1. Background/Objectives
Microglia, the resident macrophages of the central nervous system, originate from a cell lineage distinct from neurons and astrocytes. During cell turnover, terminally differentiated cells commonly exit the cell cycle, cease division, and are continually replenished through the proliferation of tissue-specific stem cells. However, the microglial population is uniquely sustained by the division of already differentiated cells. Previous studies using parabiotic mice and fate mapping analyses have revealed that adult microglia are primarily maintained through local self-renewal, with minimal contribution from circulating myeloid precursors [
1,
2].
The in vivo ablation of microglia leads to repopulation from the local expansion of residual microglia [
3,
4,
5,
6]. It has been hypothesized that CSF1R inhibitor-resistant cells which have progenitor-like features exist within heterogeneous subpopulations and contribute to repopulation [
7]. At least under physiological conditions, adult microglia generate homogeneous subpopulations during the differentiation stage, and no progenitor cells have been identified thus far [
8,
9,
10]. Several pathways involved in microglial repopulation have also been identified. CSF1R signaling is crucial for microglial proliferation during repopulation [
3]. CX3CL1-CX3CR1 signaling was reported to be critical for microglial repopulation [
11]. While P2RY12 receptors play a critical role in regulating the microglial landscape through cellular translocation, microglial repopulation is independent of P2RY12 receptor signaling [
12,
13]. Nevertheless, the mechanism driving the robust proliferation of adult microglia that is sufficient to recover from near-total ablation is not yet fully understood.
Although in vivo ablation methods are mostly employed for studying microglial repopulation [
4], utilizing in vitro models of microglial repopulation holds an advantage as it facilitates real-time observation of the process and enables the assessment of responses to various interventions. In this study, we aimed to reveal the mechanisms of repopulation using in vitro microglial removal model.
2. Materials and Methods
2.1. Mice
Cx3cr1EGFP/EGFP (B6.129P-Cx3cr1tm1Litt/J) mice, purchased from the Jackson Laboratory (Bar Harbor, ME), were inbred and maintained. Pregnant ICR and C57BL/6J wild-type mice were obtained from Japan SLC (Hamamatsu, Japan) and Charles River (Kanagawa, Japan), respectively. Cx3cr1EGFP/EGFP male mice were crossed with C57BL/6J wild-type female mice to obtain Cx3cr1+/EGFP mice. All mice were housed in a specific pathogen-free facility on a 12 h light and 12nh dark cycle with ad libitum access to food and water. All experiments were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals revised in 1996. Formal approval to conduct the experiments was obtained by the Animal Care and Use Committee of the Tokyo Metropolitan Institute of Medical Science (approval code: 24-016; approval date: 1 April 2024).
2.2. Preparation of Primary Glial Culture
Primary mixed glial cultures were prepared from the brains of P 3–5 ICR or Cx3cr1+/EGFP mice. Briefly, after the meninges were stripped off, the cerebral cortices were enzymatically (using 0.25% trypsin and 0.04% DNase-I) and mechanically (by homogenization and pipetting) dissociated. The cells obtained from 3–4 pups were suspended in Dulbecco’s Modified Eagle’s medium (DMEM, Nacalai Tesque, Kyoto, Japan) supplemented with 10% fetal bovine serum (FBS) and antibiotic–antimycotic mixed solution (1%, Nacalai Tesque) and were plated on a poly-L-lysine (Sigma-Aldrich, St. Louis, MO, USA)-coated 75 cm2 flask at 0.8–1 × 107 cells/flask. The cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2.
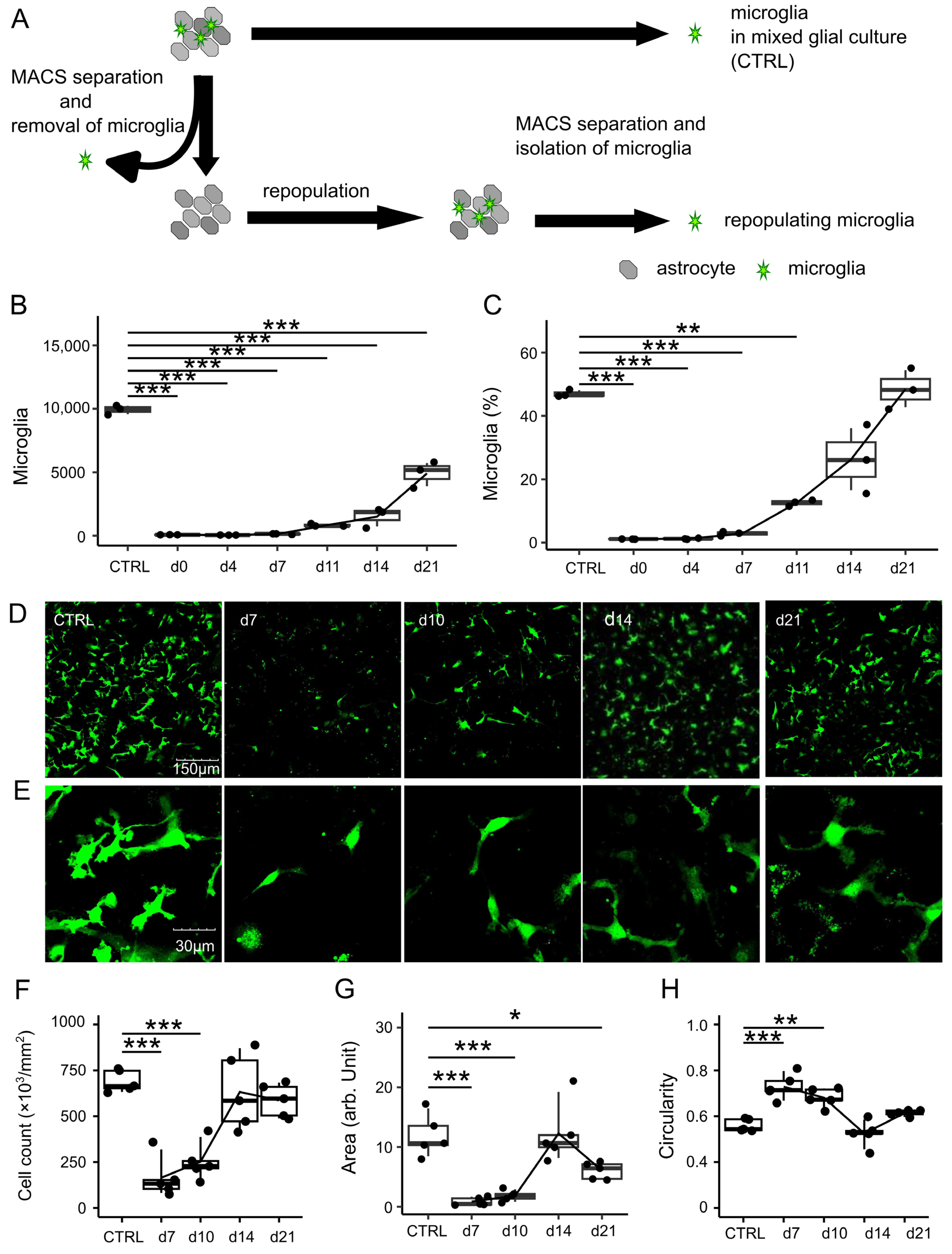
2.3. Removal of Microglia from Mixed Glial Culture Using Anti-CD11b Magnetic Particles
Within 7–12 days following the preparation of the primary glial culture (called mixed glial culture (MGC)), the cells were detached using TrypLE Express (1×) without phenol red (Thermo Fisher Scientific, Waltham, MA, USA). Subsequently, microglia were removed from the MGC using anti-mouse CD11b magnetic particles (BD Biosciences, Franklin Lakes, NJ, USA) according to the manufacturer’s protocol. Briefly, the detached cells per flask were resuspended in 50 µL of anti-CD11b magnetic particles and incubated at 4 °C for 40 min with gentle mixing every 5–10 min. Following conjugation with magnetic particles, the CD11b-positive fraction was removed using a BD DynaMag-15 Magnet (BD Biosciences). The isolated microglia-depleted cells or mixed glial cells were plated either in 24-well plates at a density of 1.0 × 105 cells/well; in Lab-Tek II 8-chamber slides (Nagle Nunc International, Naperville, IL, USA) at a density of 1 × 105 cells/well; in 35-mm, 4-compartment cell culture dishes with a glass bottom (Greiner Bio-One, Kremsmunster, Austria) at a density of 1.5 × 105 cells/well; or in 48-well plates at a density of 1 × 105 cells/well for subsequent experiments. The culture medium was replaced twice weekly.
2.4. Absolute Cell Counting by Flow Cytometry
Cells obtained from Cx3cr1
+/EGFP mice were harvested from 24-well plates using TrypLE Express (1×), without phenol red, at 4, 7, 11, 14, and 21 days after the removal of microglia. Subsequently, 5 µL of AccuCount fluorescent particles (Spherotech Inc., Lake Forest, IL, USA), at a known concentration of 1 × 10
6 particles per mL, was added and analyzed using the LSRFortessa X-20 flow cytometer (BD, Franklin Lakes, NJ, USA). A total of 1000 particles were counted, and the absolute number of microglia per 1000 particles was determined. Data analysis was performed using FlowJo software ver10 (Tree Star, Ashland, OR, USA). Detailed gating strategies are provided in
Supplementary Figure S1. This study adhered to the Guidelines for the Use of Flow Cytometry and Cell Sorting in immunological studies [
14].
2.5. Microglial Surface Marker Analysis by Flow Cytometry
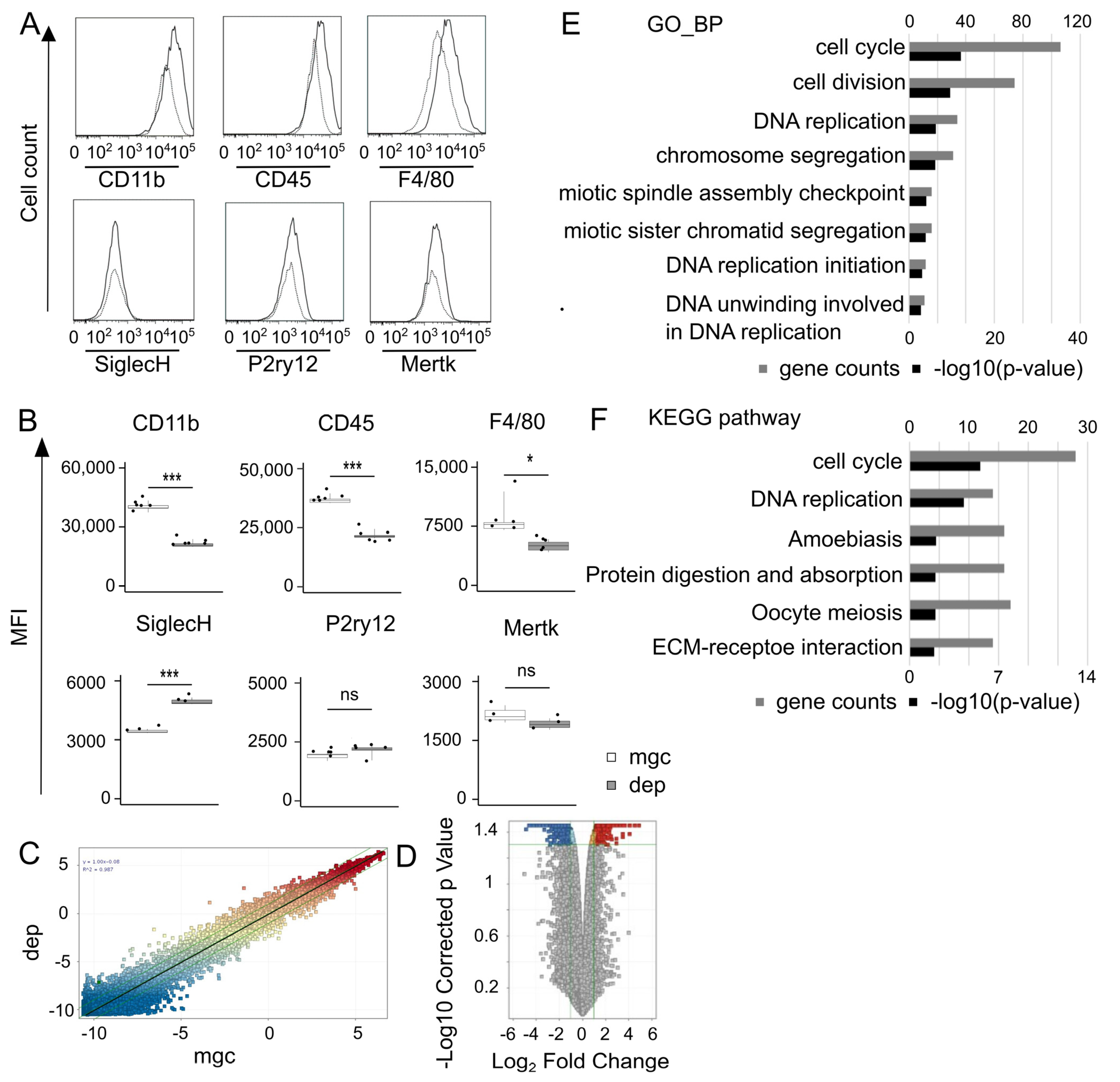
Seven days after removal, cells derived from Cx3cr1+/EGFP mice were harvested from 24-well plates using TrypLE Express. The cells were treated with Fc Block (1:50, clone 93) and then stained with phycoerythrin (PE) anti-mouse CD11b (1:50, clone M1/70), allophycocyanin (APC) anti-mouse CD45 (1:50, clone 30-F11), PE anti-mouse F4/80 (1:50, clone BM8), APC anti-mouse P2ry12 (1:50, clone S16007D), PE anti-mouse Mertk (1:50, clone 2B10C42), and APC anti-mouse SiglecH (1:50, clone M1304A01) antibodies (all obtained from BioLegend, San Diego, CA, USA). Data were acquired on an LSRFortessa X-20 flow cytometer and analyzed using FlowJo software. The threshold event number was set to 10,000 cells in the CX3CR1-EGFP-positive gate. If the acquired cells did not reach this setting, a minimum of 2000 microglia were analyzed.
2.6. Confocal Microscopy
Fluorescent images were captured using an Olympus FV 3000 confocal microscope (Olympus, Tokyo, Japan). Image J software ver1.53 (NIH, Bethesda, MD, USA) was used to autonomically calculate the cell count, occupied area, and circularity of microglial cells (circularity = 4πS/L2). In brief, 8-bit grayscale images were converted to binary by setting a minimum threshold value of 50. The binary image underwent a despeckle process to eliminate small artifacts, and then particles (size 50-inf µm2) were analyzed using the “analyze particle” command. Circularity was calculated as 4π (area/perimeter2). The cells with a circularity close to 1 were considered to have a round morphology.
2.7. Microarray and Data Analysis
Single-color microarray analysis was performed to compare the gene expression levels between repopulating and control microglia obtained from ICR mice derived from mixed glial cultures 10 days after culture. Two samples from the control microglia (nos. 1 and 2) and two samples from the repopulating microglia (nos. 3 and 4) were prepared. Total RNA was purified from microglia using the RNeasy Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. RNA purity was assessed at absorbance ratios of 260 and 280 nm, with a ratio of 1.8–2.0 indicating good RNA purity. RNA was amplified and hybridized on the Agilent Whole Mouse Genome version 2.0 ST array (G4836A), washed, and scanned using a SureScan Microarray Scanner (Agilent Technologies, Santa Clara, CA, USA). The scanned images were analyzed using Feature Extraction software version 11.5.1.5 (Agilent Technologies). The genes were normalized to the 75th percentile intensity and clustered using GeneSpring GX12.9 (Agilent Technologies).
DEGs were considered significant if they exhibited an adjusted
p-value of <0.05, determined using an unpaired
t-test adjusted by the Benjamini–Hochberg method, with a 1.5-fold or greater change between the two samples (no. 1 vs. no. 3 and no. 2 vs. no. 4). Subsequently, an enrichment analysis was performed using the DAVID Gene Functional Classification Tool (link
https://david.ncifcrf.gov/home.jsp, accessed on 24 January 2024) for DEGs, comparing repopulating microglia to controls.
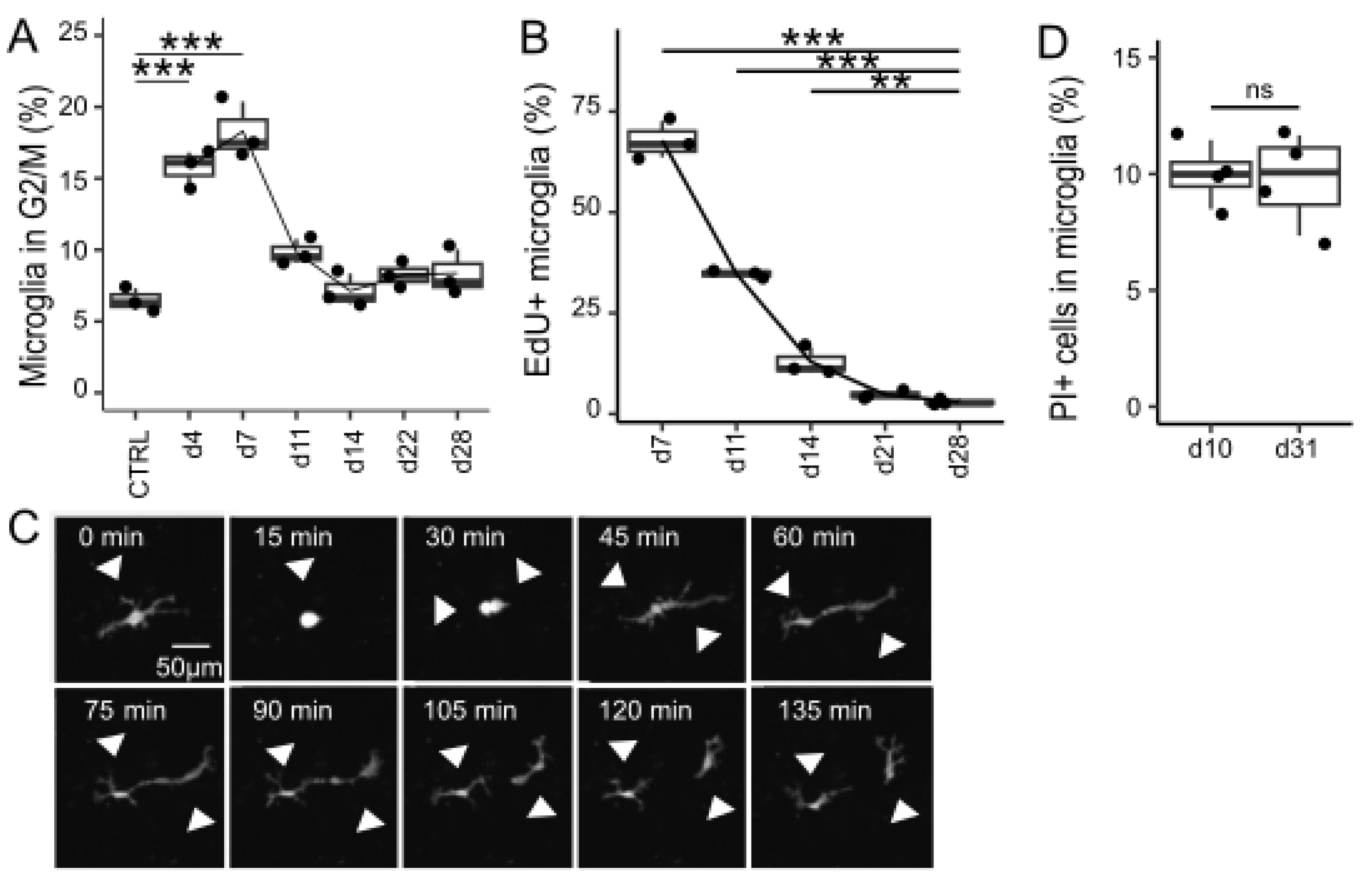
2.8. Cell Cycle Analysis
Flow cytometry using DNA-selective, cell membrane-permeant staining was used for performing an indirect cell cycle analysis on days 4, 7, 11, 14, 22, and 28 after removal, following the manufacturer’s protocol. Briefly, microglia-depleted cultures or mixed glial cultures (control) from Cx3cr1+/EGFP mice were detached from the 24-well plates. Cells were resuspended in Vybrant DyeCycle Violet stain (Thermo Fisher Scientific), which is excited at 405 nm and detected in the BV421 channel, diluted in DMEM without phenol red (Thermo Fisher Scientific) at a 1:1000 ratio. After gating GFP-positive cells, microglia in the G2/M phase were identified as BV421-high cells by flow cytometry, with the threshold determined using histogram-based gating.
2.9. EdU Cell Proliferation Assay
An EdU cell proliferation assay was performed using the Click-iT™ Plus EdU Alexa Fluor™ 647 Flow Cytometry Assay Kit (Thermo Fisher Scientific). Briefly, the cells from Cx3cr1+/EGFP mice-derived microglia-depleted cultures or mixed glial cultures (control) were labeled with 10 μM EdU for 1 h. The EdU-incorporated cells were detached from the 24-well plate, fixed with paraformaldehyde for 5 min, permeabilized with a saponin-based buffer for 10 min, and treated with a Click-iT reaction cocktail containing Alexa Fluor 647 azide for 30 min. For analysis, microglia were first gated based on GFP. EdU-positive microglia were subsequently identified as Alexa Flour 647-high cells by histogram-based gating using flow cytometry.
2.10. Live Cell Imaging of Repopulating Microglia
Microglia-depleted cells and mixed glial cells from Cx3cr1+/EGFP mice were plated and grown on 35 mm CELLview, a 4-compartment cell culture dish with a glass bottom (Greiner Bio-One), at a density of 1.5 × 105 cells/well. At 7–12 days in vitro, the medium was replaced with FluoroBite DMEM (Thermo Fisher Scientific) supplemented with 10% FBS immediately before scanning. Confocal long-term live cell imaging of repopulating microglia was performed with a confocal scanner box (Cell Voyager CV1000, Yokogawa Electric Corp., Tokyo, Japan) from 5 to 8 days after removal (15 min intervals, 240 iterations, total of 60 h). Each well was scanned using a 10× objective lens in 8 randomly selected positions.
2.11. Propidium Iodide Staining Coupled with Flow Cytometry
Microglia-depleted cultures were prepared from ICR mice as described above. After 7 or 28 days of culture, the cells were detached from the 24-well plates using TrypLE Express (1×) without phenol red. The detached cells were incubated with Fc Block (1:50, clone 93) and stained with APC anti-mouse CD11b (1:50) (BioLegend) and PI (1:10) (BioLegend) according to the manufacturer’s protocol. PI-positive microglia were analyzed using flow cytometry.
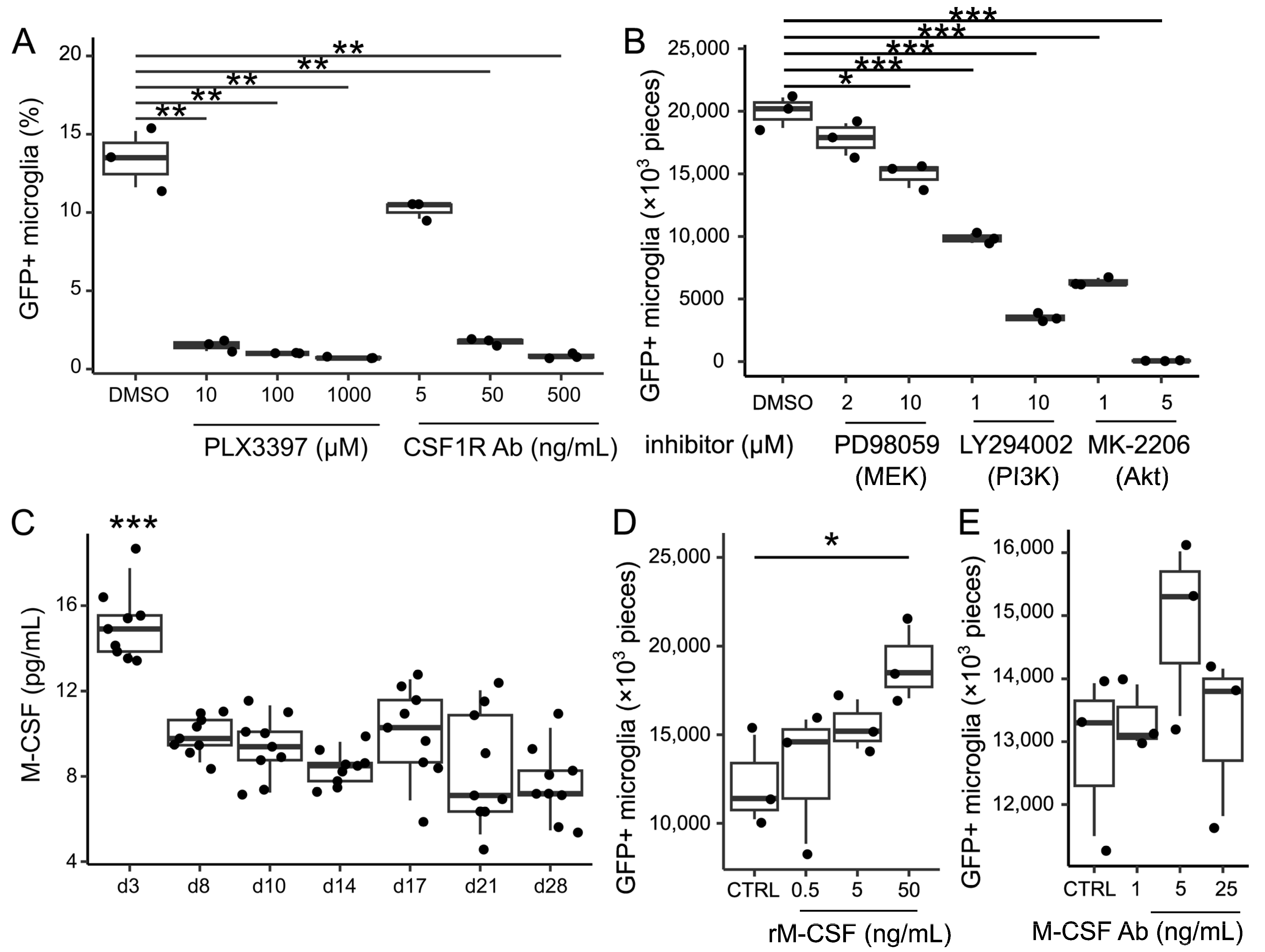
2.12. Inhibition Experiments Against Csf1r Signaling
Microglia-depleted cultures were treated with the CSF1R inhibitor (PLX3397 (pexidartinib; Cayman Chemical, Ann Arbor, MI, USA), mouse CSF1R-blocking antibody (AFS98 (BioLegend)), MEK inhibitor (PD98059, Calbiochem, San Diego, CA, USA), PI3K inhibitor (LY294002, Tocris Bioscience, Bristol, UK), or Akt inhibitor (MK2206, Tocris Bioscience). The media were replaced with DMEM containing each inhibitor or CSF1R-blocking antibody twice a week, with a total of four replacements.
2.13. Enzyme-Linked Immunosorbent Assay for M-CSF in the Supernatant of Microglia-Depleted Cultures
Microglia-depleted cultures were prepared from B6J wild-type mice. Murine M-CSF concentrations in the supernatants of microglia-depleted cultures were measured via ELISA. A 96-well plate (Corning, Corning, NY, USA) was coated with 50 µL/well of capture Ab against M-CSF (PeproTech, Cranbury, NJ, USA) at 1:200 dilution and incubated overnight at 4 °C. Subsequently, the wells were washed three times with wash buffer (BD Biosciences, Franklin Lakes, NJ, USA) and blocked with 50 µL/well of Assay Diluent (AD) (BD Biosciences) for 1 h at RT. After removal of the blocking solutions and three washes, 50 µL/well of undiluted samples, standard solutions, and the negative control (AD) was added, and the plates were incubated at room temperature for 2 h. After three washes, 50 µL/well of detection Ab (PeproTech) was added at 1:400 dilution for 1 h at RT. After another three washes, 50 µL/well of Streptavidin-HRP was added at 1:1000 dilution. After five washes, 50 µL/well of substrate solution was added. When the standard solutions turned yellow, the reaction was stopped with 25 µL/well of stop solution (BD). The absorbance was measured at 450 nm using a 2030 ARVOTMX3 Multilabel Reader (PerkinElmer, Waltham, MA, USA).
2.14. Treatment with Recombinant M-CSF or M-CSF Blocking Antibody
MGC from Cx3cr1+/EGFP mice were treated with recombinant mouse M-CSF (BioLegend) or mouse M-CSF blocking antibody (MAB4161, R&D Systems, Minneapolis, MN, USA). The medium was replaced with DMEM containing each compound twice a week, with a total of four replacements.
2.15. Coculture Experiments
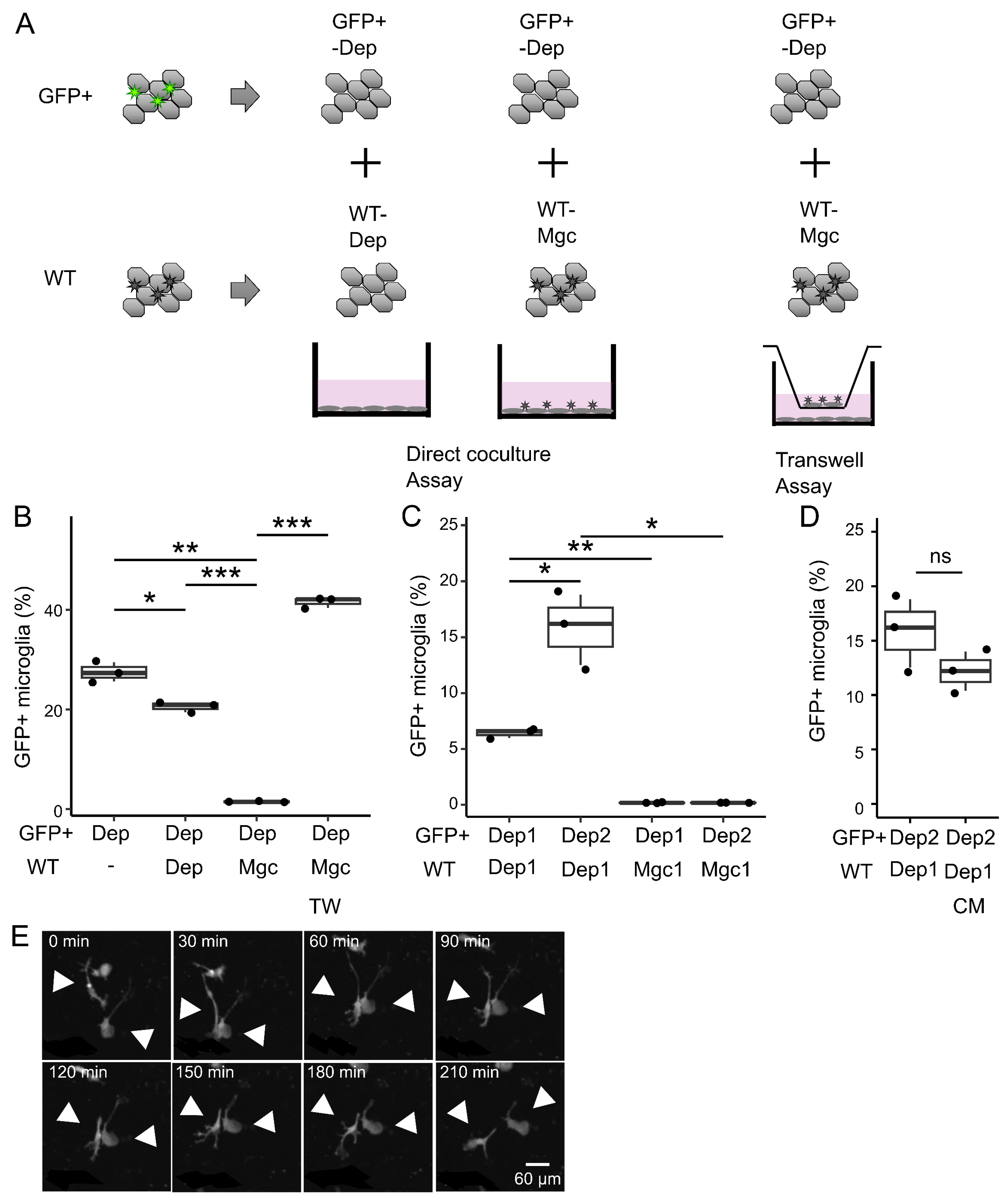
Coculture experiments were performed using cultured cells derived from Cx3cr1+/EGFP mice and ICR mice. Microglia-depleted cultures (i.e., “Dep”) derived from Cx3cr1+/EGFP mice were cocultured with either Mgc or Dep derived from ICR mice, at 1:1 or 2:1 ratios. The cells derived from both Cx3cr1+/EGFP mice and ICR mice were detached from the flasks; subsequently, the microglia were depleted (Dep), mixed at a 1:1 or 2:1 ratio, and seeded in 24-well plates. For non-contact coculture, ICR-derived cells were seeded separately from Cx3cr1+/EGFP mice-derived cells using a Transwell® insert with a 0.4 μm pore polycarbonate membrane (Corning). To assess whether secreted factors derived from mixed glial cultures suppress repopulation, the cells were cultured in the same volume of conditioned medium collected from independent mixed glial cultures which was replaced twice a week.
2.16. Statistical Analysis
All data are expressed as box-and-whisker plots, with the box depicting the median and the 25th and 75th quantiles and the whiskers showing the 5th and 95th percentiles. The line graph shows the mean values. Each experiment was independently repeated at least twice. Statistical analyses were performed using a two-tailed Welch’s
t-test and one-way analysis of variance (ANOVA) and Turkey’s post test for multiple comparisons, as indicated. The Kolmogorov–Smirnov test was used to determine the normality of data. Analyses were performed using EZR [
15] and a
p-value of <0.05 was considered significant.
4. Discussion
Models of the transient removal of microglia using chemical or genetic methods have contributed significantly to elucidating the developmental origin and differentiation mechanisms of microglia, but also the role of microglia in various disease models. However, despite the valuable insights gained from these models, certain challenges persist and have not yet been effectively addressed. For example, an in vivo microglial ablation system, which induces massive cell death, has been reported to trigger a type 1 interferon-defined inflammatory cascade, possibly resulting in neuronal cell death [
3,
16]. The effects of such cell death have been a significant obstacle in evaluating the effects of microglial ablation on immune-mediated neurological diseases, including experimental autoimmune encephalomyelitis [
16]. Furthermore, the release of damage-associated molecular patterns (DAMPs) from dying cells can induce proliferation and immune system activation in surrounding cells [
17,
18,
19]. These complexities pose significant barriers to elucidating the detailed mechanisms governing microglial repopulation.
We deemed it crucial to employ physical rather than chemical methods to remove microglia, aiming to overcome challenges associated with existing models. In our approach, microglia were transiently removed from MGCs in vitro through magnetic separation. While chemical removal has the disadvantage that the effects of cell death mentioned above cannot be ignored, physical removal offers a solution to this problem even in vitro. Utilizing anti-CD11b magnetic particles in an in vitro microglia removal system, similar to the previously reported in vivo microglia ablation system [
3,
4,
5,
6], microglia were efficiently removed, initiating a subsequent repopulation process. In the in vitro setting, the repopulation process took more than 14 days, while microglia were reported to repopulate and return to baseline levels within 5–14 days after removal in vivo [
3,
4,
5,
20]. Compared with the in vivo method, the prolonged in vitro repopulation timeframe may be attributed to cell damage resulting from the cell detachment procedure and an environment less conducive to supporting microglial survival and proliferation in vitro. In addition, cell adhesion efficiency differs between glass and plastic bottoms, which results in slower cell proliferation on microscopic examination. Microglial repopulation did not persist and reached a plateau when the number of microglia was comparable to that of MGC.
Morphological assessments revealed transient changes in repopulating microglia, such as fewer branched processes and a more rounded soma, as reflected by an increase in microglial circularity within 7–10 days after removal. Similar rounded morphologies have been reported during the microglial development in vivo [
21,
22,
23], reflecting a temporary state of immaturity. On day 21, the occupied area decreased, accompanied by an increase in circularity.
The decreased expression levels of CD11b, CD45, and F4/80 in repopulating microglia may also reflect immaturity. Conversely, the expression of microglia-specific markers, including P2ry12, Siglec-H, and Mertk, did not decrease. This indicates that repopulating cells retain their fundamental microglial characteristics regardless of their immaturity. In summary, repopulating microglia exhibited morphological and protein expression changes that transiently reflected their immaturity.
Even under physiological conditions, microglia maintain constant density through slow but stochastic proliferation and apoptosis [
8,
24]. Cell cycle analysis revealed that early repopulating microglia re-entered the cell cycle and exhibited increased proliferation. Transcriptome analysis supported this finding, indicating alterations in the expression of cell cycle-related genes in repopulating microglia. Time-lapse imaging further confirmed microglial division. In the time-lapse imaging, we were unable to observe the birth of CX3CR1-negative microglia, suggesting that, at least in this in vitro system, microglia are unlikely to be derived from CX3CR1-negative progenitors. This observation indicates that the local expansion of residual microglia contributes to repopulation [
5,
6].
Previous studies have shown that the oral administration of a CSF1R inhibitor in adult mice results in a 99% removal of microglia [
3]. CSF1R signaling is essential for microglial survival and proliferation. Inhibition experiments using both chemicals and antibodies revealed that microglial repopulation is dependent on CSF1R signaling. Although the MEK-ERK and PI3K-Akt pathways are known to be downstream of CSF1R, microglial repopulation was unaffected by MEK-ERK inhibition but was significantly suppressed by either PI3K or Akt inhibition. Phosphorylation of PI3K and Akt was evaluated by Western blotting, but bands were not well detected. The essential role of CSF1R signaling is well known, and CSF1R-PI3K-Akt signaling is also deemed to be important for repopulation, albeit indirectly. In this model, neither M-CSF nor interleukin-34, the ligands for CSF1R, were added, suggesting that microglial proliferation was sustained by endogenous CSF1R ligands produced by unknown cells. Although M-CSF was identified in the culture supernatant, changes in its concentration could not account for the observed repopulation. It is reasonable to assume that the start of microglial repopulation is additionally regulated by factors other than the CSF1R-PI3K-Akt pathway.
We then examined the phenomenon by which microglia repopulation ceases when they are sufficiently proliferated. The cessation of repopulation was not due to an increase in microglial death, as the proportion of PI-positive cells did not increase when the number of microglia reached a plateau. Therefore, we hypothesized that an unknown mechanism may inhibit microglial proliferation. In direct contact cocultures of GFP
+ and GFP
− cells, repopulation failed to occur when abundant coexisting microglia were present. This observation can be explained by a negative feedback mechanism in which microglia send inhibitory signals to each other, suppressing proliferation. The results from the experiments using conditioned media and Transwell assays support that signals inhibiting microglial proliferation are contact-dependent rather than mediated by secreted factors. Another possibility is that proliferation is inhibited by the transient, localized release of soluble factors when two microglial cells come into close proximity. In any case, such reciprocal growth inhibition could explain a variety of physiological features of microglia. In vitro, microglia have been observed to migrate, actively expand, and contract their processes. Although we did not assess the distance among microglia, microglia in the brain are arranged at regular intervals, suggesting that each cell occupies a territory within the brain parenchyma [
8,
24]. The growth inhibition against neighboring microglia may play a role in maintaining spatial homeostasis. Furthermore, the absence of known malignant tumors arising from microglia suggests an extremely potent growth suppression mechanism within these cells.
It is also known that the ability of microglia to repopulate is not infinite. If the removal of microglia was repeated in short cycles (7 days’ treatment and 7 days’ recovery) using a CSF1R inhibitor, the microglia no longer repopulated after the second removal [
25]. Unsolved questions remain regarding microglial repopulation and await elucidation of the sensing mechanism of microglial depletion.
This study has several limitations. Firstly, microglia undergo rapid changes in their properties, giving rise to heterogeneous subpopulations when examined ex vivo [
25]. Thus, it is unclear whether the observations in the present study accurately reproduced the phenomena occurring in microglia in vivo. Additionally, the current in vitro culture system differs from the living brain environment as it primarily consists of microglia, astrocytes, and oligodendrocytes, and does not include neurons or vasculature. Secondly, we used different strains of mice in different experiments, and it is possible that this difference affected the results in some experiments. However, since microglial repopulation was observed in the same manner in the two different strains of mice, we considered it unlikely that differences in mouse strain would seriously affect the outcome of the co-culture. Thirdly, we did not provide direct evidence that PI3K–AKT activation specifically occurs downstream of CSF1R signaling in the context of microglial proliferation during repopulation. The identification of the master regulators of microglial proliferation remains challenging.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}