
Neuro-Ophthalmologic Variability in Presentation of Genetically Confirmed Wolfram Syndrome: A Case Series and Review

 , , ,
, , ,
Abstract

1. Introduction
2. Cases
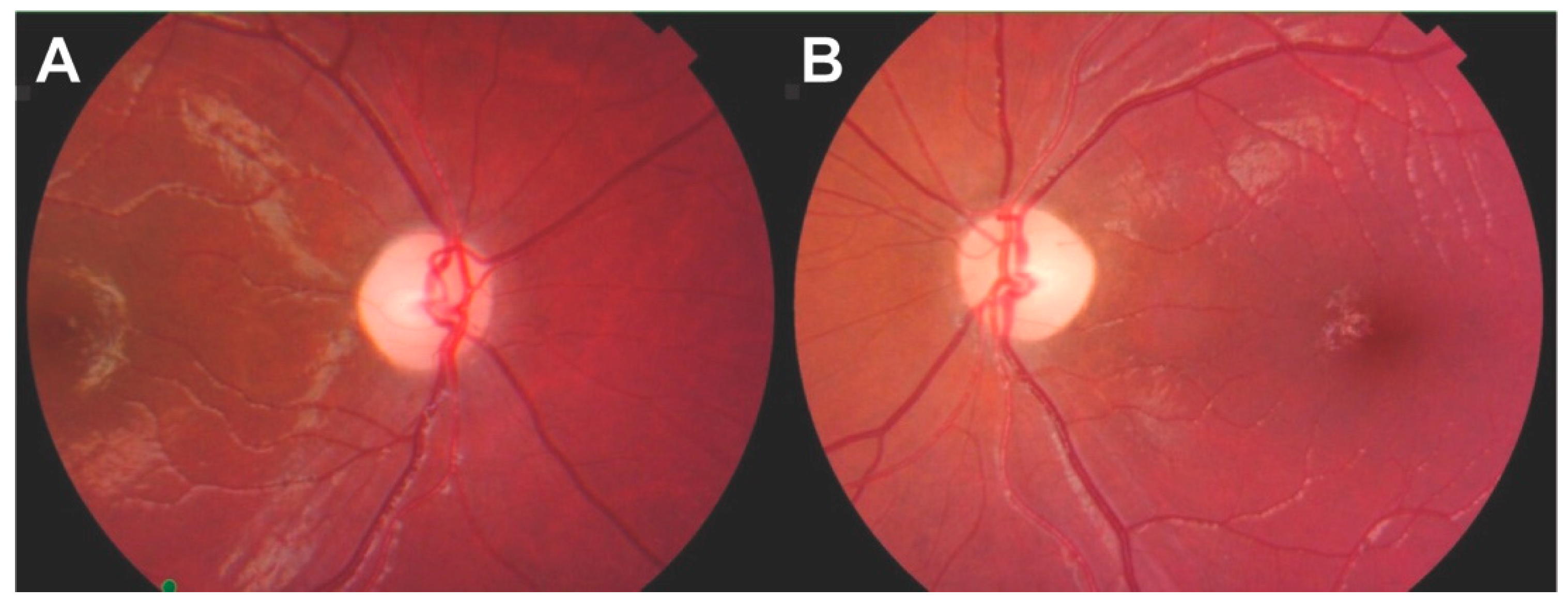
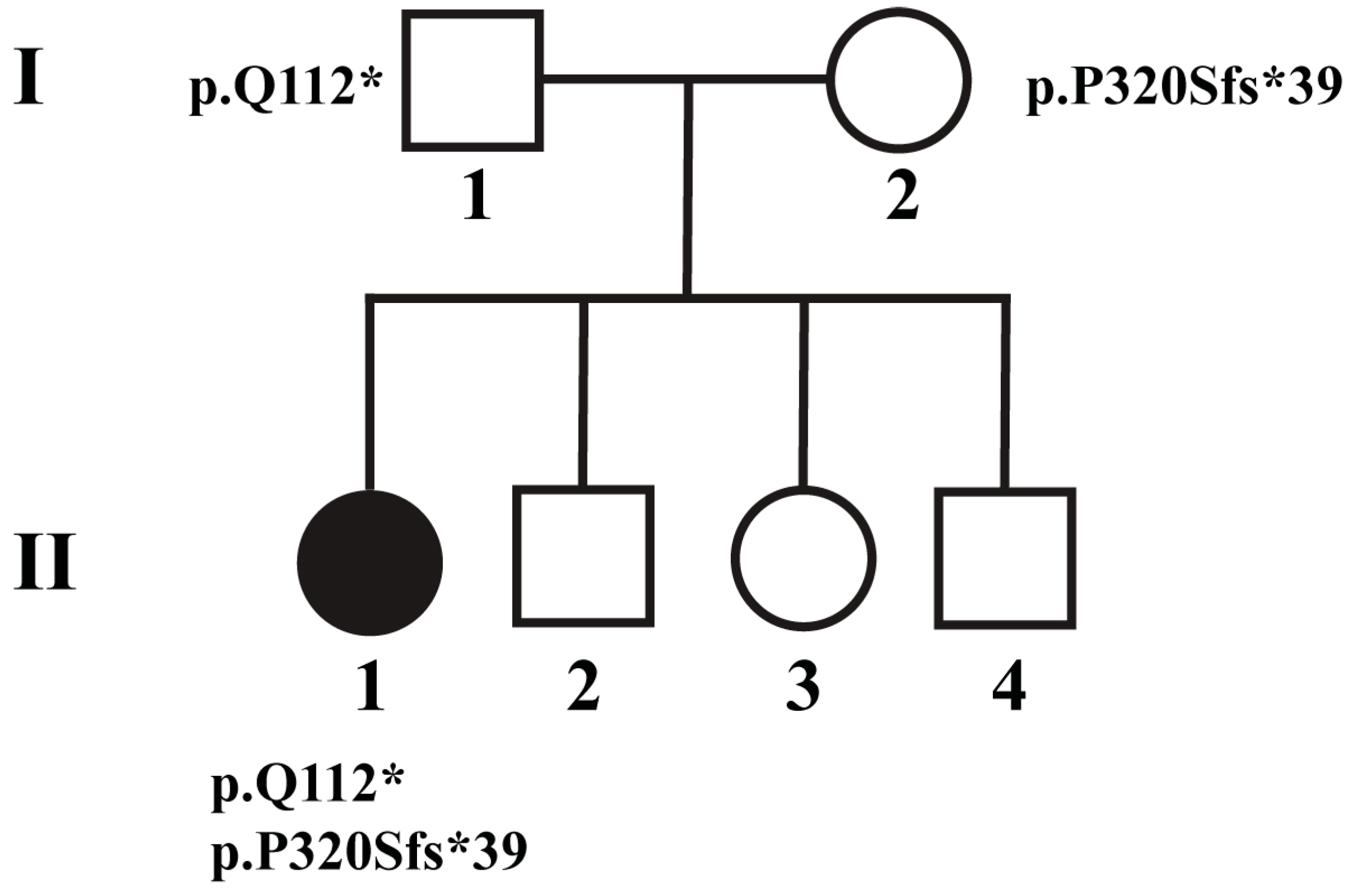
2.1. Patient 1
2.2. Patient 2
2.3. Patient 3
2.4. Patient 4
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Inoue, H.; Tanizawa, Y.; Wasson, J.; Behn, P.; Kalidas, K.; Bernal-Mizrachi, E.; Mueckler, M.; Marshall, H.; Donis-Keller, H.; Crock, P.; et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat. Genet. 1998, 20, 143–148. [Google Scholar] [CrossRef]
- Rigoli, L.; Caruso, V.; Salzano, G.; Lombardo, F. Wolfram Syndrome 1: From Genetics to Therapy. Int. J. Environ. Res. Public Health 2022, 19, 3225. [Google Scholar] [CrossRef] [PubMed]
- de Heredia, M.L.; Cleries, R.; Nunes, V. Genotypic classification of patients with Wolfram syndrome: Insights into the natural history of the disease and correlation with phenotype. Genet. Med. 2013, 15, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.; Philbrook, C.; Gerbitz, K.D.; Bauer, M.F. Wolfram syndrome: Structural and functional analyses of mutant and wild-type wolframin, the WFS1 gene product. Hum. Mol. Genet. 2003, 12, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Chaussenot, A.; Bannwarth, S.; Rouzier, C.; Vialettes, B.; Mkadem, S.A.; Chabrol, B.; Cano, A.; Labauge, P.; Paquis-Flucklinger, V. Neurologic features and genotype-phenotype correlation in Wolfram syndrome. Ann. Neurol. 2011, 69, 501–508. [Google Scholar] [CrossRef]
- Rohayem, J.; Ehlers, C.; Wiedemann, B.; Holl, R.; Oexle, K.; Kordonouri, O.; Salzano, G.; Meissner, T.; Burger, W.; Schober, E.; et al. Diabetes and neurodegeneration in Wolfram syndrome: A multicenter study of phenotype and genotype. Diabetes Care 2011, 34, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Urano, F. Wolfram Syndrome: Diagnosis, Management, and Treatment. Curr. Diab. Rep. 2016, 16, 6. [Google Scholar] [CrossRef]
- Hoekel, J.; Chisholm, S.A.; Al-Lozi, A.; Hershey, T.; Tychsen, L.; Washington University Wolfram Study Group. Ophthalmologic correlates of disease severity in children and adolescents with Wolfram syndrome. J. AAPOS 2014, 18, 461–465.e1. [Google Scholar] [CrossRef]
- Newman, N.J.; Biousse, V. Hereditary optic neuropathies. Eye 2004, 18, 1144–1160. [Google Scholar] [CrossRef]
- Waszczykowska, A.; Zmyslowska, A.; Bartosiewicz, K.; Studzian, M.; Pulaski, L.; Braun, M.; Ivask, M.; Koks, S.; Jurowski, P.; Mlynarski, W. Reduced Corneal Sensitivity With Neuronal Degeneration is a Novel Clinical Feature in Wolfram Syndrome. Am. J. Ophthalmol. 2022, 236, 63–68. [Google Scholar] [CrossRef]
- Al-Till, M.; Jarrah, N.S.; Ajlouni, K.M. Ophthalmologic findings in fifteen patients with Wolfram syndrome. Eur. J. Ophthalmol. 2002, 12, 84–88. [Google Scholar] [CrossRef]
- Kabanovski, A.; Donaldson, L.; Margolin, E. Neuro-ophthalmological manifestations of Wolfram syndrome: Case series and review of the literature. J. Neurol. Sci. 2022, 437, 120267. [Google Scholar] [CrossRef] [PubMed]
- Lessell, S.; Rosman, N.P. Juvenile diabetes mellitus and optic atrophy. Arch. Neurol. 1977, 34, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Ray, M.K.; Chen, L.; White, N.H.; Ni, R.; Hershey, T.; Marshall, B.A. Longitudinal progression of diabetes mellitus in Wolfram syndrome: The Washington University Wolfram Research Clinic experience. Pediatr. Diabetes 2022, 23, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Samara, A.; Lugar, H.M.; Hershey, T.; Shimony, J.S. Longitudinal Assessment of Neuroradiologic Features in Wolfram Syndrome. AJNR Am. J. Neuroradiol. 2020, 41, 2364–2369. [Google Scholar] [CrossRef]
- Gocmen, R.; Guler, E. Teaching NeuroImages: MRI of brain findings of Wolfram (DIDMOAD) syndrome. Neurology 2014, 83, e213–e214. [Google Scholar] [CrossRef]
- Scolding, N.J.; Kellar-Wood, H.F.; Shaw, C.; Shneerson, J.M.; Antoun, N. Wolfram syndrome: Hereditary diabetes mellitus with brainstem and optic atrophy. Ann. Neurol. 1996, 39, 352–360. [Google Scholar] [CrossRef]
- Dworak, D.P.; Nichols, J. A review of optic neuropathies. Dis. Mon. 2014, 60, 276–281. [Google Scholar] [CrossRef]
- Biousse, V.; Newman, N.J. Ischemic Optic Neuropathies. N. Engl. J. Med. 2015, 372, 2428–2436. [Google Scholar] [CrossRef]
- Bennett, J.L. Optic Neuritis. Continuum 2019, 25, 1236–1264. [Google Scholar] [CrossRef]
- Sharma, P.; Sharma, R. Toxic optic neuropathy. Indian J. Ophthalmol. 2011, 59, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J. Hereditary optic neuropathies: From the mitochondria to the optic nerve. Am. J. Ophthalmol. 2005, 140, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Rocatcher, A.; Desquiret-Dumas, V.; Charif, M.; Ferre, M.; Gohier, P.; Mirebeau-Prunier, D.; Verny, C.; Milea, D.; Lenaers, G.; Group, H.O.N.C.; et al. The top 10 most frequently involved genes in hereditary optic neuropathies in 2186 probands. Brain 2023, 146, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Rouzier, C.; Monnot, S.; Chabrol, B.; Conrath, J.; Lecomte, P.; Delobel, B.; Boileau, P.; Valero, R.; Procaccio, V.; et al. Identification of novel mutations in WFS1 and genotype-phenotype correlation in Wolfram syndrome. Am. J. Med. Genet. A 2007, 143A, 1605–1612. [Google Scholar] [CrossRef]
- Chaussenot, A.; Rouzier, C.; Quere, M.; Plutino, M.; Ait-El-Mkadem, S.; Bannwarth, S.; Barth, M.; Dollfus, H.; Charles, P.; Nicolino, M.; et al. Mutation update and uncommon phenotypes in a French cohort of 96 patients with WFS1-related disorders. Clin. Genet. 2015, 87, 430–439. [Google Scholar] [CrossRef]
- Matsunaga, K.; Tanabe, K.; Inoue, H.; Okuya, S.; Ohta, Y.; Akiyama, M.; Taguchi, A.; Kora, Y.; Okayama, N.; Yamada, Y.; et al. Wolfram syndrome in the Japanese population; molecular analysis of WFS1 gene and characterization of clinical features. PLoS ONE 2014, 9, e106906. [Google Scholar] [CrossRef]
- Hu, K.; Zatyka, M.; Astuti, D.; Beer, N.; Dias, R.P.; Kulkarni, A.; Ainsworth, J.; Wright, B.; Majander, A.; Yu-Wai-Man, P.; et al. WFS1 protein expression correlates with clinical progression of optic atrophy in patients with Wolfram syndrome. J. Med. Genet. 2022, 59, 65–74. [Google Scholar] [CrossRef]
- Wilf-Yarkoni, A.; Shor, O.; Fellner, A.; Hellmann, M.A.; Pras, E.; Yonath, H.; Shkedi-Rafid, S.; Basel-Salmon, L.; Bazak, L.; Eliahou, R.; et al. Mild Phenotype of Wolfram Syndrome Associated with a Common Pathogenic Variant Is Predicted by a Structural Model of Wolframin. Neurol. Genet. 2021, 7, e578. [Google Scholar] [CrossRef]
- Bansal, V.; Boehm, B.O.; Darvasi, A. Identification of a missense variant in the WFS1 gene that causes a mild form of Wolfram syndrome and is associated with risk for type 2 diabetes in Ashkenazi Jewish individuals. Diabetologia 2018, 61, 2180–2188. [Google Scholar] [CrossRef]
- Astuti, D.; Sabir, A.; Fulton, P.; Zatyka, M.; Williams, D.; Hardy, C.; Milan, G.; Favaretto, F.; Yu-Wai-Man, P.; Rohayem, J.; et al. Monogenic diabetes syndromes: Locus-specific databases for Alstrom, Wolfram, and Thiamine-responsive megaloblastic anemia. Hum. Mutat. 2017, 38, 764–777. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Eval. Age | Sex | WFS1 Variants | Presenting Sign/Symptom | DM | OA | DI | D | Other | MRI Findings |
|---|---|---|---|---|---|---|---|---|---|---|
| (Age in Years at Onset) | ||||||||||
| 1 | 6 | F | c.334 C>T:p.Q112* | Progressive vision loss | 4 | 5 | - | - | None | Unrevealing |
| c.958_961delinsTCC:p.P320Sfs*39 | ||||||||||
| 2 | 32 | M | c.1672C>T:p.R558C | Color vision changes | 20s | 20s | - | - | None | Optic nerve atrophy |
| c.1672C>T:p.R558C | ||||||||||
| 3 | 46 | F | c.2254G>T:p.E752* | Progressive vision loss | - | Childhood | - | - | None | Optic nerve atrophy |
| c.1673G>A:p.R558H | ||||||||||
| 4 | 45 | F | c.1230_1233del:p.V412Sfs*29 | Pupillary dilation | 9 | 20s | - | - | Neurogenic bladder | Optic nerve, brainstem, cerebellum atrophy |
| c.1672C>T:p.R558C | Progressive vision loss | Pons T2 hyper intensities | ||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jauregui, R.; Abreu, N.J.; Golan, S.; Panarelli, J.F.; Sigireddi, M.; Nayak, G.K.; Gold, D.M.; Rucker, J.C.; Galetta, S.L.; Grossman, S.N. Neuro-Ophthalmologic Variability in Presentation of Genetically Confirmed Wolfram Syndrome: A Case Series and Review. Brain Sci. 2023, 13, 1030. https://doi.org/10.3390/brainsci13071030
Jauregui R, Abreu NJ, Golan S, Panarelli JF, Sigireddi M, Nayak GK, Gold DM, Rucker JC, Galetta SL, Grossman SN. Neuro-Ophthalmologic Variability in Presentation of Genetically Confirmed Wolfram Syndrome: A Case Series and Review. Brain Sciences. 2023; 13(7):1030. https://doi.org/10.3390/brainsci13071030
Chicago/Turabian StyleJauregui, Ruben, Nicolas J. Abreu, Shani Golan, Joseph F. Panarelli, Meenakshi Sigireddi, Gopi K. Nayak, Doria M. Gold, Janet C. Rucker, Steven L. Galetta, and Scott N. Grossman. 2023. "Neuro-Ophthalmologic Variability in Presentation of Genetically Confirmed Wolfram Syndrome: A Case Series and Review" Brain Sciences 13, no. 7: 1030. https://doi.org/10.3390/brainsci13071030
APA StyleJauregui, R., Abreu, N. J., Golan, S., Panarelli, J. F., Sigireddi, M., Nayak, G. K., Gold, D. M., Rucker, J. C., Galetta, S. L., & Grossman, S. N. (2023). Neuro-Ophthalmologic Variability in Presentation of Genetically Confirmed Wolfram Syndrome: A Case Series and Review. Brain Sciences, 13(7), 1030. https://doi.org/10.3390/brainsci13071030