
Beyond Antioxidant Effects: Nature-Based Templates Unveil New Strategies for Neurodegenerative Diseases





Abstract
1. Introduction
2. Crucial Role of ROS in Physiological and Pathological Mechanisms
2.1. ROS and Mitochondrial Dysfunctions
2.2. Metal Accumulation, ROS Production, and Protein Misfolding
2.3. OS and Protein Misfolding/Accumulation
3. Nature-Based Compounds against Cellular Aging and Neurodegeneration
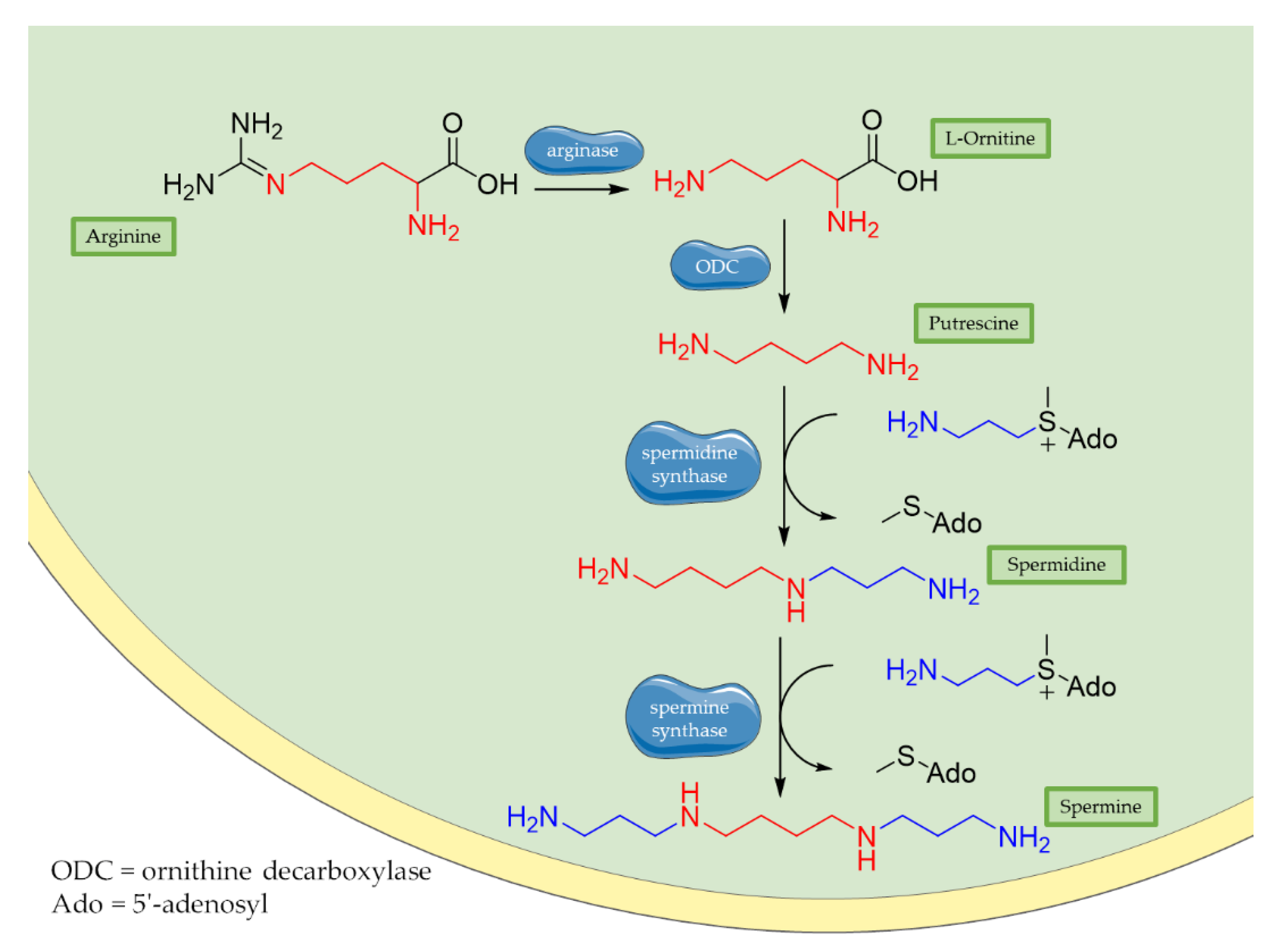
3.1. Polyamines: Spermidine and Spermine
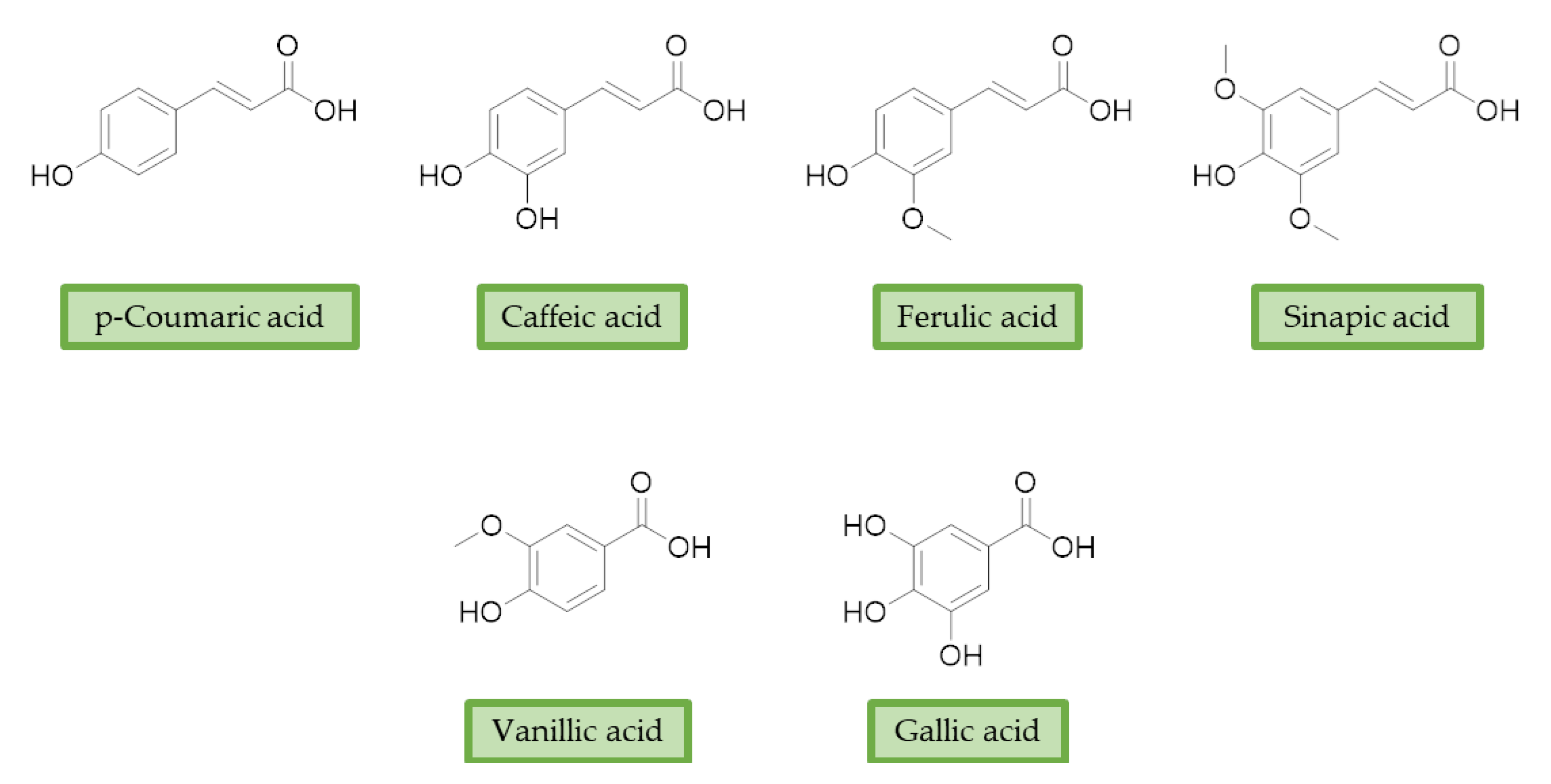
3.2. Phenolic Acids
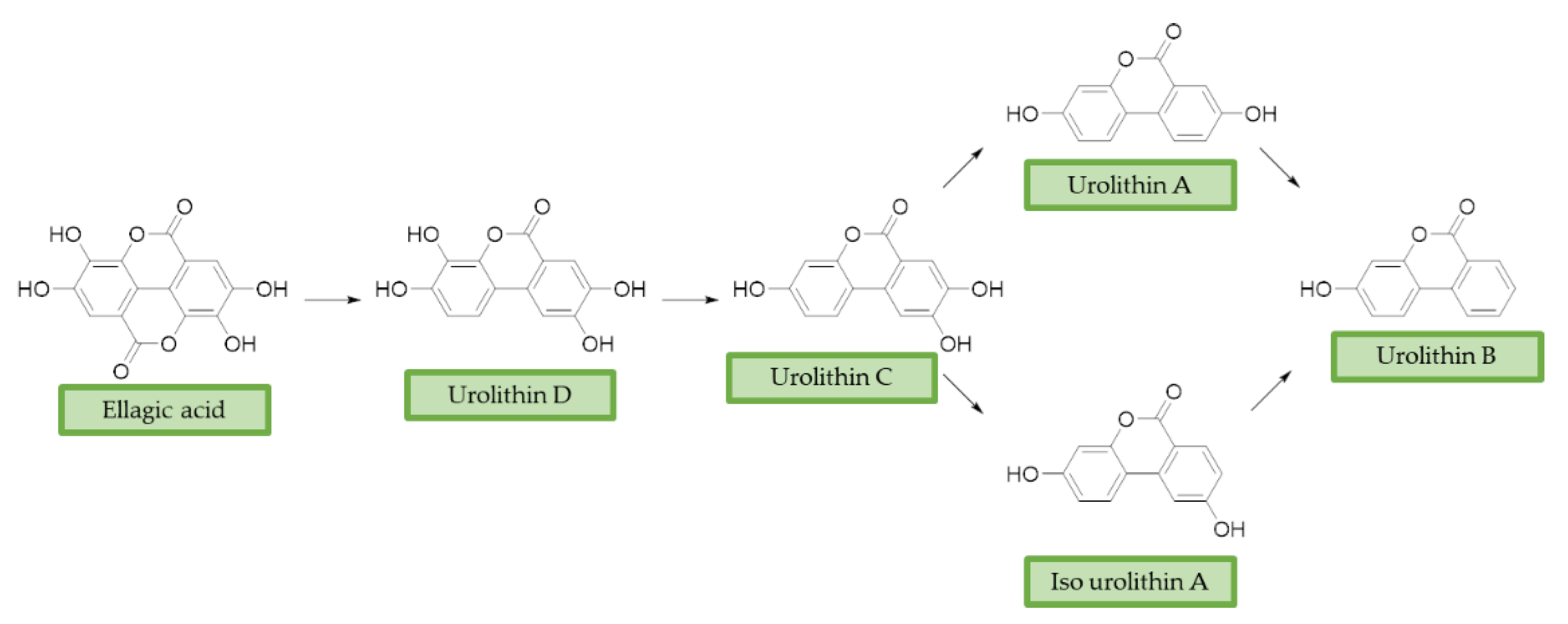
3.3. Urolithins
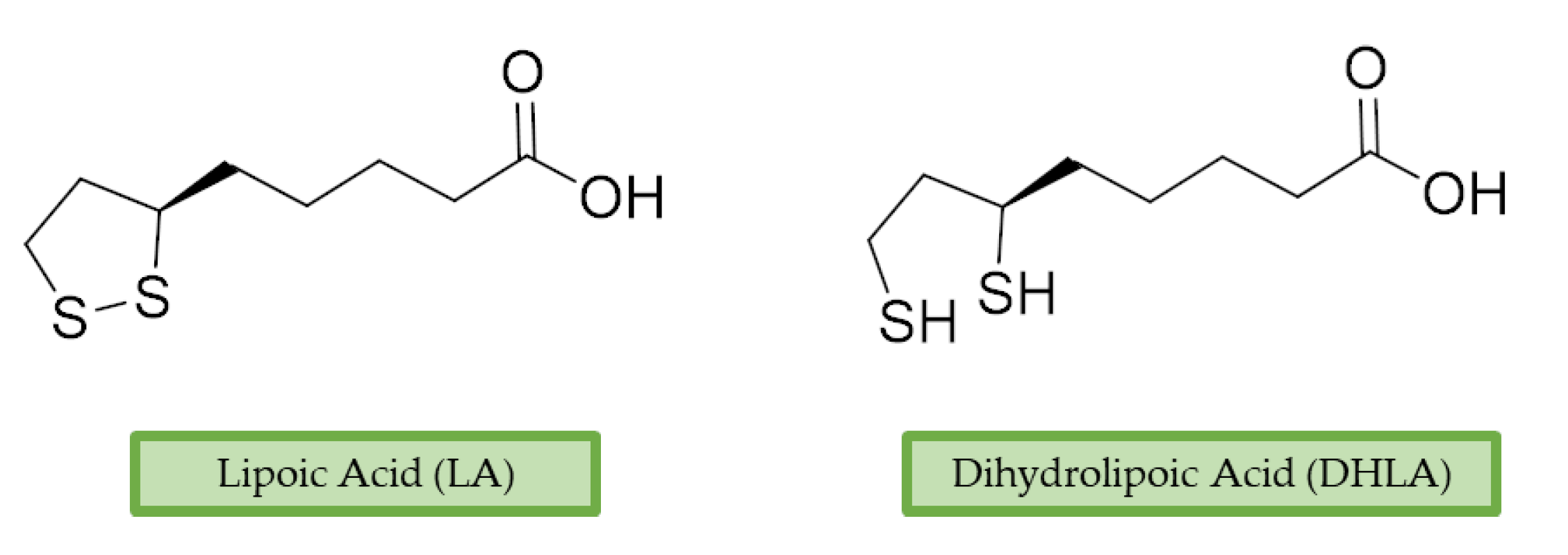
3.4. Lipoic Acid
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Johnson, I.P. Age-related neurodegenerative disease research needs aging models. Front. Aging Neurosci. 2015, 7, 168. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxidative Med. Cell. Longev. 2017, 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sidorova, Y.; Domanskyi, A. Detecting Oxidative Stress Biomarkers in Neurodegenerative Disease Models and Patients. Methods Protoc. 2020, 3, 66. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural Regen. Res. 2012, 7, 376–385. [Google Scholar]
- Nesi, G.; Sestito, S.; Digiacomo, M.; Rapposelli, S. Oxidative Stress, Mitochondrial Abnormalities and Proteins Deposition: Multitarget Approaches in Alzheimer’s Disease. Curr. Top. Med. Chem. 2017, 17, 3062–3079. [Google Scholar] [PubMed]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef]
- Fujikake, N.; Shin, M.; Shimizu, S. Association Between Autophagy and Neurodegenerative Diseases. Front. Neurosci. 2018, 12, 255. [Google Scholar] [CrossRef] [PubMed]
- Krisko, A.; Radman, M. Protein damage, ageing and age-related diseases. Open Biol. 2019, 9, 180249. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Schiavone, S.; Trabace, L. Small Molecules: Therapeutic Application in Neuropsychiatric and Neurodegenerative Disorders. Molecules 2018, 23, 411. [Google Scholar] [CrossRef] [PubMed]
- Flora, S.J.S. Structural, Chemical and Biological Aspects of Antioxidants for Strategies Against Metal and Metalloid Exposure. Oxidative Med. Cell. Longev. 2009, 2, 191–206. [Google Scholar] [CrossRef]
- Zeng, Q.; Siu, W.; Li, L.; Jin, Y.; Liang, S.; Cao, M.; Ma, M.; Wu, Z. Autophagy in Alzheimer’s disease and promising modulatory effects of herbal medicine. Exp. Gerontol. 2019, 119, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-R.; Fu, Y.-S.; Tsai, M.-J.; Cheng, H.; Weng, C.-F. Natural Compounds from Herbs that can Potentially Execute as Autophagy Inducers for Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1412. [Google Scholar] [CrossRef]
- Chung, Y.J.; Robert, C.; Gough, S.M.; Rassool, F.V.; Aplan, P.D. Oxidative stress leads to increased mutation frequency in a murine model of myelodysplastic syndrome. Leuk. Res. 2014, 38, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Luceri, C.; Bigagli, E.; Femia, A.P.; Caderni, G.; Giovannelli, L.; Lodovici, M. Aging related changes in circulating reactive oxygen species (ROS) and protein carbonyls are indicative of liver oxidative injury. Toxicol. Rep. 2018, 5, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A.; Stanley, B.A.; Sivakumaran, V.; Kembro, J.M.; O’Rourke, B.; Paolocci, N.; Cortassa, S. Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: An experimental-computational study. J. Gen. Physiol. 2012, 139, 479–491. [Google Scholar] [CrossRef]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta Bioenerg. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef]
- Dhillon, V.S.; Fenech, M. Mutations that affect mitochondrial functions and their association with neurodegenerative diseases. Mutat. Res. Rev. Mutat. Res. 2014, 759, 1–13. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Maremonti, E.; Eide, D.M.; Rossbach, L.M.; Lind, O.C.; Salbu, B.; Brede, D.A. In vivo assessment of reactive oxygen species production and oxidative stress effects induced by chronic exposure to gamma radiation in Caenorhabditis elegans. Free Radic. Biol. Med. 2019, 152, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Kauppila, J.H.; Stewart, J.B. Mitochondrial DNA: Radically free of free-radical driven mutations. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1354–1361. [Google Scholar] [CrossRef] [PubMed]
- Panel, M.; Ghaleh, B.; Morin, D. Mitochondria and aging: A role for the mitochondrial transition pore? Aging Cell 2018, 17, e12793. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A.; Cortassa, S.; Akar, F.G.; O’Rourke, B. Mitochondrial criticality: A new concept at the turning point of life or death. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Stewart, V.C.; Heales, S.J. Nitric oxide-induced mitochondrial dysfunction: Implications for neurodegeneration. Free. Radic. Biol. Med. 2003, 34, 287–303. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Brenner, C. The adenine nucleotide translocase: A central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem. 2003, 10, 1507–1525. [Google Scholar] [CrossRef]
- Wei, Y.H.; Lu, C.Y.; Wei, C.Y.; Ma, Y.S.; Lee, H.C. Oxidative stress in human aging and mitochondrial disease-consequences of defective mitochondrial respiration and impaired antioxidant enzyme system. Chin. J. Physiol. 2001, 44, 1–11. [Google Scholar] [PubMed]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef]
- Adam-Vizi, V.; Starkov, A.A. Calcium and Mitochondrial Reactive Oxygen Species Generation: How to Read the Facts. J. Alzheimer’s Dis. 2010, 20, S413–S426. [Google Scholar] [CrossRef]
- Tranah, G.J.; Nalls, M.A.; Katzman, S.M.; Yokoyama, J.S.; Lam, E.T.; Zhao, Y.; Mooney, S.; Thomas, F.; Newman, A.B.; Liu, Y.; et al. Mitochondrial DNA Sequence Variation Associated with Dementia and Cognitive Function in the Elderly. J. Alzheimer’s Dis. 2012, 32, 357–372. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Beal, M.F. Mitochondria targeted therapeutic approaches in Parkinson’s and Huntington’s diseases. Mol. Cell. Neurosci. 2013, 55, 101–114. [Google Scholar] [CrossRef]
- Reddy, P.H. Mitochondrial oxidative damage in aging and Alzheimer’s disease: Implications for mitochondrially targeted antioxidant therapeutics. J. Biomed. Biotechnol. 2006, 2006, 31372. [Google Scholar] [CrossRef]
- Kawamata, H.; Manfredi, G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech. Ageing Dev. 2010, 131, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Wells, F.R.; Lee, A.J.; Agid, F.; Agid, Y.; Jenner, P.; Marsden, C.D. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J. Neurochem. 1989, 52, 1830–1836. [Google Scholar] [CrossRef]
- Valko, M.M.H.C.M.; Morris, H.; Cronin, M.T.D. Metals, Toxicity and Oxidative Stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiao, Q.; Xu, H.; Du, X.; Shi, L.; Jia, F.; Jiang, H. Biometal Dyshomeostasis and Toxic Metal Accumulations in the Development of Alzheimer’s Disease. Front. Mol. Neurosci. 2017, 10, 339. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Bush, A.I. Metals in Alzheimer’s and Parkinson’s diseases. Curr. Opin. Chem. Biol. 2008, 12, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Baros, S.; Valko, M. Redox active metal-induced oxidative stress in biological systems. Transit. Met. Chem. 2012, 37, 127–134. [Google Scholar] [CrossRef]
- Zatta, P.; Kiss, T.; Suwalsky, M.; Berthon, G. Aluminium (III) as a promoter of cellular oxidation. Coord. Chem. Rev. 2002, 228, 271–284. [Google Scholar] [CrossRef]
- Farina, M.; Avila, D.S.; da Rocha, J.B.T.; Aschner, M. Metals, oxidative stress and neurodegeneration: A focus on iron, manganese and mercury. Neurochem. Int. 2013, 62, 575–594. [Google Scholar] [CrossRef]
- Breydo, L.; Uversky, V.N. Role of metal ions in aggregation of intrinsically disordered proteins in neurodegenerative diseases. Metallomics 2011, 3, 1163–1180. [Google Scholar] [CrossRef] [PubMed]
- Aschner, M.; Chen, P.; Martinez-Finley, E.J.; Bornhorst, J.; Chakraborty, S. Metal-induced neurodegeneration in C. elegans. Front. Aging Neurosci. 2013, 5, 18. [Google Scholar]
- Leal, S.S.; Botelho, H.M.; Gomes, C.M. Metal ions as modulators of protein conformation and misfolding in neurodegeneration. Coord. Chem. Rev. 2012, 256, 2253–2270. [Google Scholar] [CrossRef]
- Urrutia, P.J.; Mena, N.P.; Nunez, M.T. The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front. Pharmacol. 2014, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Bolognin, S.; Drago, D.; Messori, L.; Zatta, P. Chelation therapy for neurodegenerative diseases. Med. Res. Rev. 2009, 29, 547–570. [Google Scholar] [CrossRef]
- Sales, T.A.; Prandi, I.G.; de Castro, A.A.; Leal, D.H.S.; da Cunha, E.F.F.; Kuca, K.; Ramalho, T.C. Recent Developments in Metal-Based Drugs and Chelating Agents for Neurodegenerative Diseases Treatments. Int. J. Mol. Sci. 2019, 20, 1829. [Google Scholar] [CrossRef] [PubMed]
- Nedelsky, N.B.; Todd, P.K.; Taylor, J.P. Autophagy and the ubiquitin-proteasome system: Collaborators in neuroprotection. Biochim. Biophys. Acta Mol. Basis Dis. 2008, 1782, 691–699. [Google Scholar] [CrossRef]
- Valastyan, J.S.; Lindquist, S. Mechanisms of protein-folding diseases at a glance. Dis. Model. Mech. 2014, 7, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Korovila, I.; Hugo, M.; Castro, J.P.; Weber, D.; Höhn, A.; Grune, T.; Jung, T. Proteostasis, oxidative stress and aging. Redox Biol. 2017, 13, 550–567. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, N.; Bross, P. Protein Misfolding and Cellular Stress: An Overview. Adv. Struct. Saf. Stud. 2010, 648, 3–23. [Google Scholar] [CrossRef]
- Hipp, M.S.; Park, S.-H.; Hartl, F.U. Proteostasis impairment in protein-misfolding and-aggregation diseases. Trends Cell Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Yue, Z. Neuronal Aggregates: Formation, Clearance, and Spreading. Dev. Cell 2015, 32, 491–501. [Google Scholar] [CrossRef]
- Carloni, S.; Buonocore, G.; Balduini, W. Protective role of autophagy in neonatal hypoxia–ischemia induced brain injury. Neurobiol. Dis. 2008, 32, 329–339. [Google Scholar] [CrossRef]
- Giordano, S.; Darley-Usmar, V.; Zhang, J. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol. 2014, 2, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, J.R. Oxidative stress impairs autophagy through oxidation of ATG3 and ATG7. Autophagy 2018, 14, 1092–1093. [Google Scholar] [CrossRef] [PubMed]
- Kesidou, E.; Lagoudaki, R.; Touloumi, O.; Poulatsidou, K.-N.; Simeonidou, C. Autophagy and neurodegenerative disorders. Neural Regen. Res. 2013, 8, 2275–2283. [Google Scholar] [PubMed]
- Almeida, S.; Alves, M.G.; Sousa, M.; Oliveira, P.F.; Silva, B.M. Are Polyphenols Strong Dietary Agents Against Neurotoxicity and Neurodegeneration? Neurotox. Res. 2016, 30, 345–366. [Google Scholar] [CrossRef]
- Pohl, F.; Lin, P.K.T. The Potential Use of Plant Natural Products and Plant Extracts with Antioxidant Properties for the Prevention/Treatment of Neurodegenerative Diseases: In Vitro, In Vivo and Clinical Trials. Molecules 2018, 23, 3283. [Google Scholar] [CrossRef]
- Pallauf, K.; Rimbach, G. Autophagy, polyphenols and healthy ageing. Ageing Res. Rev. 2013, 12, 237–252. [Google Scholar] [CrossRef]
- Hasima, N.; Ozpolat, B. Regulation of autophagy by polyphenolic compounds as a potential therapeutic strategy for cancer. Cell Death Dis. 2014, 5, e1509. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Silva, R.F.M.; Pogačnik, L. Polyphenols from Food and Natural Products: Neuroprotection and Safety. Antioxidants 2020, 9, 61. [Google Scholar] [CrossRef]
- Koudoufio, M.; Desjardins, Y.; Feldman, F.; Spahis, S.; Delvin, E.; Levy, E. Insight into Polyphenol and Gut Microbiota Crosstalk: Are Their Metabolites the Key to Understand Protective Effects against Metabolic Disorders? Antioxidants 2020, 9, 982. [Google Scholar] [CrossRef] [PubMed]
- Bachrach, U. The early history of polyamine research. Plant Physiol. Biochem. 2010, 48, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Esparza, N.C.; Latorre-Moratalla, M.L.; Comas-Basté, O.; Toro-Funes, N.; Veciana-Nogués, M.T.; Vidal-Carou, M.C. Polyamines in Food. Front. Nutr. 2019, 6, 108. [Google Scholar] [CrossRef]
- Wallace, H.M. The polyamines: Past, present and future. Essays Biochem. 2009, 46, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pucciarelli, S.; Moreschini, B.; Micozzi, D.; de Fronzo, G.S.; Carpi, F.M.; Polzonetti, V.; Vincenzetti, S.; Mignini, F.; Napolioni, V. Spermidine and Spermine Are Enriched in Whole Blood of Nona/Centenarians. Rejuvenation Res. 2012, 15, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.K.; Tabor, C.W.; Tabor, H. Polyamine deficiency leads to accumulation of reactive oxygen species in a spe2Delta mutant of Saccharomyces cerevisiae. Yeast 2006, 23, 751–761. [Google Scholar] [CrossRef]
- Sava, I.G.; Battaglia, V.; Rossi, C.A.; Salvi, M.; Toninello, A. Free radical scavenging action of the natural polyamine spermine in rat liver mitochondria. Free. Radic. Biol. Med. 2006, 41, 1272–1281. [Google Scholar] [CrossRef]
- Jeong, J.-W.; Cha, H.-J.; Han, M.H.; Hwang, S.J.; Lee, D.-S.; Yoo, J.S.; Choi, I.-W.; Kim, S.; Kim, H.-S.; Kim, G.-Y.; et al. Spermidine Protects against Oxidative Stress in Inflammation Models Using Macrophages and Zebrafish. Biomol. Ther. 2018, 26, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I.; Sankhe, R.; Mudgal, J.; Arora, D.; Nampoothiri, M. Spermidine, an autophagy inducer, as a therapeutic strategy in neurological disorders. Neuropeptides 2020, 83, 102083. [Google Scholar] [CrossRef] [PubMed]
- Molnar, M.M.; Liddell, S.C.; Wadkins, R.M. Effects of Polyamine Binding on the Stability of DNA i-Motif Structures. ACS Omega 2019, 4, 8967–8973. [Google Scholar] [CrossRef] [PubMed]
- Iacomino, G.; Picariello, G.; d’Agostino, L. DNA and nuclear aggregates of polyamines. Biochim. Biophys. Acta Bioenerg. 2012, 1823, 1745–1755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gong, H.; Sun, Q.; Zhao, R.; Jia, Y. Spermidine-Activated Satellite Cells Are Associated with Hypoacetylation in ACVR2B and Smad3 Binding to Myogenic Genes in Mice. J. Agric. Food Chem. 2018, 66, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Lachkar, S.; Enot, D.P.; Nisosantano, M.; Pedro, J.M.B.-S.; Sica, V.; Izzo, V.; Maiuri, M.C.; Madeo, F.; Marino, G.; et al. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ. 2014, 22, 509–516. [Google Scholar] [CrossRef]
- Burgio, G.; Corona, D.F.V.; Nicotra, C.M.A.; Carruba, G.; Taibi, G. P/CAF-mediated spermidine acetylation regulates histone acetyltransferase activity. J. Enzym. Inhib. Med. Chem. 2016, 31, 75–82. [Google Scholar] [CrossRef]
- Yue, F.; Li, W.; Zou, J.; Jiang, X.; Xu, G.; Huang, H.; Liu, L. Spermidine Prolongs Lifespan and Prevents Liver Fibrosis and Hepatocellular Carcinoma by Activating MAP1S-Mediated Autophagy. Cancer Res. 2017, 77, 2938–2951. [Google Scholar] [CrossRef]
- Phadwal, K.; Kurian, D.; Salamat, M.K.F.; Macrae, V.E.; Diack, A.B.; Manson, J.C. Spermine increases acetylation of tubulins and facilitates autophagic degradation of prion aggregates. Sci. Rep. 2018, 8, 1–17. [Google Scholar] [CrossRef]
- Soda, K. Spermine and gene methylation: A mechanism of lifespan extension induced by polyamine-rich diet. Amino Acids 2019, 52, 213–224. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, H.; Zhang, J.; Yu, H.; Lin, Z.; Cai, Y. Spermidine Exhibits Protective Effects Against Traumatic Brain Injury. Cell. Mol. Neurobiol. 2020, 40, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Signor, C.; Mello, C.F.; Porto, G.P.; Ribeiro, D.A.; Rubin, M.A. Spermidine improves fear memory persistence. Eur. J. Pharmacol. 2014, 730, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Gilad, G.M.; Gilad, V.H. Novel polyamine derivatives as neuroprotective agents. J. Pharmacol. Exp. Ther. 1999, 291, 39–43. [Google Scholar] [PubMed]
- Melchiorre, C.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V. Polyamines in Drug Discovery: From the Universal Template Approach to the Multitarget-Directed Ligand Design Strategy. J. Med. Chem. 2010, 53, 5906–5914. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Cavalli, A.; Melchiorre, C. Memoquin: A multi-target-directed ligand as an innovative therapeutic opportunity for Alzheimer’s disease. Neurotherapeutics 2009, 6, 152–162. [Google Scholar] [CrossRef]
- Pan, W.; Hu, K.; Bai, P.; Yu, L.; Ma, Q.; Li, T.; Zhang, X.; Chen, C.; Peng, K.; Liu, W.; et al. Design, synthesis and evaluation of novel ferulic acid-memoquin hybrids as potential multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2016, 26, 2539–2543. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, J.; Hong, C.; Luo, W.; Wang, C. Design, synthesis and evaluation of genistein-polyamine conjugates as multi-functional anti-Alzheimer agents. Acta Pharm. Sin. B 2015, 5, 67–73. [Google Scholar] [CrossRef][Green Version]
- Simoni, E.; Caporaso, R.; Bergamini, C.; Fiori, J.; Fato, R.; Miszta, P.; Filipek, S.; Caraci, F.; Giuffrida, M.L.; Andrisano, V.; et al. Polyamine Conjugation as a Promising Strategy to Target Amyloid Aggregation in the Framework of Alzheimer’s Disease. ACS Med. Chem. Lett. 2016, 7, 1145–1150. [Google Scholar] [CrossRef]
- Zhou, Z.-Q.; Fan, H.-X.; He, R.-R.; Xiao, J.; Tsoi, B.; Lan, K.-H.; Kurihara, H.; So, K.-F.; Yao, X.-S.; Gao, H. Lycibarbarspermidines A–O, New Dicaffeoylspermidine Derivatives from Wolfberry, with Activities against Alzheimer’s Disease and Oxidation. J. Agric. Food Chem. 2016, 64, 2223–2237. [Google Scholar] [CrossRef]
- Gao, H.; Yao, X.; He, R.; Chen, G.; Zhou, Z.; Wang, C.; Hu, D.; Fan, H. Dicaffeoyl spermidine cyclized derivatives and use thereof. U.S. Patent No 10,457,702, 29 October 2019. [Google Scholar]
- Grosso, G. Effects of Polyphenol-Rich Foods on Human Health. Nutrients 2018, 10, 1089. [Google Scholar] [CrossRef] [PubMed]
- de Mello, A.J.M.; Fasolo, D. Chapter 20—Polyphenol Antioxidants from Natural Sources and Contribution to Health Promotion. In Polyphenols in Human Health and Disease; Watson, R.R., Preedy, R.V., Zibadi, S., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 253–265. [Google Scholar]
- Handique, J.; Baruah, J. Polyphenolic compounds: An overview. React. Funct. Polym. 2002, 52, 163–188. [Google Scholar] [CrossRef]
- Haque, M.E.; Javed, H.; Azimullah, S.; Khair, S.B.A.; Ojha, S. Neuroprotective potential of ferulic acid in the rotenone model of Parkinson’s disease. Drug Des. Dev. Ther. 2015, 9, 5499–5510. [Google Scholar] [CrossRef]
- Magnani, C.; Chiari, B.G.; Isaac, V.L.B.; Correa, M.A.; Salgado, H.R.N. In Vitro Safety Evaluation of Caffeic Acid. Athens J. Health 2014, 1, 181–188. [Google Scholar] [CrossRef]
- Zanwar, A.A.; Badole, S.L.; Shende, P.S.; Hegde, M.V.; Bodhankar, S.L. Role of Gallic Acid in Cardiovascular Disorders. In Polyphenols in Human Health and Disease; Elsevier BV: Amsterdam, The Netherlands, 2014; pp. 1045–1047. [Google Scholar]
- Shahidi, F.; Janitha, P.K.; Wanasundara, P.D. Phenolic antioxidants. Crit. Rev. Food Sci. Nutr. 1992, 32, 67–103. [Google Scholar] [CrossRef]
- Ren, Z.; Zhang, R.; Li, Y.; Li, Y.; Yang, Z.; Yang, H. Ferulic acid exerts neuroprotective effects against cerebral ischemia/reperfusion-induced injury via antioxidant and anti-apoptotic mechanisms in vitro and in vivo. Int. J. Mol. Med. 2017, 40, 1444–1456. [Google Scholar] [CrossRef]
- Chandrasekhar, Y.; Kumar, G.P.; Ramya, E.M.; Anilakumar, K.R. Gallic Acid Protects 6-OHDA Induced Neurotoxicity by Attenuating Oxidative Stress in Human Dopaminergic Cell Line. Neurochem. Res. 2018, 43, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Kantham, S.; Rao, V.M.; Palanivelu, M.K.; Pham, H.L.; Shaw, P.N.; McGeary, R.P.; Ross, B.P. Metal chelation, radical scavenging and inhibition of Aβ42 fibrillation by food constituents in relation to Alzheimer’s disease. Food Chem. 2016, 199, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Andjelkovic, M.; Vancamp, J.; Demeulenaer, B.; Depaemelaere, G.; Socaciu, C.; Verloo, M.; Verhe, R. Iron-chelation properties of phenolic acids bearing catechol and galloyl groups. Food Chem. 2006, 98, 23–31. [Google Scholar] [CrossRef]
- Yu, M.; Chen, X.; Liu, J.; Ma, Q.; Zhuo, Z.; Chen, H.; Zhou, L.; Yang, S.; Zheng, L.; Hou, S.T.; et al. Gallic acid disruption of Abeta1-42 aggregation rescues cognitive decline of APP/PS1 double transgenic mouse. Neurobiol. Dis. 2019, 124, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Ardah, M.T.; Paleologou, K.E.; Elv, G.; Khair, S.B.A.; Kazim, A.S.; Minhas, S.T.; Al-Tel, T.H.; Al-Hayani, A.A.; Haque, M.E.; Eeliezer, D.; et al. Structure activity relationship of phenolic acid inhibitors of α-synuclein fibril formation and toxicity. Front. Aging Neurosci. 2014, 6, 197. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-L.; Chang, J.-C.; Lin, W.-Y.; Li, C.-C.; Hsieh, M.; Chen, H.-W.; Wang, T.-S.; Wu, W.-T.; Liu, C.-S.; Liu, K.-L. Caffeic acid and resveratrol ameliorate cellular damage in cell and Drosophila models of spinocerebellar ataxia type 3 through upregulation of Nrf2 pathway. Free. Radic. Biol. Med. 2018, 115, 309–317. [Google Scholar] [CrossRef]
- Askar, M.H.; Hussein, A.M.; Al-Basiony, S.F.; Meseha, R.K.; Metias, E.F.; Salama, M.M.; Antar, A.; El-Sayed, A. Effects of Exercise and Ferulic Acid on Alpha Synuclein and Neuroprotective Heat Shock Protein 70 in An Experimental Model of Parkinsonism Disease. CNS Neurol. Disord. Drug Targets 2019, 18, 156–169. [Google Scholar] [CrossRef]
- Chen, J.-L.; Duan, W.-J.; Luo, S.; Li, S.; Ma, X.-H.; Hou, B.-N.; Cheng, S.-Y.; Fang, S.-H.; Wang, Q.; Huang, S.-Q.; et al. Ferulic acid attenuates brain microvascular endothelial cells damage caused by oxygen-glucose deprivation via punctate-mitochondria-dependent mitophagy. Brain Res. 2017, 1666, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Osseni, R.A.; Debbasch, C.; Christen, M.O.; Rat, P.; Warnet, J.M. Tacrine-induced Reactive Oxygen Species in a Human Liver Cell Line: The Role of Anethole Dithiolethione as a Scavenger. Toxicol. Vitr. 1999, 13, 683–688. [Google Scholar] [CrossRef]
- Pi, R.; Mao, X.; Chao, X.; Cheng, Z.; Liu, M.; Duan, X.; Ye, M.; Chen, X.; Mei, Z.; Han, Y.; et al. Tacrine-6-ferulic acid, a novel multifunctional dimer, inhibits amyloid-beta-mediated Alzheimer’s disease-associated pathogenesis in vitro and in vivo. PLoS ONE 2012, 7, e31921. [Google Scholar] [CrossRef]
- Zhu, J.; Yang, H.; Chen, Y.; Lin, H.; Li, Q.; Mo, J.; Bian, Y.; Pei, Y.; Sun, H. Synthesis, pharmacology and molecular docking on multifunctional tacrine-ferulic acid hybrids as cholinesterase inhibitors against Alzheimer’s disease. J. Enzym. Inhib. Med. Chem. 2018, 33, 496–506. [Google Scholar] [CrossRef]
- Takahashi, T.; Miyazawa, M. Serotonin derivatives as inhibitors of beta-secretase (BACE 1). Pharmacie 2011, 66, 301–305. [Google Scholar]
- Dhiman, P.; Malik, N.; Khatkar, A. Hybrid caffeic acid derivatives as monoamine oxidases inhibitors: Synthesis, radical scavenging activity, molecular docking studies and in silico ADMET analysis. Chem. Central J. 2018, 12, 1–17. [Google Scholar] [CrossRef]
- Estrada, M.; Herrera-Arozamena, C.; Pérez, C.; Viña, D.; Romero, A.; Morales-García, J.A.; Pérez-Castillo, A.; Rodríguez-Franco, M.I. New cinnamic—N-benzylpiperidine and cinnamic—N,N-dibenzyl(N-methyl)amine hybrids as Alzheimer-directed multitarget drugs with antioxidant, cholinergic, neuroprotective and neurogenic properties. Eur. J. Med. Chem. 2016, 121, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Benchekroun, M.; Pachón-Angona, I.; Luzet, V.; Martin, H.; Oset-Gasque, M.J.; Marco-Contelles, J.; Ismaili, L. Synthesis, antioxidant and Aβ anti-aggregation properties of new ferulic, caffeic and lipoic acid derivatives obtained by the Ugi four-component reaction. Bioorg. Chem. 2019, 85, 221–228. [Google Scholar] [CrossRef]
- Nesi, G.; Chen, Q.; Sestito, S.; Digiacomo, M.; Yang, X.; Wang, S.; Pi, R.; Rapposelli, S. Nature-based molecules combined with rivastigmine: A symbiotic approach for the synthesis of new agents against Alzheimer’s disease. Eur. J. Med. Chem. 2017, 141, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Simoni, E.; Serafini, M.M.; Bartolini, M.; Caporaso, R.; Pinto, A.; Necchi, D.; Fiori, J.; Andrisano, V.; Minarini, A.; Rosini, M.; et al. Nature-Inspired Multifunctional Ligands: Focusing on Amyloid-Based Molecular Mechanisms of Alzheimer’s Disease. ChemMedChem 2016, 11, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Sang, Z.; Wang, K.; Han, X.; Cao, M.; Tan, Z.; Liu, W. Design, Synthesis, and Evaluation of Novel Ferulic Acid Derivatives as Multi-Target-Directed Ligands for the Treatment of Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 1008–1024. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Choubey, P.K.; Sharma, P.; Seth, A.; Saraf, P.; Shrivastava, S.K. Design, synthesis, and biological evaluation of ferulic acid based 1,3,4-oxadiazole hybrids as multifunctional therapeutics for the treatment of Alzheimer’s disease. Bioorg. Chem. 2020, 95, 103506. [Google Scholar] [CrossRef]
- Landete, J. Ellagitannins, ellagic acid and their derived metabolites: A review about source, metabolism, functions and health. Food Res. Int. 2011, 44, 1150–1160. [Google Scholar] [CrossRef]
- Llorach, R.; Cerdá, B.; Cerón, J.J.; Espín, J.C.; Tomás-Barberán, F.A. Evaluation of the bioavailability and metabolism in the rat of punicalagin, an antioxidant polyphenol from pomegranate juice. Eur. J. Nutr. 2003, 42, 18–28. [Google Scholar] [CrossRef]
- Jurgoński, A.; Juśkiewicz, J.; Fotschki, B.; Kołodziejczyk, K.; Milala, J.; Kosmala, M.; Grzelak-Błaszczyk, K.; Markiewicz, L. Metabolism of strawberry mono- and dimeric ellagitannins in rats fed a diet containing fructo-oligosaccharides. Eur. J. Nutr. 2017, 56, 853–864. [Google Scholar] [CrossRef]
- Tomás-Barberán, F.A.; García-Villalba, R.; González-Sarrías, A.; Selma, M.V.; Espín, J.C. Ellagic Acid Metabolism by Human Gut Microbiota: Consistent Observation of Three Urolithin Phenotypes in Intervention Trials, Independent of Food Source, Age, and Health Status. J. Agric. Food Chem. 2014, 62, 6535–6538. [Google Scholar] [CrossRef]
- Cerdá, B.; Periago, P.M.; Espín, A.J.C.; Tomás-Barberán, F.A. Identification of Urolithin A as a Metabolite Produced by Human Colon Microflora from Ellagic Acid and Related Compounds. J. Agric. Food Chem. 2005, 53, 5571–5576. [Google Scholar] [CrossRef] [PubMed]
- Heilman, J.; Andreux, P.; Tran, N.; Rinsch, C.; Blanco-Bose, W. Safety assessment of Urolithin A, a metabolite produced by the human gut microbiota upon dietary intake of plant derived ellagitannins and ellagic acid. Food Chem. Toxicol. 2017, 108, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Félix, A.A.; Williams, E.G.; Jha, P.; Sasso, G.L.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Seeram, N.P.; Zhang, Y.; McKeever, R.; Henning, S.M.; Lee, R.-P.; Suchard, M.A.; Li, Z.; Chen, S.; Thames, G.; Zerlin, A.; et al. Pomegranate Juice and Extracts Provide Similar Levels of Plasma and Urinary Ellagitannin Metabolites in Human Subjects. J. Med. Food 2008, 11, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Bialonska, D.; Kasimsetty, S.G.; Khan, S.I.; Ferreira, D. Urolithins, Intestinal Microbial Metabolites of Pomegranate Ellagitannins, Exhibit Potent Antioxidant Activity in a Cell-Based Assay. J. Agric. Food Chem. 2009, 57, 10181–10186. [Google Scholar] [CrossRef]
- Cásedas, G.; Les, F.; Choya-Foces, C.; Hugo, M.; López, V. The Metabolite Urolithin-A Ameliorates Oxidative Stress in Neuro-2a Cells, Becoming a Potential Neuroprotective Agent. Antioxidants 2020, 9, 177. [Google Scholar] [CrossRef]
- Saha, P.; Yeoh, B.S.; Singh, R.; Chandrasekar, B.; Vemula, P.K.; Haribabu, B.; Vijay-Kumar, M.; Jala, V.R. Gut Microbiota Conversion of Dietary Ellagic Acid into Bioactive Phytoceutical Urolithin a Inhibits Heme Perox-idases. PLoS ONE 2016, 11, e0156811. [Google Scholar] [CrossRef] [PubMed]
- Boakye, Y.D.; Groyer, L.; Heiss, E.H. An increased autophagic flux contributes to the anti-inflammatory potential of urolithin A in macrophages. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 61–70. [Google Scholar] [CrossRef]
- Zhao, W.; Shi, F.; Guo, Z.; Zhao, J.; Song, X.; Yang, H. Metabolite of ellagitannins, urolithin A induces autophagy and inhibits metastasis in human sw620 colorectal cancer cells. Mol. Carcinog. 2018, 57, 193–200. [Google Scholar] [CrossRef]
- Velagapudi, R.; Lepiarz, I.; El-Bakoush, A.; Katola, F.O.; Bhatia, H.; Fiebich, B.L.; Olajide, O.A. Induction of Autophagy and Activation of SIRT-1 Deacetylation Mechanisms Mediate Neuroprotection by the Pomegranate Metabolite Urolithin A in BV2 Microglia and Differentiated 3D Human Neural Progenitor Cells. Mol. Nutr. Food Res. 2019, 63, e1801237. [Google Scholar] [CrossRef]
- Ahsan, A.; Zheng, Y.; Wu, X.; Tang, W.; Liu, M.; Ma, S.; Jiang, L.; Hu, W.; Zhang, X.; Chen, Z. Urolithin A-activated autophagy but not mitophagy protects against ischemic neuronal injury by inhibiting ER stress in vitro and in vivo. CNS Neurosci. Ther. 2019, 25, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liu, Z.; Zhou, Y.; Hou, N.; Yan, W.; Qin, Y.; Ye, Q.; Cheng, X.-Y.; Xiao, Q.; Wu, X.; et al. Urolithin B, a gut microbiota metabolite, protects against myocardial ischemia/reperfusion injury via p62/Keap1/Nrf2 signaling pathway. Pharmacol. Res. 2020, 153, 104655. [Google Scholar] [CrossRef]
- Gulcan, H.O.; Unlu, S.; Esiringu, I.; Ercetin, T.; Sahin, Y.; Oz, D.; Sahin, M.F. Design, synthesis and biological evaluation of novel 6H-benzo[c]chromen-6-one, and 7,8,9,10-tetrahydro-benzo[c]chromen-6-one derivatives as potential cholinesterase inhibitors. Bioorg. Med. Chem. 2014, 22, 5141–5154. [Google Scholar] [CrossRef]
- Norouzbahari, M.; Burgaz, E.V.; Erçetin, T.; Fallah, A.; Foroumadi, A.; Firoozpour, L.; Sahin, M.F.; Gazi, M.; Gulcan, H.O. Design, Synthesis and Characterization of Novel Urolithin Derivatives as Cholinesterase Inhibitor Agents. Lett. Drug Des. Discov. 2018, 15, 1131–1140. [Google Scholar] [CrossRef]
- Blanquet, P. Casein kinase 2 as a potentially important enzyme in the nervous system. Prog. Neurobiol. 2000, 60, 211–246. [Google Scholar] [CrossRef]
- Perez, D.I.; Gil, C.; Martinez, A. Protein kinases CK1 and CK2 as new targets for neurodegenerative diseases. Med. Res. Rev. 2010, 31, 924–954. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Gianoncelli, A.; Bonvini, P.; Zorzi, E.; Pasquale, R.; Rosolen, A.; Pinna, L.A.; Meggio, F.; Zagotto, G.; Moro, S. Urolithin as a converging scaffold linking ellagic acid and coumarin analogues: Design of potent protein kinase CK2 inhibitors. ChemMedChem 2011, 6, 2273–2286. [Google Scholar] [CrossRef]
- Xie, S.-S.; Lan, J.-S.; Wang, X.; Wang, Z.-M.; Jiang, N.; Li, F.; Wu, J.-J.; Wang, J.; Kong, L.-Y. Design, synthesis and biological evaluation of novel donepezil–coumarin hybrids as multi-target agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2016, 24, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Walgren, J.L.; Amani, Z.; McMillan, J.M.; Locher, M.; Buse, M.G. Effect of R(+)alpha-lipoic acid on pyruvate metabolism and fatty acid oxidation in rat hepatocytes. Metabolism 2004, 53, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Packer, L.; Witt, E.H.; Tritschler, H.J. Alpha-lipoic acid as a biological antioxidant. Free. Radic. Biol. Med. 1995, 19, 227–250. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, Y.; Yu, W.; Jiang, S. Scavenging ability on ROS of alpha-lipoic acid (ALA). Food Chem. 2004, 84, 563–567. [Google Scholar] [CrossRef]
- Moraes, T.B.; Zanin, F.; da Rosa, A.; de Oliveira, A.; Coelho, J.; Petrillo, F.; Wajner, M.; Dutra-Filho, C.S. Lipoic acid prevents oxidative stress in vitro and in vivo by an acute hyperphenylalaninemia chemically-induced in rat brain. J. Neurol. Sci. 2010, 292, 89–95. [Google Scholar] [CrossRef]
- Suh, J.H.; Wang, H.; Liu, R.-M.; Liu, J.; Hagen, T.M. (R)-alpha-lipoic acid reverses the age-related loss in GSH redox status in post-mitotic tissues: Evidence for increased cysteine requirement for GSH synthesis. Arch. Biochem. Biophys. 2004, 423, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Ersahin, M.; Toklu, H.Z.; Çetinel, Ş.; Yüksel, M.; Erzik, C.; Berkman, M.Z.; Yeğen, B.Ç.; Sener, G.; Yeǧen, B.Ç. Alpha Lipoic Acid Alleviates Oxidative Stress and Preserves Blood Brain Permeability in Rats with Subarachnoid Hemorrhage. Neurochem. Res. 2009, 35, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Camiolo, G.; Tibullo, D.; Giallongo, C.; Romano, A.; Parrinello, N.L.; Musumeci, G.; di Rosa, M.; Vicario, N.; Brundo, M.V.; Amenta, F.; et al. α-Lipoic Acid Reduces Iron-induced Toxicity and Oxidative Stress in a Model of Iron Overload. Int. J. Mol. Sci. 2019, 20, 609. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, F.; Nomier, M.A.; Sabik, L.M.E.; Shaheen, M.A. Manganese-induced neurotoxicity and the potential protective effects of lipoic acid and Spirulina platensis. Toxicol. Mech. Methods 2020, 30, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Al-Otaibi, S.S.; Arafah, M.M.; Sharma, B.; Alhomida, A.S.; Siddiqi, N.J. Synergistic Effect of Quercetin and α-Lipoic Acid on Aluminium Chloride Induced Neurotoxicity in Rats. J. Toxicol. 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Smirnova, J.; Kabin, E.; Järving, I.; Bragina, O.; Tõugu, V.; Plitz, T.; Palumaa, P. Copper(I)-binding properties of de-coppering drugs for the treatment of Wilson disease. α-Lipoic acid as a potential anti-copper agent. Sci. Rep. 2018, 8, 1463. [Google Scholar] [CrossRef]
- Khalaf, A.A.; Zaki, A.R.; Galal, M.K.; Ogaly, H.A.; Ibrahim, M.A.; Hassan, A. The potential protective effect of α-lipoic acid against nanocopper particle–induced hepatotoxicity in male rats. Hum. Exp. Toxicol. 2016, 36, 881–891. [Google Scholar] [CrossRef]
- Bjørklund, G.; Crisponi, G.; Nurchi, V.M.; Cappai, R.; Djordjevic, A.B.; Aaseth, J. A Review on Coordination Properties of Thiol-Containing Chelating Agents Towards Mercury, Cadmium, and Lead. Molecules 2019, 24, 3247. [Google Scholar] [CrossRef]
- Androne, L.; Gavan, N.A.; Veresiu, I.A.; Orasan, R. In vivo effect of lipoic acid on lipid peroxidation in patients with diabetic neuropathy. Vivo 2000, 14, 327–330. [Google Scholar]
- Freitas, R. The evaluation of effects of lipoic acid on the lipid peroxidation, nitrite formation and antioxidant enzymes in the hippocampus of rats after pilocarpine-induced seizures. Neurosci. Lett. 2009, 455, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Haugaard, N.; Levin, R.M.; Surname, F. Regulation of the activity of choline acetyl transferase by lipoic acid. Mol. Cell. Biochem. 2000, 213, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.F.; Bussiere, J.R.; Hammond, R.S.; Montine, T.J.; Henson, E.; Jones, R.E.; Stackman, R.W. Chronic dietary α-lipoic acid reduces deficits in hippocampal memory of aged Tg2576 mice. Neurobiol. Aging 2007, 28, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Hager, K.; Kenklies, M.; McAfoose, J.; Engel, J.; Münch, G. Alpha-lipoic acid as a new treatment option for Alzheimer’s disease—A 48 months follow-up analysis. J. Neural. Transm. Suppl. 2007, 72, 189–193. [Google Scholar]
- Fava, A.; Pirritano, D.; Plastino, M.; Cristiano, D.; Puccio, G.; Colica, C.; Ermio, C.; de Bartolo, M.; Mauro, G.; Bosco, D. The Effect of Lipoic Acid Therapy on Cognitive Functioning in Patients with Alzheimer’s Disease. J. Neurodegener. Dis. 2013, 2013, 454253. [Google Scholar] [CrossRef]
- Rosini, M.; Andrisano, V.; Bartolini, M.; Bolognesi, M.L.; Hrelia, P.; Minarini, A.; Tarozzi, A.; Melchiorre, C. Rational Approach to Discover Multipotent Anti-Alzheimer Drugs. J. Med. Chem. 2005, 48, 360–363. [Google Scholar] [CrossRef]
- Rosini, M.; Simoni, E.; Bartolini, M.; Tarozzi, A.; Matera, R.; Milelli, A.; Hrelia, P.; Andrisano, V.; Bolognesi, M.L.; Melchiorre, C. Exploiting the lipoic acid structure in the search for novel multitarget ligands against Alzheimer’s disease. Eur. J. Med. Chem. 2011, 46, 5435–5442. [Google Scholar] [CrossRef] [PubMed]
- Koufaki, M.; Kiziridi, C.; Nikoloudaki, F.; Alexis, M.N. Design and synthesis of 1,2-dithiolane derivatives and evaluation of their neuroprotective activity. Bioorg. Med. Chem. Lett. 2007, 17, 4223–4227. [Google Scholar] [CrossRef]
- Estrada, M.; Pérez, C.; Soriano, E.; Laurini, E.; Romano, M.; Pricl, S.; Morales-García, A.J.; Pérez-Castillo, A.; Rodríguez-Franco, M.I. New neurogenic lipoic-based hybrids as innovative Alzheimer’s drugs with sigma-1 agonism and beta-secretase inhibition. Future Med. Chem. 2016, 8, 1191–1207. [Google Scholar] [CrossRef]
- Tu, Y.-L.; Chen, Q.-H.; Wang, S.-N.; Uri, A.; Yang, X.-H.; Chu, J.-Q.; Chen, J.-K.; Luo, B.-L.; Chen, X.-H.; Wen, S.-J.; et al. Discovery of lipoic acid-4-phenyl-1H-pyrazole hybrids as novel bifunctional ROCK inhibitors with antioxidant activity. RSC Adv. 2016, 6, 58516–58520. [Google Scholar] [CrossRef]
- Jones, M.; Wang, J.; Harmon, S.; Kling, B.; Heilmann, J.; Gilmer, J.F. Novel Selective Butyrylcholinesterase Inhibitors Incorporating Antioxidant Functionalities as Potential Bimodal Therapeutics for Alzheimer’s Disease. Molecules 2016, 21, 440. [Google Scholar] [CrossRef]
- Jalili-Baleh, L.; Forootanfar, H.; Küçükkılınç, T.T.; Nadri, H.; Abdolahi, Z.; Ameri, A.; Jafari, M.; Ayazgok, B.; Baeeri, M.; Foroumadi, A.; et al. Design, synthesis and evaluation of novel multi-target-directed ligands for treatment of Alzheimer’s disease based on coumarin and lipoic acid scaffolds. Eur. J. Med. Chem. 2018, 152, 600–614. [Google Scholar] [CrossRef]
- Pagoni, A.; Marinelli, L.; di Stefano, A.; Ciulla, M.; Turkez, H.; Mardinoglu, A.; Vassiliou, S.; Cacciatore, I. Novel anti-Alzheimer phenol-lipoyl hybrids: Synthesis, physico-chemical characterization, and biological evaluation. Eur. J. Med. Chem. 2020, 186, 111880. [Google Scholar] [CrossRef] [PubMed]
- Michalska, P.; Tenti, G.; Satriani, M.; Cores, A.; Ramos, M.T.; García, A.G.; Menéndez, J.C.; León, R. Aza-CGP37157-lipoic hybrids designed as novel Nrf2-inducers and antioxidants exert neuroprotection against oxidative stress and show neuroinflammation inhibitory properties. Drug Dev. Res. 2019, 81, 283–294. [Google Scholar] [CrossRef]
- Pachón-Angona, I.; Martin, H.; Chhor, S.; Oset-Gasque, M.J.; Refouvelet, B.; Marco-Contelles, J.; Ismaili, L. Synthesis of new ferulic/lipoic/comenic acid-melatonin hybrids as antioxidants and Nrf2 activators via Ugi reaction. Futur. Med. Chem. 2019, 11, 3097–3108. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, L.; Liao, R.; Li, Q.; Pi, R.; Yang, X. N2L, a novel lipoic acid-niacin dimer protects HT22 cells against beta-amyloid peptide-induced damage through attenuating apoptosis. Metab. Brain Dis. 2019, 34, 1761–1770. [Google Scholar] [CrossRef] [PubMed]
- Uppakara, K.; Jamornwan, S.; Duan, L.-X.; Yue, K.-R.; Sunrat, C.; Dent, E.W.; Wan, S.-B.; Saengsawang, W. Novel α-Lipoic Acid/3-n-Butylphthalide Conjugate Enhances Protective Effects against Oxidative Stress and 6-OHDA Induced Neuronal Damage. ACS Chem. Neurosci. 2020, 11, 1634–1642. [Google Scholar] [CrossRef]
- Syed, Y.Y. Correction to: Sodium Oligomannate: First Approval. Drugs 2020, 80, 445–446. [Google Scholar] [CrossRef] [PubMed]
- Al Zahrani, N.A.; El-Shishtawy, R.M.; Asiri, A.M. Recent developments of gallic acid derivatives and their hybrids in medicinal chemistry: A review. Eur. J. Med. Chem. 2020, 204, 112609. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physiological Role of ROS | Ref. | Pathological Role of ROS | Ref. |
|---|---|---|---|
| Signalling between mitochondria and surrounding cells | [20] | mDNA damage, deletion, and mutation | [21] |
| Regulation of cellular proliferation, differentiation and apoptosis | [18,19] | Mitochondrial membrane permeability alteration and mitochondrial failure | [22,25,27,31] |
| Induction of MAPKs activation in cardiovascular system. | [20] | Lipid peroxidation | [27] |
| Influence on pro-survival transcription factors (i.g. Nrf2 and NF-κB). | [21] | ETC enzymes malfunctions | [29] |
| Adaption and regulation of hypoxia | [20] | Promotion of inflammation | [16] |
| Regulation of immune functions | [19,20] | Metallostasis and metal accumulation | [40,42] |
| Induction of autophagy | [20] | Proteostasis and misfolded proteins clearance impairment | [51,53] |
| Entry | Scaffolds Combination | Structure | Effects | Ref. |
|---|---|---|---|---|
| 1 | 1-aminoindan beared with polyamine scaffold |  | Neuroprotection against NMDA toxicity and ischemia damages No neurotoxicity | [85] |
| 2 | 1,4-benzoquinone and polyamine structure of caproctamine |  | ↓Aβ aggregation ↓tau phosphorylation ↑antioxidant activity ↓AChE ↓BACE-1 | [87] |
| 3 | Ferulic acid-memoquin hybrids |  | ↓AChE ↓BuChE ↓self-induced Aβ1-42 aggregation no cytotoxicity in SH-SY5Y cells good BBB predicted permeability | [88] |
| 4 | Genistein with polyamines |  | ↓AChE ↓BuChE Fe3+/Cu2+/Zn2+ chelation no HepG-2 cell cytotoxicity | [89] |
| 5 | 3,5-dibenzylidenepiperidin-4- one functionalized with spermine |  | ↓Aβ42 aggregation no antioxidant properties in T67 cells neuroprotection and no cytoxicity in vitro | [90] |
| 6 | Dicaffeoylsper-midine cyclized derivatives |  | Antioxidant activity ↑memory and learning in fruit flies model | [92] |
| Entry | Scaffolds Combination | Structure | Effects | Ref. |
|---|---|---|---|---|
| 7 | Tacrine linked with ferulic acid |  | ↓ Aβ-aggregation ↓ ROS production ↓ AChE ↑ cognitive functions ↑ SOD/ChAT | [110] |
| 8 | Tacrine and functionalized ferulic acid |  | ↓ Aβ-self aggregation ↓ AChE ↓ BuChE ↑ memory no hepatotoxicity | [111] |
| 9 | Ferulic and caffeic merged with serotonin |  | ↑ antioxidant activity ↓ BACE-1 | [112] |
| 10 | Aromatic amides and esters of caffeic acid |  | ↓MAO-A/MAO-B ↑ antioxidant activity | [113] |
| 11 | Hydroxycinnamic acids and NBP (donepezil) |  | ↓MAO-A/MAO-B ↓AChE ↓BuChE ↑ antioxidant activity | [114] |
| 12 | Hydroxycinnamic scaffolds and DBMA (AP2238) |  | ↓MAO-A/MAO-B ↓AChE ↓BuChE ↑ antioxidant activity | [114] |
| 13 | Caffeic acid with hydrophobic moieties |  | ↓Aβ1-40 self-aggregation ↑ antioxidant activity neuroprotection in SH-SY5Y cells | [115] |
| 14 | Rivastigmine with GA |  | ↑ antioxidant activity Cu2+ chelating properties ↓ChEs ↓Aβ self-aggregation neuroprotective effects in vitro no cytotoxicity | [116] |
| 15 | Caffeic acid and diallyl sulfide |  | ↓Aβ42 self-aggregation ↑cytoprotection against H2O2-induced damages ↓p53 alteration induced by Aβ | [117] |
| 16 | Ferulic core merged with 1,2,3,4-tetrahydroisoquinoline and (benzyl(ethyl)amino)butoxy scaffold |  | ↑antioxidant activity ↓AChE ↓BuChE ↓ MAO-A/MAO-B ↓ Aβ self-aggregation ↑ self-induced Aβ1-42 fibrils disaggregation ↑neuroprotective effect in SH-5YSY cells ↑autophagy in U87 cells ↑motility in Zebrafish model ↓ Aβ1-40-induced vascular injury in Zebrafish model ↑In vivo cognitive functions | [118] |
| 17 | Ferulic acid merged with 1,3,4-oxadiazole scaffold |  | ↓ChEs ↓BACE-1 ↓ Aβ self-aggregation ↓ Aβ AChE-induced aggregation neuroprotective effects in vitro ↑In vivo cognitive functions | [119] |
| Entry | Scaffolds Combination | Structure | Effects | Ref. |
|---|---|---|---|---|
| 18 | Urolithin scaffold with rivastigmine portion |  | ↓AChE ↓BuChE | [136] |
| 19 | Urolithin scaffold with donepezil-like moieties |  | ↓AChE ↓BuChE | [136] |
| 20 | Donepezil-like urolithin and tetrahydrourolithin derivatives |  | ↓AChE ↓BuChE ↓ AChE induced Aβ aggregation | [137] |
| 21 | Nitro- and bromo-derivatives of urolithins |  | CK2 inhibition Selectivity in other kinases panel | [140] |
| 22 | Tetrahydrourolithin scaffold linked with donepezil moiety |  | AChE/BuChE inhibition MAO-B inhibition BBB permeability no cytotoxicity in brain and liver cells | [141] |
| Entry | Scaffold Combination | Structure | Effects | Ref. |
|---|---|---|---|---|
| 23 | Lipoic acid and tacrine |  | ↑ROS protection ↓AChE ↓BuChE ↓ AChE-induced Aβ aggregation | [160] |
| 24 | Dopamine and LA linked by tetrazole ring |  | ↑antioxidant activity neuroprotection in vitro | [162] |
| 25 | LA-NBP and LA-DBMA conjugation |  | ↓AChE ↓BuChE ↓BACE-1 ↑antioxidant activity σ1R agonism good BBB permeability prediction neuroprotection in vitro | [163] |
| 26 | LA-4-Phenyl-1H-pyrazole derivatives |  | ROCK1/ROCK2 inhibition ↓ROS ↑GSH vasorelaxant activity | [164] |
| 27 | Lipoic isosorbide-2-benzylcarbamate |  | ↓ROS ↓BuChE ↓cytotoxicity in treated HT-22 cells | [165] |
| 28 | LA and coumarin scaffold linked bridged with triazole |  | ↓AChE ↓BuChE ↓ Aβ peptide aggregation ↓intracellular ROS neuroprotection against H2O2− or Aβ1-42-induced cytotoxicity in SH-SY5Y cell lines Selective Cu/Fe chelation | [166] |
| 29 | FA/CA-LA hybrids |  | ↓Aβ1-42-induced neurotoxicity in SH-SY5Y cells ↑protection in H2O2-insulted cells no cytotoxicity | [167] |
| 30 | Lipoic-functionalized benzodiazepine |  | ↑ROS scavenging ↑Nrf2-ARE pathway ↑HO-1/GCLc neuroprotection in in vitro model no cytotoxicity no hepatotoxicity | [168] |
| 31 | Lipoic-melatonin hybrids |  | ↑ROS scavenging ↑Nrf2-ARE pathway antioxidant activity and neuroprotection in vitro no cytotoxicity | [169] |
| 32 | LA-niacin hybrids |  | ↓Aβ1-42-induced cytotoxicity in HT22 cells ↓mitochondrial dysfunctions ↓intracellular ROS ↑SOD, CAT, GPx ↓apoptosis in Aβ1-42treated cells | [170] |
| 33 | LA-3-n-butylphthalide amide |  | ↓intracellular ROS ↑direct ROS-scavenger ↓ H2O2-induced cell death ↑GSH ↓ H2O2-induced damage in cortical neurons ↓6-OHDA-induced neuronal damage in SH-5YSY cells | [171] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bacci, A.; Runfola, M.; Sestito, S.; Rapposelli, S. Beyond Antioxidant Effects: Nature-Based Templates Unveil New Strategies for Neurodegenerative Diseases. Antioxidants 2021, 10, 367. https://doi.org/10.3390/antiox10030367
Bacci A, Runfola M, Sestito S, Rapposelli S. Beyond Antioxidant Effects: Nature-Based Templates Unveil New Strategies for Neurodegenerative Diseases. Antioxidants. 2021; 10(3):367. https://doi.org/10.3390/antiox10030367
Chicago/Turabian StyleBacci, Andrea, Massimiliano Runfola, Simona Sestito, and Simona Rapposelli. 2021. "Beyond Antioxidant Effects: Nature-Based Templates Unveil New Strategies for Neurodegenerative Diseases" Antioxidants 10, no. 3: 367. https://doi.org/10.3390/antiox10030367
APA StyleBacci, A., Runfola, M., Sestito, S., & Rapposelli, S. (2021). Beyond Antioxidant Effects: Nature-Based Templates Unveil New Strategies for Neurodegenerative Diseases. Antioxidants, 10(3), 367. https://doi.org/10.3390/antiox10030367