1. Introduction
Optical sensing applied to the human intracellular environment represents the best way for the analysis of cell functioning and its behavior in response to specific physiological conditions. Cell ultrastructure, embryogenic development [
1], pH–calcium–chloride–oxygen monitoring [
2] and, most importantly, metastasis proliferation [
3] are a few examples of what this type of optical sensors would help to analyze.
Intracellular optical sensing works by the introduction of a luminescent (fluorescent or chemiluminescent) probe inside the target cell and its subsequent detection.
The configuration of an optical sensing strategy, generally, starts with the fluorophore choice (considering its optical and biochemical properties) and its characterization, mainly consisting of testing cells survival after treatment and photostability. Once defined, the fluorescent dye must be transferred through the membrane of the investigated cell model by an uptake procedure that often requires fluorophore structural modifications, carriers and cell permeabilization [
4,
5,
6]. Finally, a high performances optical sensing system for the fluorescence detection must be chosen.
Nowadays, research efforts are moving towards the miniaturization, time/cost-saving and user-friendly sensing detection systems, in order to provide the basis for portable Point-of-Care platforms and patient self-monitoring. In this sense, one of the most representative examples is the use of photomultiplier-based systems having the greatest advantage, with respect to traditional detection instruments, of being highly miniaturizable, integrable and cheap hence, perfectly matching with the goal of a portable application, by using solid state photomultipliers (e.g. silicon photomultipliers, SiPM). However, this type of technology coupled with traditionally employed dyes, usually, implies the issue of the electronic management for luminescence analysis, which is quite sophisticated due to the chemical-physical properties of the luminescent probe used. Prolonged exposure times and high irradiation powers, affect conventional dyes emission efficiency, resulting in a bleaching process that reduces the fluorescence intensity and, sometimes, brings a series of toxic intracellular effects such as free radicals production [
7]. Moreover, room temperature storage degrades the organic dyes normally employed for cells labeling [
8,
9].
Cyanine dyes, for example, suffer from self-quenching of fluorescence [
10], caused by the proximity and overlap of absorption and emission’s peaks, and is photobleached after prolonged exposure to the laser beam [
11,
12], hence dark conditions are required during the labeling experiment, which is problematic for fluorescence sensing, especially in time-lapse applications. Moreover, its short lifetime implies a complex electronic management of the fluorescence detection, thus increasing the complexity of sensing technology. Other evidences report a Rhodamine 6G, Fluorescein, Pyrene and Indo-1 bleaching process caused by an increase in the molecules intrinsic temperature, following the light absorption and the source irradiation power [
13]. There are, also, some examples of luminescent quantum dots (nanocrystals composed by heavy metal atoms like Cd, Hg, Pb and As) causing cytotoxicity after the cell uptake, due to the release of metallic ions that can bind the sulfhydryl groups of many biomolecules, such as proteins, leading to the decrease in various sub-cellular organelles activity [
14].
In order to surpass all of the limitations related to conventional optical intracellular sensing methods, we investigated an approach that used a miniaturizable photomultiplier-based system in combination with a cell labeling method performed by the use of fluorophore Tris(2,2’bipyridyl)ruthenium(II) (Ru(bpy)
32+), whose structure is schematically shown in
Figure 1. This molecule is an octahedral coordination complex of transition metal ruthenium bounded to three heteroaromatic bypiridine units.
Its optical properties would allow for the solving of some issues, related to the use of the conventional fluorophores described before. Ru(bpy)
32+ has two absorption peaks at 290 nm and 450 nm, ligand-center (LC) and metal-ligand (MLCT) electronic transitions, respectively, and a lifetime of 360 ns suspended in water. Absorption peaks are far away from the emission one at 630 nm [
15,
16] and 100 nm to the closest absorption peak, thus strongly reducing the self-quenching of fluorescence since the absorption and emission curves do not overlap. Therefore, the incident radiation can be shielded using a simple and cheap band-pass filter. Moreover, the molecular properties do not change even in stressful environmental conditions [
12]. We already proved the Ru(bpy)
32+ stability by monitoring its absorption, emission and lifetime under very unsustainable chemical-physical conditions [
12]. More precisely, we tested fluorophore in a variable range of temperatures, from RT to 100 °C; pH, from 1 to 11; light irradiation, up to 24 hours. All of the experiments that were performed indicated that Ru(bpy)
32+ could be used in a wide range of conditions, therein including the environmental light.
Most of the evidences of Ru-based complexes for intracellular labeling report examples other than the commercial Ru(bpy)
32+, as a result of long and expensive chemical functionalization [
17,
18,
19,
20] including, sometimes, carrier mediations [
21]. As an example, in [
17] uptake in cells was performed using a Ru(bpy)
2dppz
2+ molecule modified by changing bypiridine with 4,7-diphenyl-1,10-phenanthroline ligand in order to make the fluorophore more hydrophobic for uptake, but strongly increasing the costs and dye preparation time. Moreover, characterizations of cell uptake processes of modified Ru-based complexes in human cells revealed that the bigger the aromatic surface of the N-N ligand in the [Ru(bpy)
2 (N-N)]
2+ starting molecule the higher the cytotoxicity of the final compound was, thus affecting the cell viability [
22].
For these reasons, we introduced a different approach to the intracellular analysis of human cells, based on the Ru(bpy)32+ performances and stability and its suitability for the development of an innovative intracellular optical sensing system.
We performed a full characterization of the labeling and sensing approaches by introducing the fluorophore Ru(bpy)32+ in human dermal fibroblasts (HDF) and, subsequently, detected its fluorescence by a sensitive and miniaturizable laser beam-photomultiplier based system. HDFs uptake of Ru(bpy)32+ was confirmed by Fluorescence Microscopy (FM) and Confocal Laser Scanning Microscopy (CLSM). Cells survival to the labeling method was checked by MTT assays of cells exposed to the fluorophore.
A further investigation of cell sensitivity to Ru(bpy)32+ was performed on a human tumor cell model, performing the fluorophore uptake within the human colon adenocarcinoma (LoVo) cells and monitoring their vitality and fluorescence intensity across time.
2. Materials and Methods
2.1. Materials
Human Dermal Fibroblasts adult- 106-05A, HDF, were from Sigma. Human colon adenocarcinoma, LoVo, were from ATCC® CCL-229™. Medium for HDF line was Dulbecco Modified Eagle Medium (DMEM) with 4.5 g/L Glucose with L-Glutamine. Medium for LoVo was RPMI-1640 with L-glutamine and sodium bicarbonate. Media were supplemented with antibiotic-antimycotic (10,000 U/mL Penicillin, 10 μg/ml Streptomycin, 25 μg/ml Fungizon) and 10% Fetal Bovine Serum. To detach cells, we used Trypsin - EDTA·4Na (0.05%/0.02% w/v). All products were purchased from GIBCO, Thermo Fisher Scientific. Cuvettes for spectrophotometric analysis were UV-transparent disposable Ultra-Micro, 2 × 3.5 mm2 with 10 mm optical path. Slides for fluorescence microscopy were micro coverglasses by Electron Microscopy Sciences (EMS), mounted on a glass slide 25 mm × 75 mm (EMS). Powder of tris(2,2′-bipyridyl)dichlororuthenium(II) hexahydrate was from Sigma-Aldrich. Dulbecco’s Phosphate buffer saline 1× (PBS) was from GIBCO, Thermo Fisher Scientific.
2.2. Cell Culture and Labeling
HDF and LoVo cells were expanded in 75 cm3 flasks in medium and maintained at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. Medium changes were performed after 3–4 days and, upon confluence (80%), cells were trypsinized and prepared for experiments. Vital cell count was performed in a Bürker chamber with Trypan blue method. The solution of 13.5 µM (corresponding to 10 mg/mL) Ru(bpy)32+ was prepared by dissolving the fluorophore in PBS 1×. Subsequently, the solution was sonicated for 20 min in order to avoid the formation of fluorophore molecules aggregates. 13.5 µM Ru(bpy)32+ solution was, then, diluted to 0.14 µM, 0.4 µM, 0.7 µM, 1 µM, 1.4 µM, corresponding to 0.1 mg/mL, 0.25 mg/mL, 0.5 mg/mL, 0.75 mg/mL, 1 mg/mL, respectively. Finally, cells were incubated in modified medium for 24, 48, or 96 h, to perform the fluorophore uptake for labeling.
As control samples for all assays, we prepared both HDF and LoVo cell using the same medium without fluorophore, and keeping the same experimental conditions reported before.
2.3. Fluorescence and Confocal Microscopy
In fluorescence (FM) and confocal laser scanning microscopy (CLSM) experiments, 1x10
4 cells were seeded on micro coverglasses. Treated samples were exposed to 0.4, 0.7, or 1 µM solution of Ru(bpy)
32+ in specific cell medium and analyzed after 24, 48, and 96 h, while control samples were growth in medium without fluorophore. Then, all samples were washed three times in PBS, in order to remove all of the fluorophore excess, and fixed using 4% paraformaldehyde/PBS. After washing, slides were dried, mounted with Fluor Gel (EMS) and observed at 488 nm wavelength with an Olympus BX50 fluorescence microscope equipped with a DC500 camera (Leica). Emission bandwidths of 515–580 nm (for intrinsic fluorescence) and above 590 nm (long passband filter for Ru(bpy)
32+) were monitored. The images of the same samples were acquired with a CLSM (Zeiss LSM700) and analyzed using the ZEN 2011 software [
23]. The fluorescence signal intensity of the cells in the serial sections were evaluated using ImageJ software (NIH, Bethesda, MD; available at
http://rsb.info.nih.gov/ij/index.html).
2.4. Optical Measurement for Bio-Sensing Applications
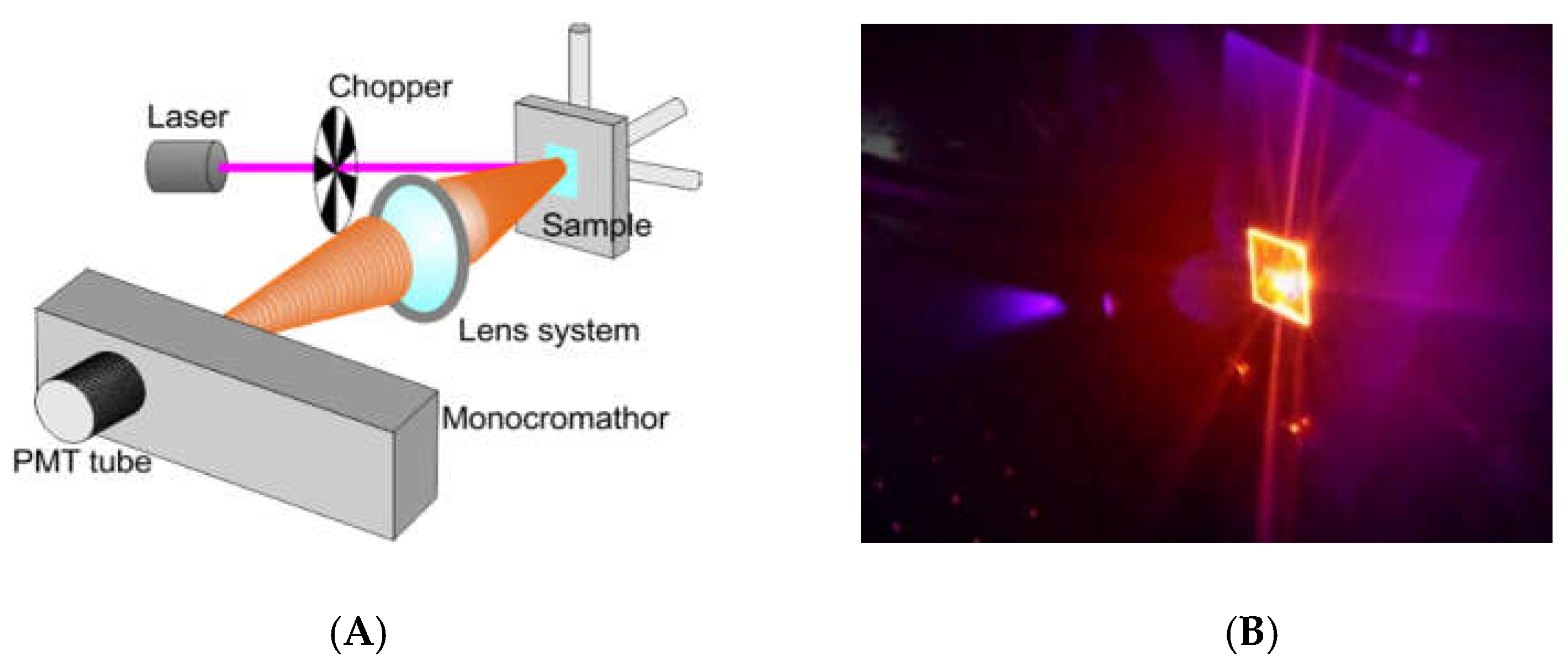
We tested the optical behavior of Ru(bpy)
32+ based labeling with a non-conventional system for bio-analysis, reported in
Figure 2A. We performed a fluorescence emission analysis of HDFs treated with 0.4, 0.7, and 1 µM of fluorophore for 24, 48 and 96 h (see experimental section for details). The system used for analysis included: A laser source (Coherent) operating at 405 nm to a power of 50 mW. This laser wavelength fell within the Ru(bpy)
32+ excitation curve. A chopper; a monochromator; a PMT Hamamatsu R-908; a lock-in, and a series of lenses to collect the signal at the monochromator entrance slits, completing the detection system. Measurements were collected from 550 nm to 730 nm by using the monochromator to select the wavelength with an error of 0.1 nm. Cells were fixed and dried on a glass coverslip, blocked in a sample holder (
Figure 2B). A computerized system for instruments management and data acquisition completed the system. A laser spot of about 3 mm in diameter was used, to a surface covered of ~7 mm
2. The average number of cells measured was ~3 × 10
5. Each spectrum was collected and, using an off-line analysis, the peak maximum recorded.
2.5. MTT Assay on HDF and LoVo
We tested Ru(bpy)32+ effect on the cells vitality by MTT assay. HDF cells were exposed to fluorophore concentrations ranging from 0.14 µM to 14 µM for 24 h and incubated with 0.05 mg/mL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide salt. After 2.30 h of incubation at 37 °C, formazan salts, produced by succinate dehydrogenase activity in live cells, were solubilized with 0.1 N isopropanol/HCl and quantified spectrophotometrically by a Cary 50, Varian. The absorbance values were converted to optical density (Δ OD: λ570-λ650). All samples were measured in triplicate. LoVo cells were exposed to 0.14 µM of Ru(bpy)32+ for 24 h and MTT assays. The same procedure described above was performed 1, 2 or 5 days after exposure.
4. Discussion
We investigated a strategy aimed for the implementation of a miniaturized intracellular optical sensing for human cells. The approach is based on the use of a cell labeling process, performed through the fluorophore Tris(2,2’-bipyridyl)ruthenium(II), in combination with a miniaturizable detection system suitable for portable optical sensing applications.
Ru(bpy)32+ fluorophore for cell labeling brings a series of optimizations to both sample preparation and optical detection. Conventional dyes share some chemical-physical properties that are quite limiting towards a possible integration and miniaturization of the sensing system.
Literature report some evidences of photobleaching and phototoxicity processes for some dyes stressed by prolonged laser exposures, so that dark condition and low energy laser beam are needed. Other evidences highlight, also, the risk of self-quenching for most of the conventional dyes, due to their absorption and emission peaks short gap and overlap, which imply a complex architecture and electronic management of the luminescence detection system.
The use of Ru(bpy)
32+ for cell labeling allows overpassing all these limitations, thanks to its structural and optical properties. The fluorophore, in fact, is robust and able to keep its structural and functional integrity under very unsustainable chemical-physical conditions [
12], and exhibits long gap absorption/emission peaks, that avoid the risk of self-quenching, thus simplifying the detection technology. Moreover, the molecule is commercially available, thus reducing costs and time related to structural modifications, carriers functionalization and/or cell permeabilization.
The characterization of the sensing strategy, in terms of labeling efficiency and detection sensitivity, was performed on HDFs, representative of the human cell model. FM and CLSM revealed that Ru(bpy)
32+ passed through the HDFs membrane to reach the intracellular environment with a cytosolic localization. The specific uptake mechanism was not characterized, since there are some studies in the literature [
17]. The optical characterization of labeled HDFs by the unconventional photoluminescence system (
Figure 2) proved the suitability of our method for the integration in an optical biosensor device; the use of miniaturized photomultipliers, already employed in sensing applications [
25,
26,
27] may result in the fabrication of a portable sensing system that could be used also within incubators. Results from optical testing reported a sensitive Ru(bpy)
32+ fluorescence detection, highlighting a strong correlation with both fluorophore concentration and labeling duration. Finally, cell vitality was not affected by the labeling technique. In fact, MTT assays revealed that HDF cells treated with different fluorophore concentrations (0.14 µM to 1.4 µM) kept their vitality, if compared to controls.
Once characterized and validated, we investigated the versatility of this strategy studying other cell models such as the human colon adenocarcinoma cells, LoVo. We performed the uptake of Ru(bpy)
32+, as reported in
Figure 8, and tested the intracellular fluorescence of LoVo cells and their vitality at 1, 2, and 5 days after uptake, emulating a fluorescent time-lapse analysis of tumor cell proliferation. Results reported in
Figure 9 proved that the labeling method did not affect LoVo viability, since there were no divergences between control and treated samples, and the fluorescence signal was perfectly detectable across time and cell generations, thanks to the robustness of Ru(bpy)
32+. This result led us to infer the possible application of this sensing strategy in oncogenesis analysis, where a combination of the labeling and detection properties with an appropriate imaging software would enable one to distinguish between healthy and tumor cells in a population, following the speed of growth and the spatial localization.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









