
Activation of NRF2 and ATF4 Signaling by the Pro-Glutathione Molecule I-152, a Co-Drug of N-Acetyl-Cysteine and Cysteamine

 ,
,  ,
,  ,
,  ,
,  and
and
Abstract
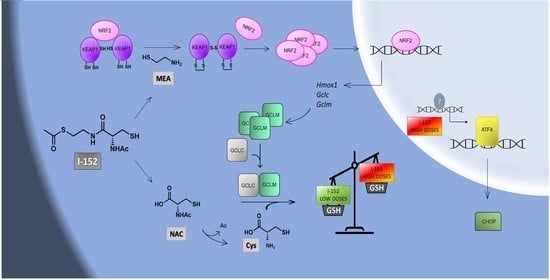
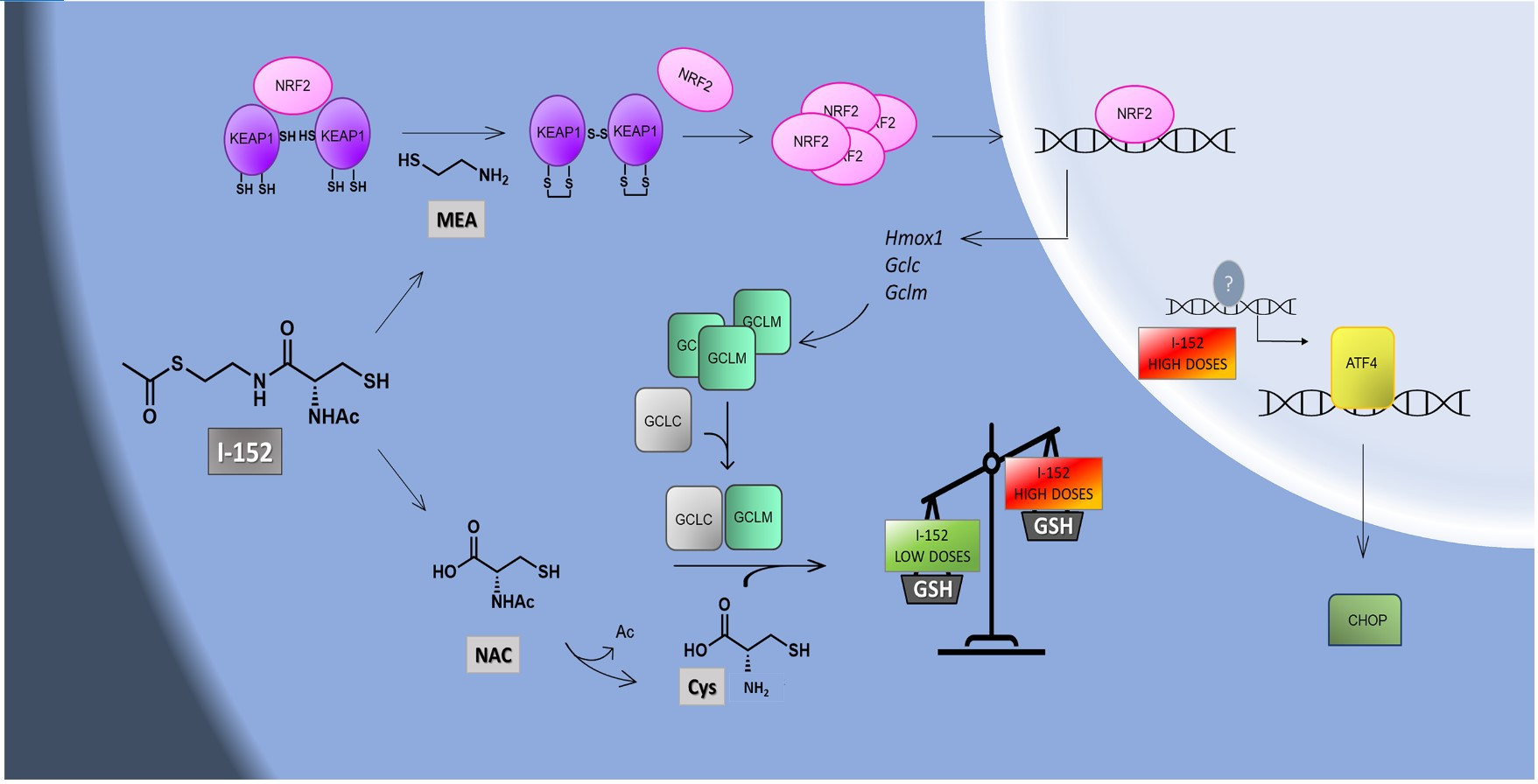
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Cell Lysates
2.3. SDS-PAGE and Western Immunoblotting
2.4. Cycloheximide (CHX) Chase Assay and Relative Half-Life Determination
2.5. NEM-Alkylated Redox Western Blotting
2.6. RNA Isolation and cDNA Synthesis
2.7. Quantitative Real-Time PCR
2.8. Thiol Content Determination by High Performance Liquid Chromatography (HPLC)
2.9. Lactate Dehydrogenase (LDH)-Based Cytotoxicity Assay
2.10. Statistical Analysis
3. Results
3.1. I-152 Increases the Levels of NRF2
3.2. I-152 Induces KEAP1 Oxidation and NRF2 Stabilization
3.3. I-152 Activates NRF2-Dependent Gene Transcription and Increases GCLM Protein Levels
3.4. I-152 Increases Intracellular Thiol Content and Dose-Dependently Modulates GSH Levels
3.5. I-152 Activates the ATF4 Signaling Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ATF4 | activating transcription factor 4 |
| CA | carnosinic acid |
| CHAC1 | cation transport regulator-like protein 1 |
| CHOP | C/EBP-homologous protein (CHOP10/GADD153) |
| CHX | cycloheximide |
| DTT | dithiothreitol |
| eIF2α | eukaryotic translation initiation factor 2α |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GCLC | ɣ-glutamyl-cysteine ligase catalytic subunit |
| GCLM | ɣ-glutamyl-cysteine ligase modifier subunit |
| GSH | glutathione |
| GUSB | β-D-glucuronidase |
| HMOX1 | heme oxygenase 1 |
| HPLC | high performance liquid chromatography |
| IRS | integrated stress response |
| KEAP1 | kelch-like ECH-associated protein 1 |
| LDH | lactate dehydrogenase |
| MEA | cysteamine/β-mercaptoethylamine |
| MEAs-s | cystamine |
| NAC | N-acetyl-cysteine |
| NEM | N-ethylmaleimide |
| NRF2 | nuclear factor E2-related factor 2 |
| SMEA | S-acetyl-cysteamine |
References
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shertzer, H.G.; Schneider, S.N.; Nebert, D.W.; Dalton, T.P. Glutamate cysteine ligase catalysis: Dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J. Biol. Chem. 2005, 280, 33766–33774. [Google Scholar] [CrossRef] [PubMed]
- Franklin, C.C.; Backos, D.S.; Mohar, I.; White, C.C.; Forman, H.J.; Kavanagh, T.J. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol. Asp. Med. 2009, 30, 86–98. [Google Scholar] [CrossRef]
- Wild, A.C.; Moinova, H.R.; Mulcahy, R.T. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J. Biol. Chem. 1999, 274, 33627–33636. [Google Scholar] [CrossRef] [PubMed]
- Moinova, H.R.; Mulcahy, R.T. Up-regulation of the human gamma-glutamylcysteine synthetase regulatory subunit gene involves binding of Nrf-2 to an electrophile responsive element. Biochem. Biophys. Res. Commun. 1999, 261, 661–668. [Google Scholar] [CrossRef]
- He, X.; Ma, Q. NRF2 cysteine residues are critical for oxidant/electrophile-sensing, Kelch-like ECH-associated protein-1-dependent ubiquitination-proteasomal degradation, and transcription activation. Mol. Pharmacol. 2009, 76, 1265–1278. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed]
- Karpinski, B.A.; Morle, G.D.; Huggenvik, J.; Uhler, M.D.; Leiden, J.M. Molecular cloning of human CREB-2: An ATF/CREB transcription factor that can negatively regulate transcription from the cAMP response element. Proc. Natl. Acad. Sci. USA 1992, 89, 4820–4824. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed]
- Ehren, J.L.; Maher, P. Concurrent regulation of the transcription factors Nrf2 and ATF4 mediates the enhancement of glutathione levels by the flavonoid fisetin. Biochem. Pharmacol. 2013, 85, 1816–1826. [Google Scholar] [CrossRef]
- Mimura, J.; Inose-Maruyama, A.; Taniuchi, S.; Kosaka, K.; Yoshida, H.; Yamazaki, H.; Kasai, S.; Harada, N.; Kaufman, R.J.; Oyadomari, S.; et al. Concomitant Nrf2- and ATF4-activation by Carnosic Acid Cooperatively Induces Expression of Cytoprotective Genes. Int. J. Mol. Sci. 2019, 20, 1706. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Mimura, J.; Okada, T.; Sato, H.; Liu, T.; Maruyama, A.; Ohyama, C.; Itoh, K. Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol. Cell. Biol. 2014, 34, 3421–3434. [Google Scholar] [CrossRef] [PubMed]
- He, C.H.; Gong, P.; Hu, B.; Stewart, D.; Choi, M.E.; Choi, A.M.; Alam, J. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J. Biol. Chem. 2001, 276, 20858–20865. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Nomura, S.; Maebara, K.; Sato, K.; Tamba, M.; Bannai, S. Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochem. Biophys. Res. Commun. 2004, 325, 109–116. [Google Scholar] [CrossRef]
- Siu, F.; Bain, P.J.; LeBlanc-Chaffin, R.; Chen, H.; Kilberg, M.S. ATF4 is a mediator of the nutrient-sensing response pathway that activates the human asparagine synthetase gene. J. Biol. Chem. 2002, 277, 24120–24127. [Google Scholar] [CrossRef]
- Crawford, R.R.; Prescott, E.T.; Sylvester, C.F.; Higdon, A.N.; Shan, J.; Kilberg, M.S.; Mungrue, I.N. Human CHAC1 Protein Degrades Glutathione, and mRNA Induction Is Regulated by the Transcription Factors ATF4 and ATF3 and a Bipartite ATF/CRE Regulatory Element. J. Biol. Chem. 2015, 290, 15878–15891. [Google Scholar] [CrossRef]
- Bachhawat, A.K.; Kaur, A. Glutathione Degradation. Antioxid. Redox Signal. 2017, 27, 1200–1216. [Google Scholar] [CrossRef]
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 2009, 390, 191–214. [Google Scholar] [CrossRef]
- Kode, A.; Rajendrasozhan, S.; Caito, S.; Yang, S.R.; Megson, I.L.; Rahman, I. Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L478–L488. [Google Scholar] [CrossRef]
- Wei, Y.; Lu, M.; Mei, M.; Wang, H.; Han, Z.; Chen, M.; Yao, H.; Song, N.; Ding, X.; Ding, J.; et al. Pyridoxine induces glutathione synthesis via PKM2-mediated Nrf2 transactivation and confers neuroprotection. Nat. Commun. 2020, 11, 941. [Google Scholar] [CrossRef]
- Oiry, J.; Mialocq, P.; Puy, J.Y.; Fretier, P.; Clayette, P.; Dormont, D.; Imbach, J.L. NAC/MEA conjugate: A new potent antioxidant which increases the GSH level in various cell lines. Bioorg. Med. Chem. Lett. 2001, 11, 1189–1191. [Google Scholar] [CrossRef]
- Crinelli, R.; Zara, C.; Smietana, M.; Retini, M.; Magnani, M.; Fraternale, A. Boosting GSH Using the Co-Drug Approach: I-152, a Conjugate of N-acetyl-cysteine and β-mercaptoethylamine. Nutrients 2019, 11, 1291. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Diotallevi, A.; De Santi, M.; Ceccarelli, M.; Vitale, F.; Brandi, G.; Magnani, M. Leishmania infantum Induces Mild Unfolded Protein Response in Infected Macrophages. PLoS ONE 2016, 11, e0168339. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Amatore, D.; Celestino, I.; Brundu, S.; Galluzzi, L.; Coluccio, P.; Checconi, P.; Magnani, M.; Palamara, A.T.; Fraternale, A.; Nencioni, L. Glutathione increase by the n-butanoyl glutathione derivative (GSH-C4) inhibits viral replication and induces a predominant Th1 immune profile in old mice infected with influenza virus. FASEB Bioadv. 2019, 1, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Wu, J.; Murray, J.K.; Gellman, S.H.; Wozniak, M.A.; Keely, P.J.; Boyer, M.E.; Gomez, T.M.; Hasso, S.M.; Fallon, J.F.; et al. An antiangiogenic neurokinin-B/thromboxane A2 regulatory axis. J. Cell Biol. 2006, 174, 1047–1058. [Google Scholar] [CrossRef]
- Fraternale, A.; Crinelli, R.; Casabianca, A.; Paoletti, M.F.; Orlandi, C.; Carloni, E.; Smietana, M.; Palamara, A.T.; Magnani, M. Molecules altering the intracellular thiol content modulate NF-kB and STAT-1/IRF-1 signalling pathways and IL-12 p40 and IL-27 p28 production in murine macrophages. PLoS ONE 2013, 8, e57866. [Google Scholar] [CrossRef]
- Smith, S.M.; Wunder, M.B.; Norris, D.A.; Shellman, Y.G. A simple protocol for using a LDH-based cytotoxicity assay to assess the effects of death and growth inhibition at the same time. PLoS ONE 2011, 6, e26908. [Google Scholar] [CrossRef]
- Beutler, E. Lactate Deydrogenase (LDH). In Red Cell Metabolism. A Manual of Biochemical Method, 3th ed.; Beutler, E., Ed.; Grune and Stratton, Inc.: New York, NY, USA, 1984; pp. 65–66. [Google Scholar]
- Brundu, S.; Palma, L.; Picceri, G.G.; Ligi, D.; Orlandi, C.; Galluzzi, L.; Chiarantini, L.; Casabianca, A.; Schiavano, G.F.; Santi, M.; et al. Glutathione Depletion Is Linked with Th2 Polarization in Mice with a Retrovirus-Induced Immunodeficiency Syndrome, Murine AIDS: Role of Proglutathione Molecules as Immunotherapeutics. J. Virol. 2016, 90, 7118–7130. [Google Scholar] [CrossRef]
- Calkins, M.J.; Townsend, J.A.; Johnson, D.A.; Johnson, J.A. Cystamine protects from 3-nitropropionic acid lesioning via induction of nf-e2 related factor 2 mediated transcription. Exp. Neurol. 2010, 224, 307–317. [Google Scholar] [CrossRef]
- Wang, L.L.; Huang, Y.H.; Yan, C.Y.; Wei, X.D.; Hou, J.Q.; Pu, J.X.; Lv, J.X. N-acetylcysteine Ameliorates Prostatitis via miR-141 Regulating Keap1/Nrf2 Signaling. Inflammation 2016, 39, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Choo, Y.S.; Lesort, M. Cystamine and cysteamine prevent 3-NP-induced mitochondrial depolarization of Huntington’s disease knock-in striatal cells. Eur. J. Neurosci. 2006, 23, 1701–1710. [Google Scholar] [CrossRef]
- Wilmer, M.J.; Kluijtmans, L.A.; van der Velden, T.J.; Willems, P.H.; Scheffer, P.G.; Masereeuw, R.; Monnens, L.A.; van den Heuvel, L.P.; Levtchenko, E.N. Cysteamine restores glutathione redox status in cultured cystinotic proximal tubular epithelial cells. Biochim. Biophys. Acta 2011, 1812, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Ezeriņa, D.; Takano, Y.; Hanaoka, K.; Urano, Y.; Dick, T.P. N-Acetyl Cysteine Functions as a Fast-Acting Antioxidant by Triggering Intracellular H2S and Sulfane Sulfur Production. Cell Chem. Biol. 2018, 25, 447–459. [Google Scholar] [CrossRef]
- Stewart, D.; Killeen, E.; Naquin, R.; Alam, S.; Alam, J. Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J. Biol. Chem. 2003, 278, 2396–2402. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar] [CrossRef]
- Xiao, W.; Loscalzo, J. Metabolic Responses to Reductive Stress. Antioxid. Redox Signal. 2020, 32, 1330–1347. [Google Scholar] [CrossRef]
- Oslowski, C.M.; Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011, 490, 71–92. [Google Scholar] [CrossRef]
- Lee, J.I.; Kang, J.; Stipanuk, M.H. Differential regulation of glutamate-cysteine ligase subunit expression and increased holoenzyme formation in response to cysteine deprivation. Biochem. J. 2006, 393, 181–190. [Google Scholar] [CrossRef]
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta 2013, 1830, 4117–4129. [Google Scholar] [CrossRef]
- Whillier, S.; Raftos, J.E.; Chapman, B.; Kuchel, P.W. Role of N-acetylcysteine and cystine in glutathione synthesis in human erythrocytes. Redox Rep. 2009, 14, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Khomenko, T.; Kolodney, J.; Pinto, J.T.; McLaren, G.D.; Deng, X.; Chen, L.; Tolstanova, G.; Paunovic, B.; Krasnikov, B.F.; Hoa, N.; et al. New mechanistic explanation for the localization of ulcers in the rat duodenum: Role of iron and selective uptake of cysteamine. Arch. Biochem. Biophys. 2012, 525, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Palanski, B.A.; Khosla, C. Cystamine and Disulfiram Inhibit Human Transglutaminase 2 via an Oxidative Mechanism. Biochemistry 2018, 57, 3359–3363. [Google Scholar] [CrossRef] [PubMed]
- Fourquet, S.; Guerois, R.; Biard, D.; Toledano, M.B. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J. Biol. Chem. 2010, 285, 8463–8471. [Google Scholar] [CrossRef]
- Chen, Y.T.; Shi, D.; Yang, D.; Yan, B. Antioxidant sulforaphane and sensitizer trinitrobenzene sulfonate induce carboxylesterase-1 through a novel element transactivated by nuclear factor-E2 related factor-2. Biochem. Pharmacol. 2012, 84, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, K.; Wang, N.; Zhang, H. N-acetylcysteine induces apoptosis via the mitochondria-dependent pathway but not via endoplasmic reticulum stress in H9c2 cells. Mol. Med. Rep. 2017, 16, 6626–6633. [Google Scholar] [CrossRef] [PubMed]
- Alam, K.; Ghousunnissa, S.; Nair, S.; Valluri, V.L.; Mukhopadhyay, S. Glutathione-redox balance regulates c-rel-driven IL-12 production in macrophages: Possible implications in antituberculosis immunotherapy. J. Imunol. 2010, 184, 2918–2929. [Google Scholar] [CrossRef]
- Finn, N.A.; Kemp, M.L. Pro-oxidant and antioxidant effects of N-acetylcysteine regulate doxorubicin-induced NF-kappa B activity in leukemic cells. Mol. Biosyst. 2012, 8, 650–662. [Google Scholar] [CrossRef]
- Singh, F.; Charles, A.L.; Schlagowski, A.I.; Bouitbir, J.; Bonifacio, A.; Piquard, F.; Krähenbühl, S.; Geny, B.; Zoll, J. Reductive stress impairs myoblasts mitochondrial function and triggers mitochondrial hormesis. Biochim. Biophys. Acta 2015, 1853, 1574–1585. [Google Scholar] [CrossRef]
- Perra, L.; Balloy, V.; Foussignière, T.; Moissenet, D.; Petat, H.; Mungrue, I.N.; Touqui, L.; Corvol, H.; Chignard, M.; Guillot, L. CHAC1 Is Differentially Expressed in Normal and Cystic Fibrosis Bronchial Epithelial Cells and Regulates the Inflammatory Response Induced by Pseudomonas aeruginosa. Front. Immunol. 2018, 9, 2823. [Google Scholar] [CrossRef]
- Wortel, I.; van der Meer, L.T.; Kilberg, M.S.; van Leeuwen, F.N. Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol. Metab. 2017, 28, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Miyazaki, S.; Matsuyama, S.; Takeda, M.; Kawano, M.; Nakagawa, H.; Nishimura, K.; Matsuo, S. Selection of autophagy or apoptosis in cells exposed to ER-stress depends on ATF4 expression pattern with or without CHOP expression. Biol. Open 2013, 2, 1084–1090. [Google Scholar] [CrossRef] [PubMed]
- Teske, B.F.; Fusakio, M.E.; Zhou, D.; Shan, J.; McClintick, J.N.; Kilberg, M.S.; Wek, R.C. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell 2013, 24, 2477–2490. [Google Scholar] [CrossRef] [PubMed]
- B’chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef]
- Afonyushkin, T.; Oskolkova, O.V.; Philippova, M.; Resink, T.J.; Erne, P.; Binder, B.R.; Bochkov, V.N. Oxidized phospholipids regulate expression of ATF4 and VEGF in endothelial cells via NRF2-dependent mechanism: Novel point of convergence between electrophilic and unfolded protein stress pathways. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1007–1013. [Google Scholar] [CrossRef]
- Cho, H.; Wu, M.; Zhang, L.; Thompson, R.; Nath, A.; Chan, C. Signaling dynamics of palmitate-induced ER stress responses mediated by ATF4 in HepG2 cells. BMC Syst. Biol. 2013, 7, 9. [Google Scholar] [CrossRef]
- Lange, P.S.; Chavez, J.C.; Pinto, J.T.; Coppola, G.; Sun, C.W.; Townes, T.M.; Geschwind, D.H.; Ratan, R.R. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J. Exp. Med. 2008, 205, 1227–1242. [Google Scholar] [CrossRef]
- Cacciatore, I.; Cornacchia, C.; Pinnen, F.; Mollica, A.; Di Stefano, A. Prodrug approach for increasing cellular glutathione levels. Molecules 2010, 15, 1242–1264. [Google Scholar] [CrossRef]
- Kals, J.; Starkopf, J.; Zilmer, M.; Pruler, T.; Pulges, K.; Hallaste, M.; Kals, M.; Pulges, A.; Soomets, U. Antioxidant UPF1 attenuates myocardial stunning in isolated rat hearts. Int. J. Cardiol. 2008, 125, 133–135. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target mRNA | Accession Number | Forward Primer (5′–3′) | Reverse Primer (5′–3′) |
|---|---|---|---|
| Atf4 | NM_001287180 | GCAGTGTTGCTGTAACGGAC | ATCTCGGTCATGTTGTGGGG |
| Chac1 | NM_026929 | TATAGTGACAGCCGTGTGGG | GCTCCCCTCGAACTTGGTAT |
| Chop | NM_007837 | GAGTCCCTGCCTTTCACCTT | TTCCTCTTCGTTTCCTGGGG |
| Gclc | NM_010295 | GGAGAGGACAAACCCCAACC | CTCAGACATCGTTCCTCCGT |
| Gclm | NM_008129 | GGAACCTGCTCAACTGGGG | GGTCTTTTGGATACAGTCCCGA |
| Hmox1 | NM_010442 | TTAAGCTGGTGATGGCTTCCT | AGTGGGGCATAGACTGGGTT |
| Nrf2 | NM_010902 | CACATTCCCAAACAAGATGCCT | TATCCAGGGCAAGCGACTCA |
| Gusb | NM_010368 | GGGTGTGGTATGAACGGGAA | CCATTCACCCACACAACTGC |
| Gapdh | NM_001289726 | TGCCCCCATGTTTGTGATG | TGTGGTCATGAGCCCTTCC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crinelli, R.; Zara, C.; Galluzzi, L.; Buffi, G.; Ceccarini, C.; Smietana, M.; Mari, M.; Magnani, M.; Fraternale, A. Activation of NRF2 and ATF4 Signaling by the Pro-Glutathione Molecule I-152, a Co-Drug of N-Acetyl-Cysteine and Cysteamine. Antioxidants 2021, 10, 175. https://doi.org/10.3390/antiox10020175
Crinelli R, Zara C, Galluzzi L, Buffi G, Ceccarini C, Smietana M, Mari M, Magnani M, Fraternale A. Activation of NRF2 and ATF4 Signaling by the Pro-Glutathione Molecule I-152, a Co-Drug of N-Acetyl-Cysteine and Cysteamine. Antioxidants. 2021; 10(2):175. https://doi.org/10.3390/antiox10020175
Chicago/Turabian StyleCrinelli, Rita, Carolina Zara, Luca Galluzzi, Gloria Buffi, Chiara Ceccarini, Michael Smietana, Michele Mari, Mauro Magnani, and Alessandra Fraternale. 2021. "Activation of NRF2 and ATF4 Signaling by the Pro-Glutathione Molecule I-152, a Co-Drug of N-Acetyl-Cysteine and Cysteamine" Antioxidants 10, no. 2: 175. https://doi.org/10.3390/antiox10020175
APA StyleCrinelli, R., Zara, C., Galluzzi, L., Buffi, G., Ceccarini, C., Smietana, M., Mari, M., Magnani, M., & Fraternale, A. (2021). Activation of NRF2 and ATF4 Signaling by the Pro-Glutathione Molecule I-152, a Co-Drug of N-Acetyl-Cysteine and Cysteamine. Antioxidants, 10(2), 175. https://doi.org/10.3390/antiox10020175