang, N.; Xu, R.M. Structural basis for allosteric, substratedependent stimulation of SIRT1 activity by resveratrol. Genes Dev. 2015, 29, 1316–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranganathan, S.; Bowser, R. p53 and cell cycle proteins participate in spinal motor neuron cell death in ALS. Open Pathol. J. 2010, 4, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, Y.C.; Jeong, S.G.; Kim, S.H.; Cho, G.W. Reduced sirtuin 1/adenosine monophosphate-activated protein kinase in amyotrophic lateral sclerosis patient-derived mesenchymal stem cells can be restored by resveratrol. J. Tissue Eng. Regen. Med. 2019, 13, 110–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, B.; Milbrandt, J. Resveratrol stimulates AMP kinase activity in neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 7217–7222. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, E.; Rajasekaran, R. Quantum chemical and molecular mechanics studies on the assessment of interactions between resveratrol and mutant SOD1 (G93A) protein. J. Comput. Aided Mol. Des. 2018, 32, 1347–1361. [Google Scholar] [CrossRef]
- Zhuang, X.; Li, X.; Zhao, B.; Liu, Z.; Song, F.; Lu, J. Native mass spectrometry based method for studying the interactions between superoxide dismutase 1 and stilbenoids. ACS Chem. Neurosci. 2020, 11, 184–190. [Google Scholar] [CrossRef]
- Adami, R.; Bottai, D. Curcumin and neurological diseases. Nutr. Neurosci. 2020, 1–21. [Google Scholar] [CrossRef]
- Soo, S.K.; Rudich, P.D.; Traa, A.; Harris-Gauthier, N.; Shields, H.J.; Van Raamsdonk, J.M. Compounds that extend longevity are protective in neurodegenerative diseases and provide a novel treatment strategy for these devastating disorders. Mech. Ageing Dev. 2020, 190, 111297. [Google Scholar] [CrossRef]
- Bhatia, N.K.; Srivastava, A.; Katyal, N.; Jain, N.; Khan, M.A.I.; Kundu, B.; Deep, S. Curcumin binds to the pre-fibrillar aggregates of Cu/Zn superoxide dismutase (SOD1) and alters its amyloidogenic pathway resulting in reduced cytotoxicity. Biochim. Biophys. Acta Proteins Proteom. 2015, 1854, 426–436. [Google Scholar] [CrossRef]
- Rane, J.S.; Bhaumik, P.; Panda, D. Curcumin inhibits tau aggregation and disintegrates preformed tau filaments in vitro. J. Alzheimer’s Dis. 2017, 60, 999–1014. [Google Scholar] [CrossRef]
- den Haan, J.; Morrema, T.H.J.; Rozemuller, A.J.; Bouwman, F.H.; Hoozemans, J.J.M. Different curcumin forms selectively bind fibrillar amyloid beta in post mortem Alzheimer’s disease brains: Implications for in-vivo diagnostics. Acta Neuropathol. Commun. 2018, 6, 75. [Google Scholar] [CrossRef]
- Ma, Z.; Wang, N.; He, H.; Tang, X. Pharmaceutical strategies of improving oral systemic bioavailability of curcumin for clinical application. J. Control. Release 2019, 316, 359–380. [Google Scholar] [CrossRef]
- Lu, J.; Duan, W.; Guo, Y.; Jiang, H.; Li, Z.; Huang, J.; Hong, K.; Li, C. Mitochondrial dysfunction in human TDP-43 transfected NSC34 cell lines and the protective effect of dimethoxy curcumin. Brain Res. Bull. 2012, 89, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Xu, L.; Wu, L.; Wang, X.; Duan, W.; Li, H.; Li, C. Curcumin abolishes mutant TDP-43 induced excitability in a motoneuron-like cellular model of ALS. Neuroscience 2014, 272, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Guo, Y.; Xiao, J.; Chen, X.; Li, Z.; Han, H.; Li, C. Neuroprotection by monocarbonyl dimethoxycurcumin C: Ameliorating the toxicity of mutant TDP-43 via HO-1. Mol. Neurobiol. 2014, 49, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Tripodo, G.; Chlapanidas, T.; Perteghella, S.; Vigani, B.; Mandracchia, D.; Trapani, A.; Galuzzi, M.; Tosca, M.C.; Antonioli, B.; Gaetani, P.; et al. Mesenchymal stromal cells loading curcumin-INVITE-micelles: A drug delivery system for neurodegenerative diseases. Colloids Surf. B Biointerfaces 2015, 125, 300–308. [Google Scholar] [CrossRef]
- Ahmadi, M.; Agah, E.; Nafissi, S.; Jaafari, M.R.; Harirchian, M.H.; Sarraf, P.; Faghihi-Kashani, S.; Hosseini, S.J.; Ghoreishi, A.; Aghamollaii, V.; et al. Safety and efficacy of nanocurcumin as add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: A pilot randomized clinical trial. Neurotherapeutics 2018, 15, 430–438. [Google Scholar] [CrossRef] [Green Version]
- Pervin, M.; Unno, K.; Takagaki, A.; Isemura, M.; Nakamura, Y. Function of green tea catechins in the brain: Epigallocatechin gallate and its metabolites. Int. J. Mol. Sci. 2019, 20, 3630. [Google Scholar] [CrossRef] [Green Version]
- Koh, S.H.; Kwon, H.; Kim, K.S.; Kim, J.; Kim, M.H.; Yu, H.J.; Kim, M.; Lee, K.W.; Do, B.R.; Jung, H.K.; et al. Epigallocatechin gallate prevents oxidative-stress-induced death of mutant Cu/Zn-superoxide dismutase (G93A) motoneuron cells by alteration of cell survival and death signals. Toxicology 2004, 202, 213–225. [Google Scholar] [CrossRef]
- Koh, S.H.; Lee, S.M.; Kim, H.Y.; Lee, K.Y.; Lee, Y.J.; Kim, H.T.; Kim, J.; Kim, M.H.; Hwang, M.S.; Song, C.; et al. The effect of epigallocatechin gallate on suppressing disease progression of ALS model mice. Neurosci. Lett. 2006, 395, 103–107. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, S.; Li, X.; Luo, G.; Li, L.; Le, W. Neuroprotective effects of (-)-epigallocatechin-3-gallate in a transgenic mouse model of amyotrophic lateral sclerosis. Neurochem. Res. 2006, 31, 1263–1269. [Google Scholar] [CrossRef]
- Che, F.; Wang, G.; Yu, J.; Wang, X.; Lu, Y.; Fu, Q.; Su, Q.; Jiang, J.; Du, Y. Effects of epigallocatechin-3-gallate on iron metabolismin spinal cord motor neurons. Mol. Med. Rep. 2017, 16, 3010–3014. [Google Scholar] [CrossRef]
- Srinivasan, E.; Rajasekaran, R. Probing the inhibitory activity of epigallocatechin-gallate on toxic aggregates of mutant (L84F) SOD1 protein through geometry based sampling and steered molecular dynamics. J. Mol. Graph. Model. 2017, 74, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Zhuang, X.; Pi, Z.; Liu, S.; Liu, Z.; Song, F. Determining the effect of catechins on SOD1 conformation and aggregation by ion mobility mass spectrometry combined with optical spectroscopy. J. Am. Soc. Mass Spectrom. 2018, 29, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.F.; Chang, H.Y.; Hou, S.C.; Liou, G.G.; Der Way, T.; James Shen, C.K. The self-interaction of native TDP-43 C terminus inhibits its degradation and contributes to early proteinopathies. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, S.; Suzuki, N.; Masuda, M.; Hisanaga, S.I.; Iwatsubo, T.; Goedert, M.; Hasegawa, M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 2005, 280, 7614–7623. [Google Scholar] [CrossRef] [Green Version]
- Winter, A.N.; Ross, E.K.; Wilkins, H.M.; Stankiewicz, T.R.; Wallace, T.; Miller, K.; Linseman, D.A. An anthocyanin-enriched extract from strawberries delays disease onset and extends survival in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. Nutr. Neurosci. 2018, 21, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.H.; Wang, S.Y.; Wang, X.D.; Jiang, H.Q.; Yang, Y.Q.; Wang, Y.; Cheng, J.L.; Zhang, C.T.; Liang, W.W.; Feng, H.L. Fisetin exerts antioxidant and neuroprotective effects in multiple mutant hSOD1 models of amyotrophic lateral sclerosis by activating ERK. Neuroscience 2018, 379, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, E.; Rajasekaran, R. Comparative binding of kaempferol and kaempferide on inhibiting the aggregate formation of mutant (G85R) SOD1 protein in familial amyotrophic lateral sclerosis: A quantum chemical and molecular mechanics study. BioFactors 2018, 44, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Inden, M.; Shirai, K.; Sekine, S.I.; Masaki, Y.; Kurita, H.; Ichihara, K.; Inuzuka, T.; Hozumi, I. The effects of Brazilian green propolis that contains flavonols against mutant copper-zinc superoxide dismutase-mediated toxicity. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Said Ahmed, M.; Hung, W.Y.; Zu, J.S.; Hockberger, P.; Siddique, T. Increased reactive oxygen species in familial amyotrophic lateral sclerosis with mutations in SOD1. J. Neurol. Sci. 2000, 176, 88–94. [Google Scholar] [CrossRef]
- Ip, P.; Sharda, P.R.; Cunningham, A.; Chakrabartty, S.; Pande, V.; Chakrabartty, A. Quercitrin and quercetin 3-β-d-glucoside as chemical chaperones for the A4V SOD1 ALS-causing mutant. Protein Eng. Des. Sel. 2017, 30, 431–440. [Google Scholar] [CrossRef]
- Bhatia, N.K.; Modi, P.; Sharma, S.; Deep, S. Quercetin and baicalein act as potent antiamyloidogenic and fibril destabilizing agents for SOD1 fibrils. ACS Chem. Neurosci. 2020, 11, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.R.; Wani, W.Y.; Sunkaria, A.; Kandimalla, R.J.; Sharma, R.K.; Verma, D.; Bal, A.; Gill, K.D. Quercetin attenuates neuronal death against aluminum-induced neurodegeneration in the rat hippocampus. Neuroscience 2016, 324, 163–176. [Google Scholar] [CrossRef]
- Korkmaz, O.T.; Aytan, N.; Carreras, I.; Choi, J.K.; Kowall, N.W.; Jenkins, B.G.; Dedeoglu, A. 7,8-Dihydroxyflavone improves motor performance and enhances lower motor neuronal survival in a mouse model of amyotrophic lateral sclerosis. Neurosci. Lett. 2014, 566, 286–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trieu, V.N.; Uckun, F.M. Genistein is neuroprotective in murine models of familial amyotrophic lateral sclerosis and stroke. Biochem. Biophys. Res. Commun. 1999, 258, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Fu, J.; Li, S.; Li, Z. Neuroprotective effects of genistein in a SOD1-G93A transgenic mouse model of amyotrophic lateral sclerosis. J. Neuroimmune Pharmacol. 2019, 14, 688–696. [Google Scholar] [CrossRef]
- Assogna, M.; Casula, E.P.; Borghi, I.; Bonnì, S.; Samà, D.; Motta, C.; Di Lorenzo, F.; D’Acunto, A.; Porrazzini, F.; Minei, M.; et al. Effects of palmitoylethanolamide combined with luteoline on frontal lobe functions, high frequency oscillations, and GABAergic transmission in patients with frontotemporal dementia. J. Alzheimer Dis. 2020, 76, 1297–1308. [Google Scholar] [CrossRef]
- Koza, L.A.; Winter, A.N.; Holsopple, J.; Baybayon-Grandgeorge, A.N.; Pena, C.; Olson, J.R.; Mazzarino, R.C.; Patterson, D.; Linseman, D.A. Protocatechuic acid extends survival, improves motor function, diminishes gliosis, and sustains neuromuscular junctions in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. Nutrients 2020, 12, 1824. [Google Scholar] [CrossRef]
- Fontanilla, C.V.; Wei, X.; Zhao, L.; Johnstone, B.; Pascuzzi, R.M.; Farlow, M.R.; Du, Y. Caffeic acid phenethyl ester extends survival of a mouse model of amyotrophic lateral sclerosis. Neuroscience 2012, 205, 185–193. [Google Scholar] [CrossRef]
- Maya, S.; Prakash, T.; Goli, D. Evaluation of neuroprotective effects of wedelolactone and gallic acid on aluminium-induced neurodegeneration: Relevance to sporadic amyotrophic lateral sclerosis. Eur. J. Pharmacol. 2018, 835, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Maya, S.; Prakash, T.; Goli, D. Effect of wedelolactone and gallic acid on quinolinic acid-induced neurotoxicity and impaired motor function: Significance to sporadic amyotrophic lateral sclerosis. Neurotoxicology 2018, 68, 1–12. [Google Scholar] [CrossRef]
- Shimojo, Y.; Kosaka, K.; Noda, Y.; Shimizu, T.; Shirasawa, T. Effect of rosmarinic acid in motor dysfunction and life span in a mouse model of familial amyotrophic lateral sclerosis. J. Neurosci. Res. 2010, 88, 896–904. [Google Scholar] [CrossRef]
- Seo, J.-S.; Choi, J.; Leem, Y.-H.; Han, P.-L. Rosmarinic acid alleviates neurological symptoms in the G93A-SOD1 transgenic mouse model of amyotrophic lateral sclerosis. Exp. Neurobiol. 2015, 24, 341–350. [Google Scholar] [CrossRef] [Green Version]
- West, M.; Mhatre, M.; Ceballos, A.; Floyd, R.A.; Grammas, P.; Gabbita, S.P.; Hamdheydari, L.; Mai, T.; Mou, S.; Pye, Q.N.; et al. The arachidonic acid 5-lipoxygenase inhibitor nordihydroguaiaretic acid inhibits tumor necrosis factor α activation of microglia and extends survival of G93A-SOD1 transgenic mice. J. Neurochem. 2004, 91, 133–143. [Google Scholar] [CrossRef]
- Blacher, E.; Bashiardes, S.; Shapiro, H.; Rothschild, D.; Mor, U.; Dori-Bachash, M.; Kleimeyer, C.; Moresi, C.; Harnik, Y.; Zur, M.; et al. Potential Roles of Gut Microbiome and Metabolites in Modulating ALS in Mice. Nature 2019, 572, 474–480. [Google Scholar] [CrossRef]
- Casani-Cubel, J.; Benlloch, M.; Sanchis-Sanchis, C.E.; Marin, R.; Lajara-Romance, J.M.; de la Rubia Orti, J.E. The impact of microbiota on the pathogenesis of amyotrophic lateral sclerosis and the possible benefits of polyphenols. An overview. Metabolites 2021, 11, 120. [Google Scholar] [CrossRef] [PubMed]
- Unno, T.; Takeo, T. Absorption of (–)-epigallocatechin gallate into the circulation system of rats. Biosci. Biotechnol. Biochem. 1995, 59, 1558–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.B.; Tao, S.; Lee, M.-J.; Hu, Q.; Meng, X.; Lin, Y.; Yang, C.S. Effects of gut microbiota and time of treatment on tissue levels of green tea polyphenols in mice. BioFactors 2018, 44, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Gasperotti, M.; Passamonti, S.; Tramer, F.; Masuero, D.; Guella, G.; Mattivi, F.; Vrhovsek, U. Fate of microbial metabolites of dietary polyphenols in rats: Is the brain their target destination? ACS Chem. Neurosci. 2015, 6, 1341–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pogačnik, L.; Pirc, K.; Palmela, I.; Skrt, M.; Kim, K.S.; Brites, D.; Brito, M.A.; Ulrih, N.P.; Silva, R.F.M. Potential for brain accessibility and analysis of stability of selected flavonoids in relation to neuroprotection in vitro. Brain Res. 2016, 1651, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Pervin, M.; Unno, K.; Nakagawa, A.; Takahashi, Y.; Iguchi, K.; Yamamoto, H.; Hoshino, M.; Hara, A.; Takagaki, A.; Nanjo, F.; et al. Blood brain barrier permeability of (−)-epigallocatechin gallate, its proliferation-enhancing activity of human neuroblastoma SH-SY5Y cells, and its preventive effect on age-related cognitive dysfunction in mice. Biochem. Biophys. Rep. 2017, 9, 180–186. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).