Membrane-Assisted Crystallization: A Molecular View of NaCl Nucleation and Growth


 ,
,  ,
,  ,
,  ,
, 

Abstract
Featured Application
Abstract
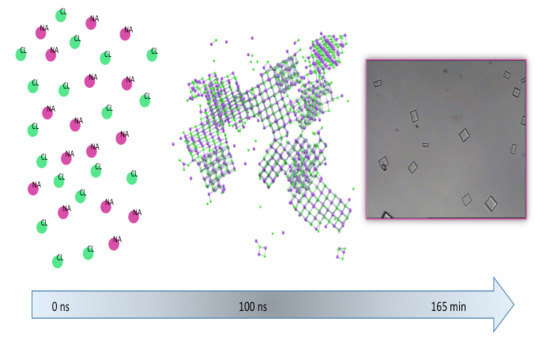
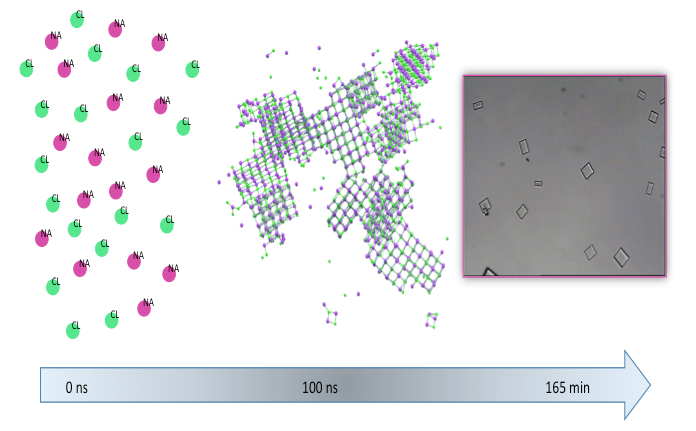
1. Introduction
2. Materials and Methods
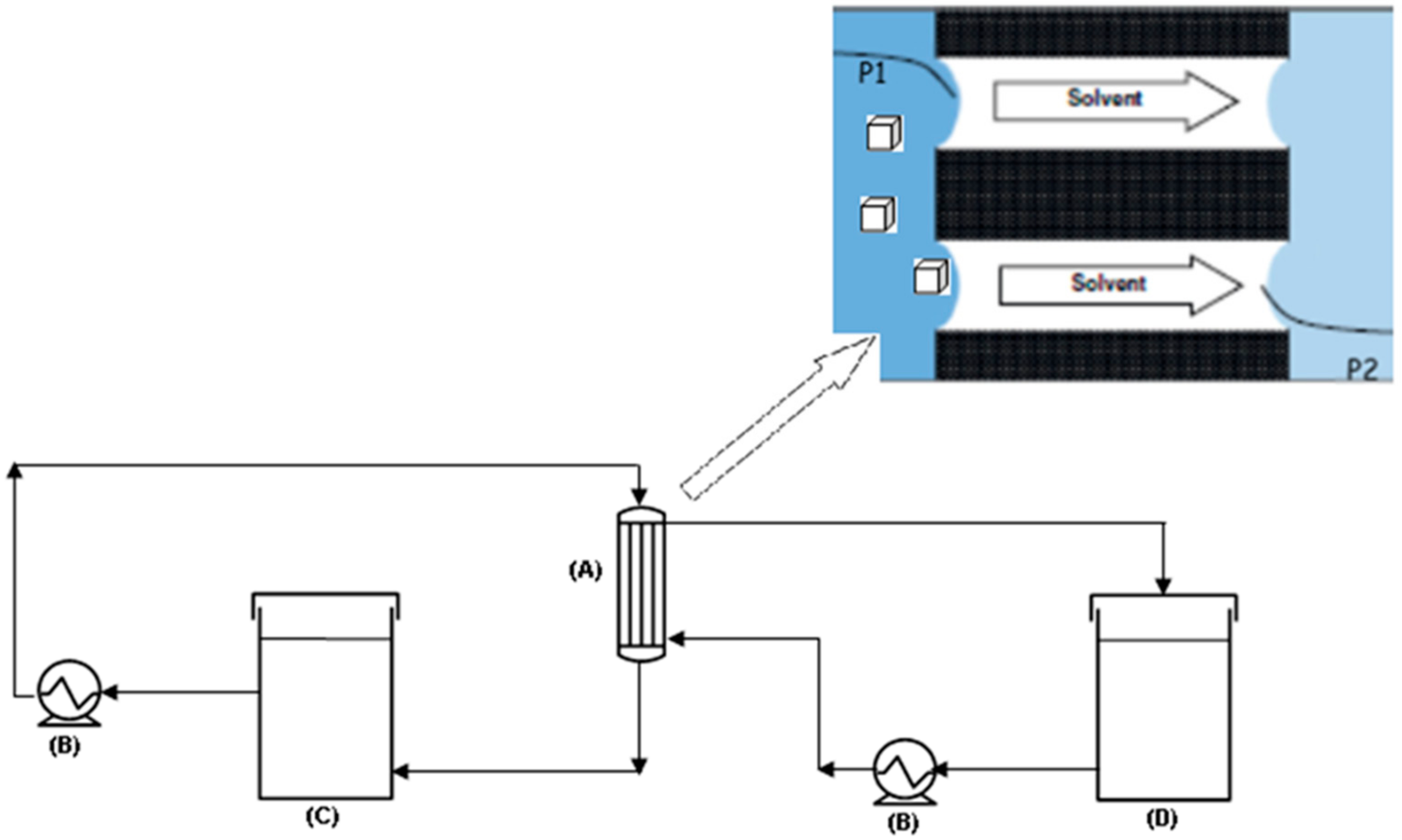
2.1. Experiments
2.2. Modelling
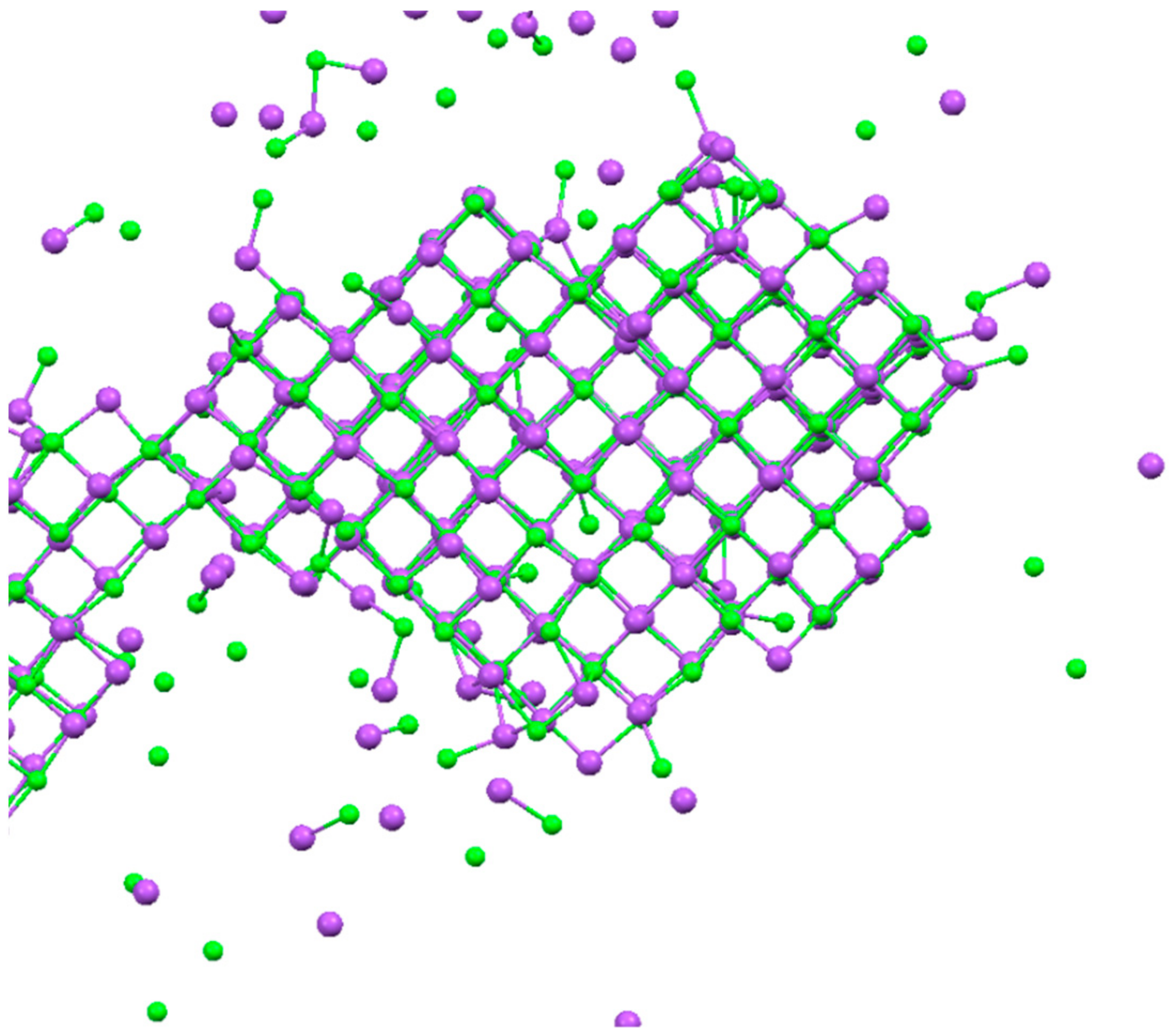
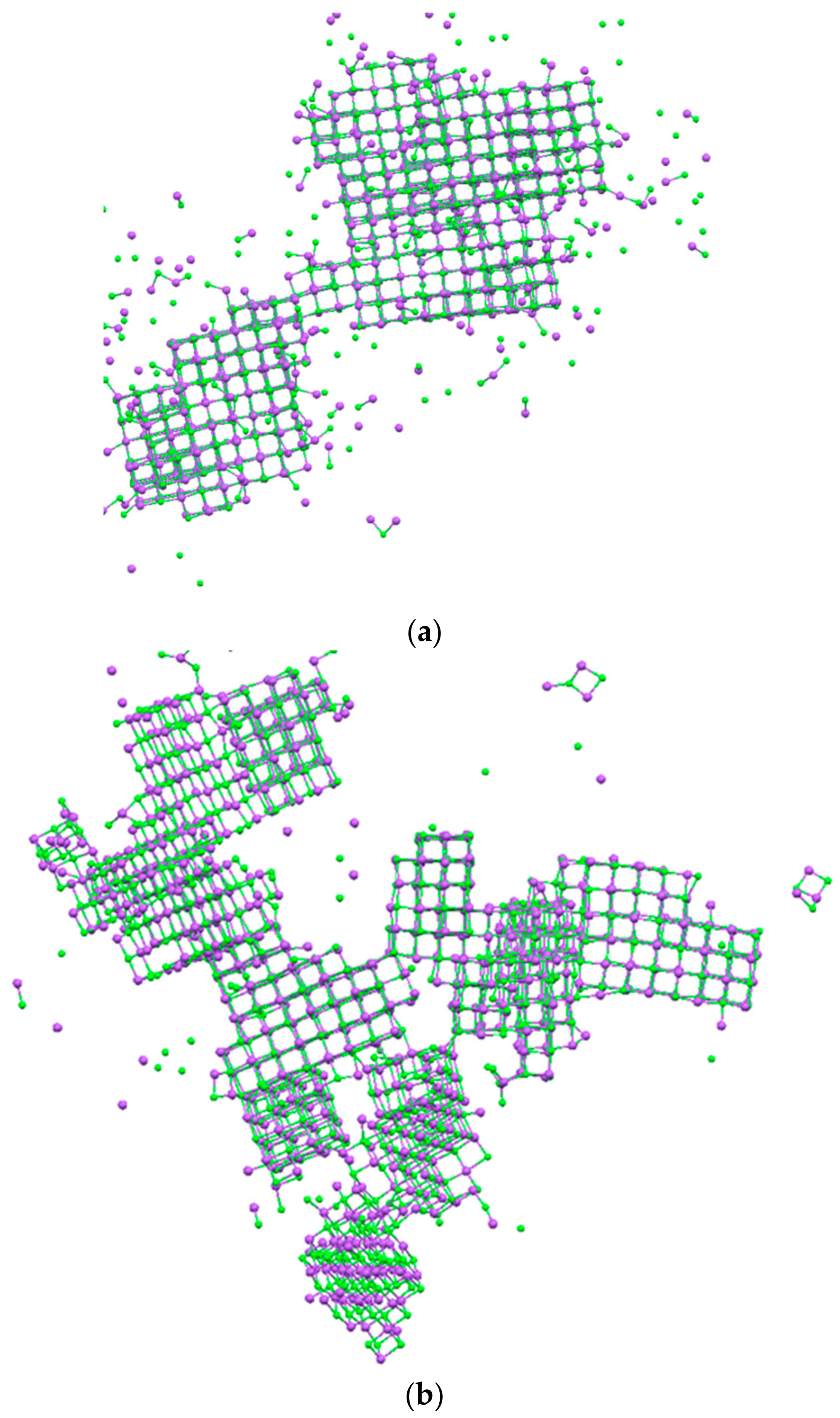
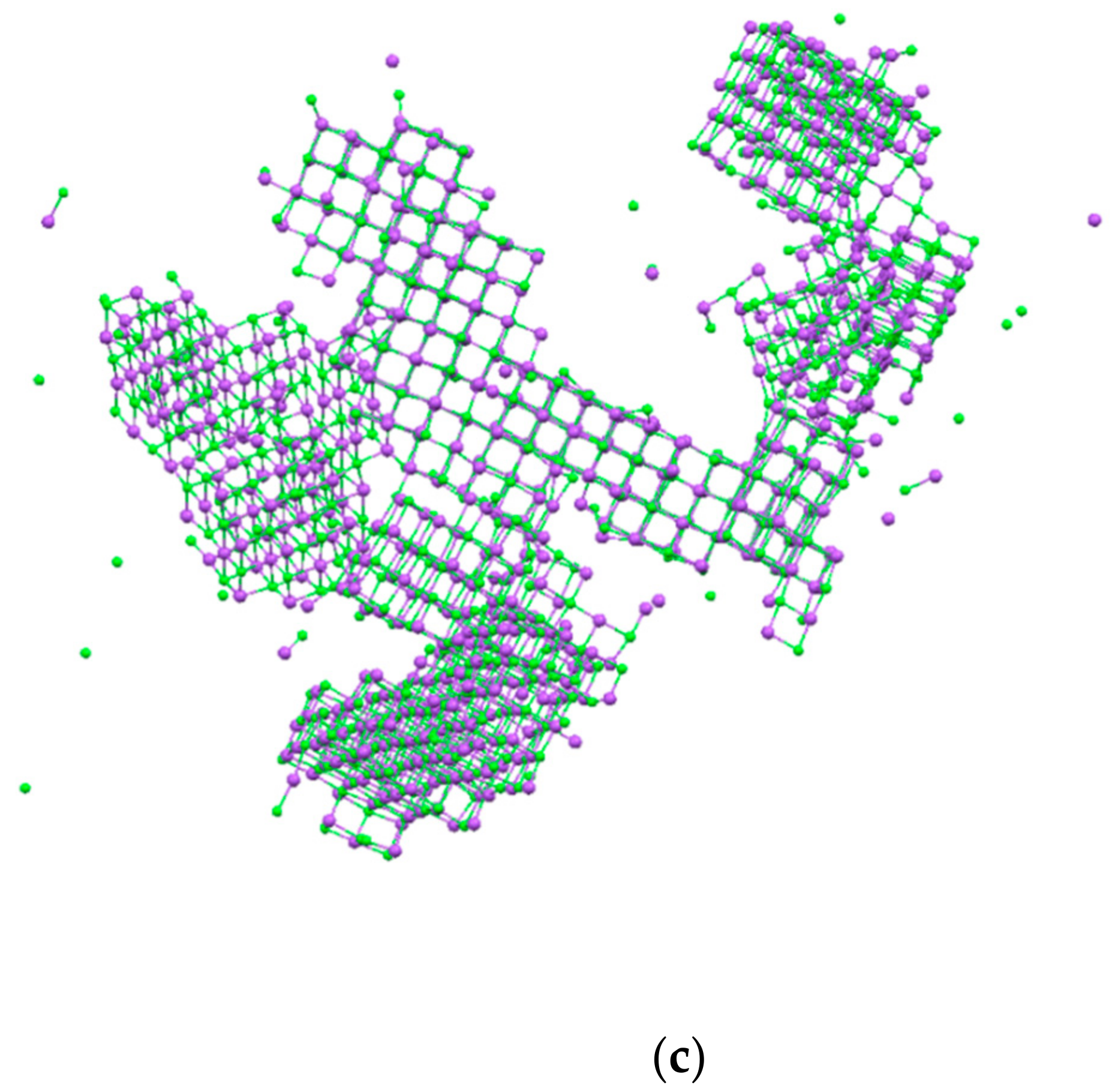
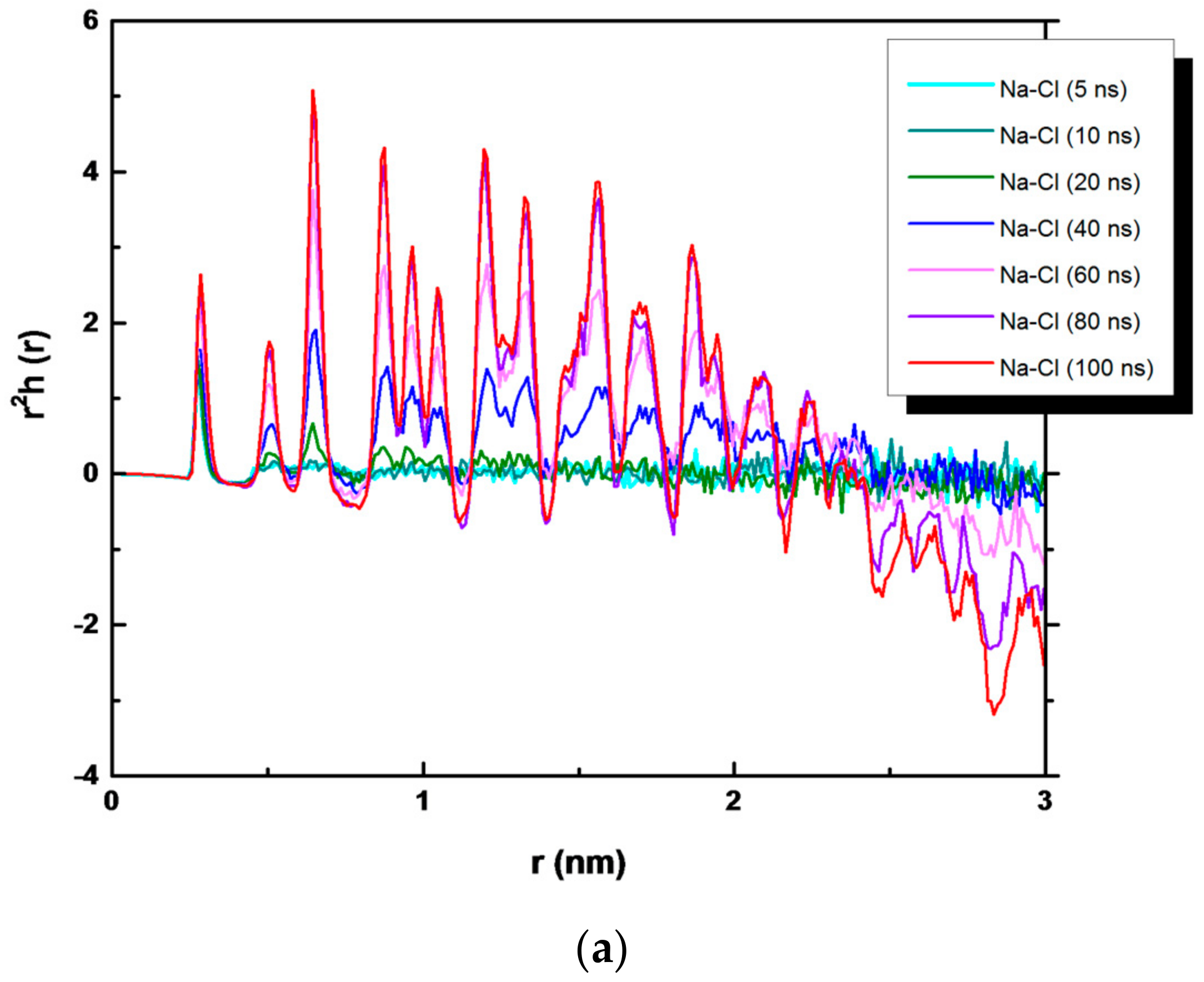
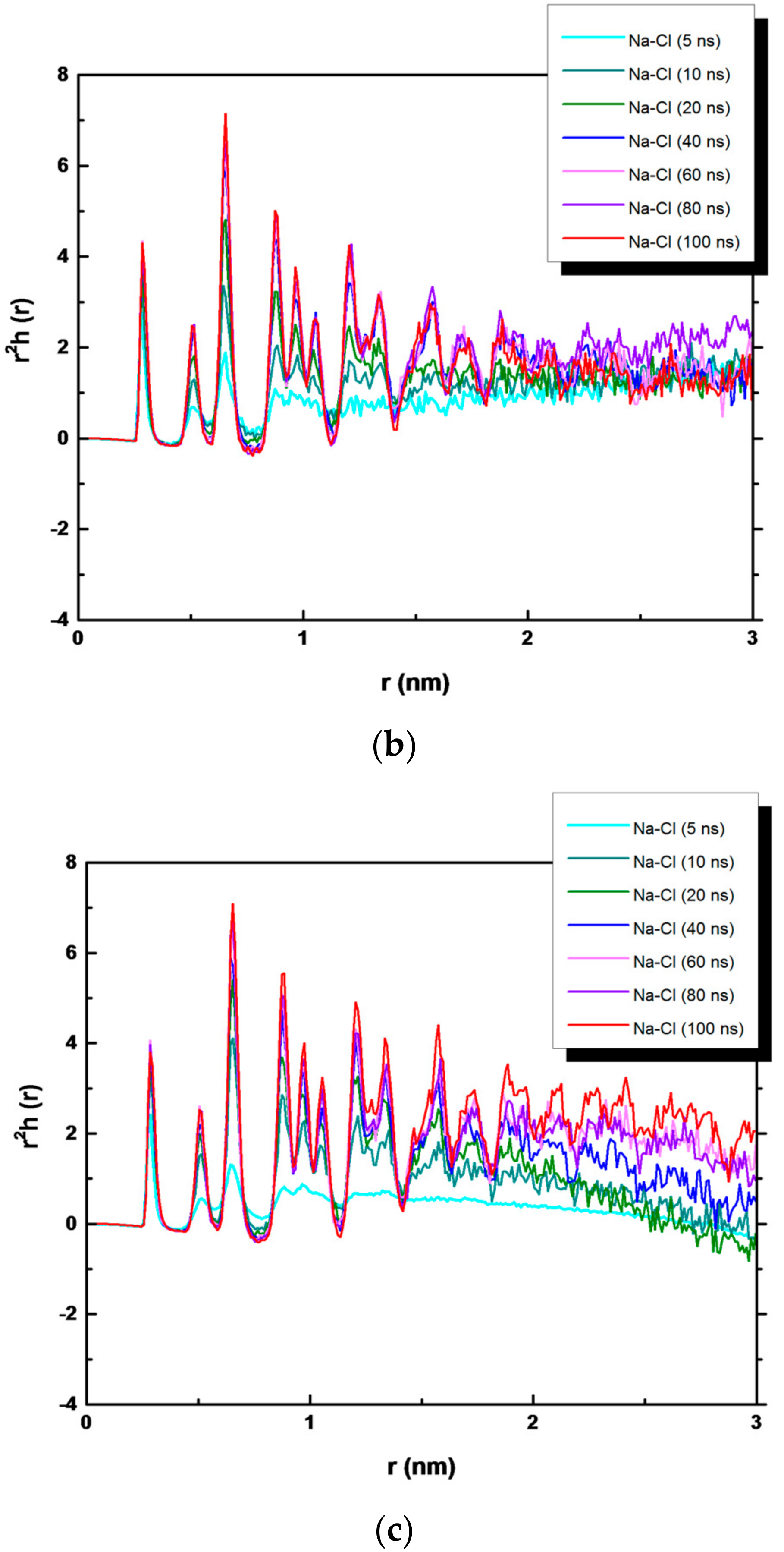
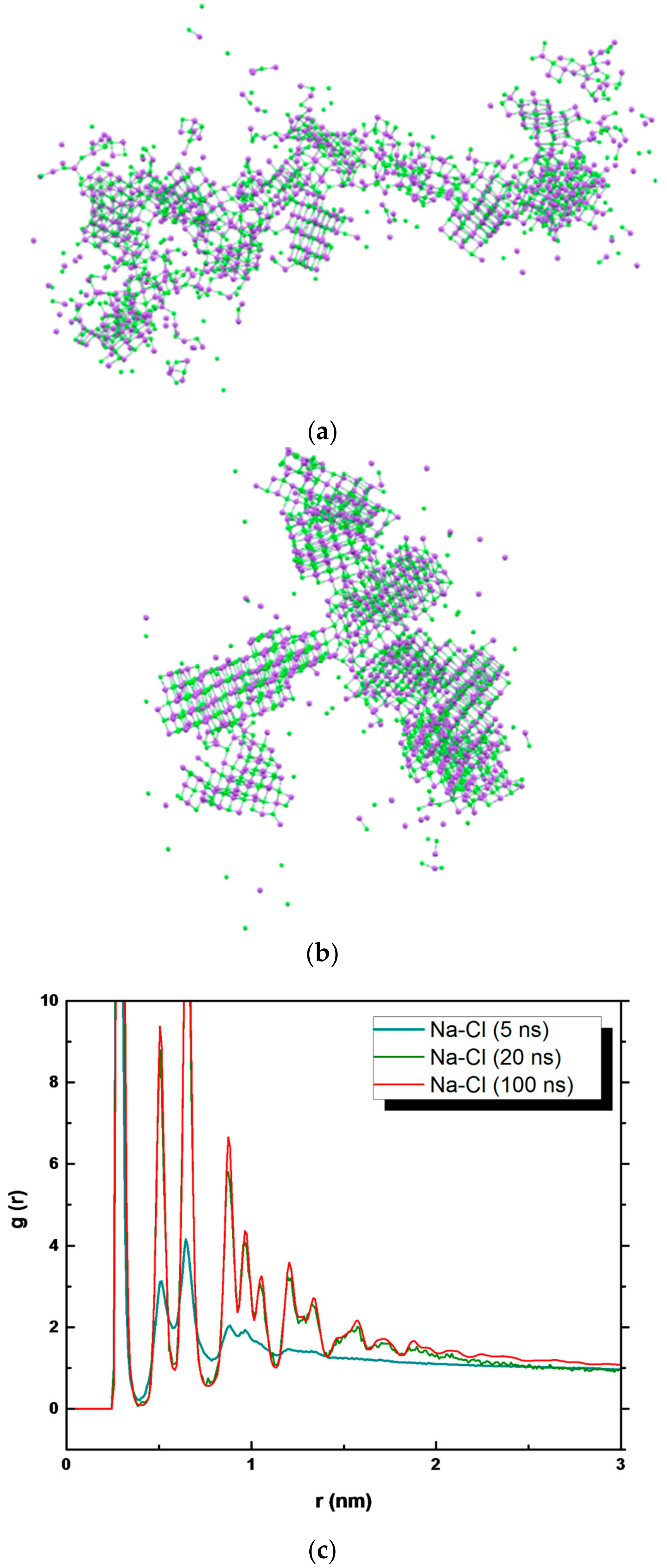
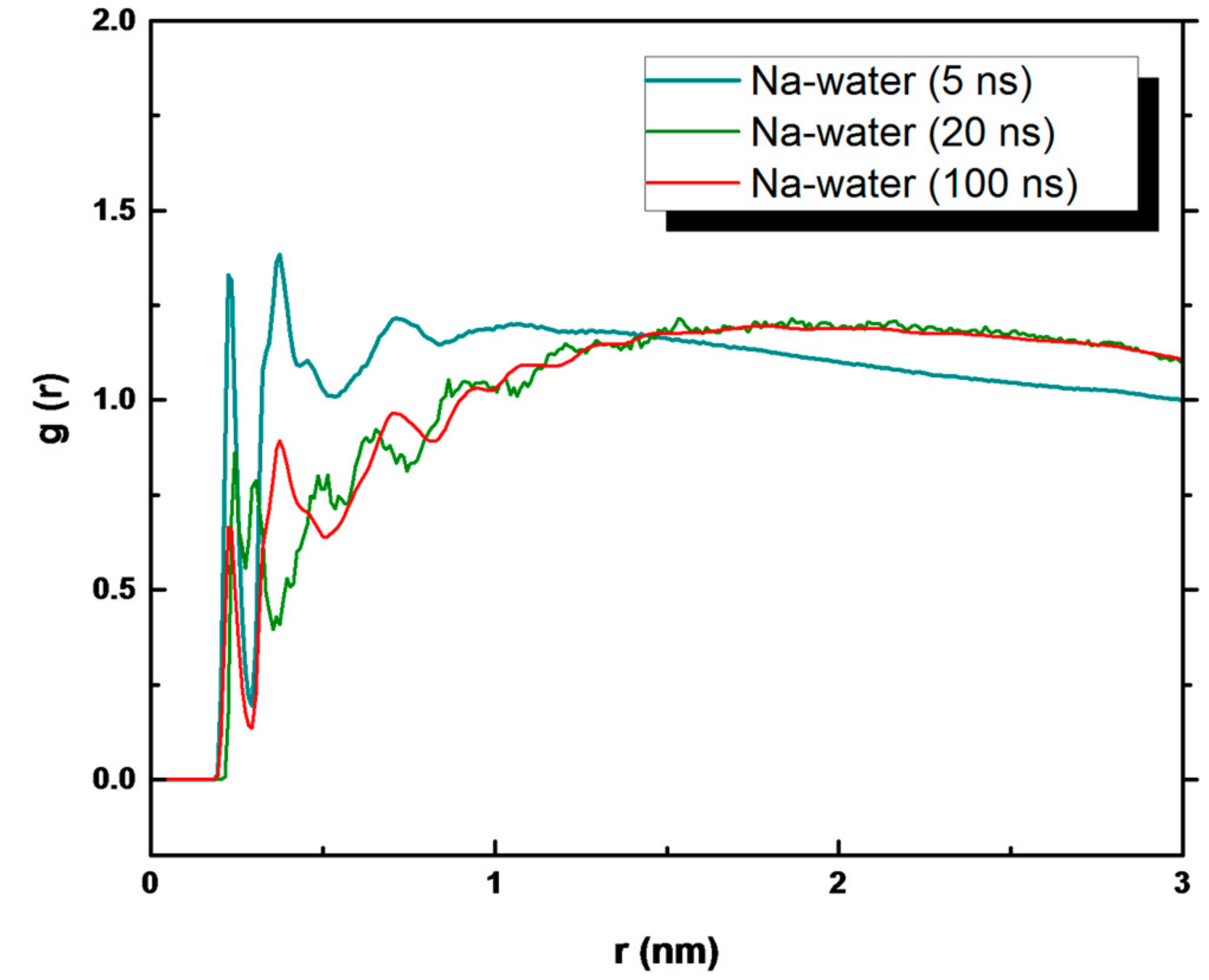
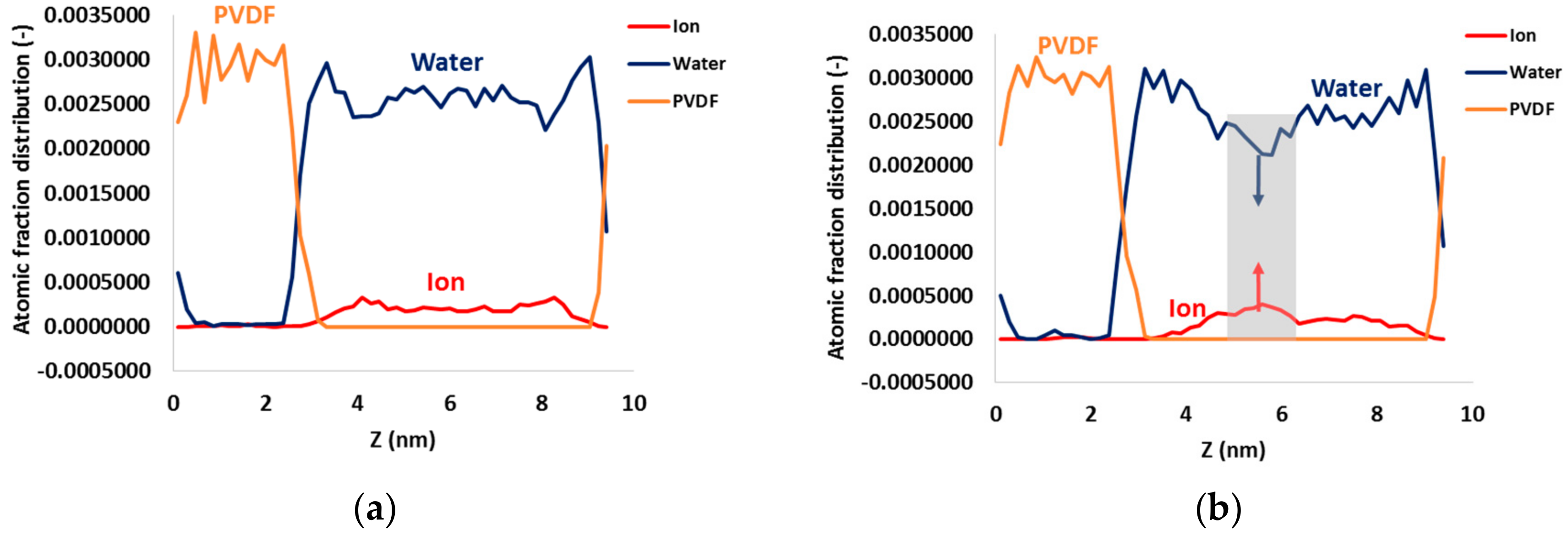
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Curcio, E.; Criscuoli, A.; Drioli, E. Membrane crystallizers. Ind. Eng. Chem. Res. 2001, 40, 2679–2684. [Google Scholar] [CrossRef]
- Drioli, E.; Criscuoli, A.; Curcio, E. Membrane Contactors: Fundamentals, Applications and Potentialities; Membrane Science and Technology Series, 11; Elsevier: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Macedonio, F.; Drioli, E. Editorial–Special issue of desalination journal on membrane engineering for desalination in the mining and extraction industry. Desalination 2018, 440, 1. [Google Scholar] [CrossRef]
- Quist-Jensen, C.A.; Macedonio, F.; Drioli, E. Integrated membrane desalination systems with membrane crystallization units for resource recovery: A new approach for mining from the sea. Crystals 2016, 6, 36. [Google Scholar] [CrossRef]
- Chabanon, E.; Mangin, D.; Charcosset, C. Membranes and crystallization processes: State of the art and prospects. J. Membr. Sci. 2016, 509, 57–67. [Google Scholar] [CrossRef]
- Curcio, E.; Fontananova, E.; Di Profio, G.; Drioli, E. Influence of the structural properties of poly(vinylidene fluoride) membranes on the heterogeneous nucleation rate of protein crystals. J. Phys. Chem. B 2006, 110, 12438–12445. [Google Scholar] [CrossRef] [PubMed]
- Di Profio, G.; Curcio, E.; Drioli, E. Membrane Crystallization Technology, Chapter 4.02, Comprehensive Membrane Science and Engineering, 1st ed.; Drioli, E., Giorno, L., Fontananova, E., Eds.; Elsevier: Kidlington, UK, 2010. [Google Scholar]
- Ji, X.; Curcio, E.; Al Obaidani, S.; Di Profio, G.; Fontananova, E.; Drioli, E. Membrane distillation-crystallization of seawater reverse osmosis brines. Sep. Purif. Technol. 2010, 71, 76–82. [Google Scholar] [CrossRef]
- Di Profio, G.; Curcio, E.; Cassetta, A.; Lamba, D.; Drioli, E. Membrane crystallization of lysozyme: Kinetic aspects. J. Cryst. Growth 2003, 257, 359–369. [Google Scholar] [CrossRef]
- Gugliuzza, A.; Fabiani, R.; Garavaglia, M.G.; Spisso, A.; Drioli, E. Study of the surface character as responsible for controlling interfacial forces at membrane-feed interface. J. Colloid Interface Sci. 2006, 303, 388–403. [Google Scholar] [CrossRef] [PubMed]
- Gugliuzza, A.; Aceto, M.C.; Drioli, E. Interactive functional poly(vinylidene fluoride) membranes with modulated lysozyme affinity: A promising class of new interfaces for contactor crystallizers. Polym. Int. 2009, 58, 1452–1464. [Google Scholar] [CrossRef]
- Vekilov, P.G. The two-step mechanism of nucleation of crystals in solution. Nanoscale 2010, 2, 2346–2357. [Google Scholar] [CrossRef] [PubMed]
- Anwar, J.; Zahn, D. Uncovering molecular processes in crystal nucleation and growth by using molecular simulation. Angew. Chem. Int. Ed. 2011, 50, 1996–2013. [Google Scholar] [CrossRef] [PubMed]
- Sleutel, M.; Lutsko, J.; Van Driessche, A.E.S.; Duran-Olivencia, M.A.; Maes, D. Observing classical nucleation theory at work by monitoring phase transitions with molecular precision. Nat. Commun. 2014, 5, 5598–5606. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Kimura, Y.; Vekilov, P.G.; Furukawa, E.; Shirai, M.; Matsumoto, H.; Van Driessche, A.E.S.; Tsukamoto, K. Two types of amorphous protein particles facilitate crystal nucleation. Proc. Natl. Acad. Sci. USA 2017, 114, 2154–2159. [Google Scholar] [CrossRef] [PubMed]
- Van Driessche, A.E.S.; Van Gerven, N.; Bomans, P.H.H.; Joosten, R.R.M.; Friedrich, H.; Gil-Carton, D.; Sommerdijk, N.A.J.M.; Sleutel, M. Molecular nucleation mechanisms and control strategies for crystal polymorph selection. Nature 2018, 556, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Shin, S.-H.; Bertozzi, C.R.; De Yoreo, J.J. Self-catalyzed growth of S layers via an amorphous-to-crystalline transition limited by folding kinetics. Proc. Natl. Acad. Sci. USA 2010, 107, 16536–16541. [Google Scholar] [CrossRef] [PubMed]
- Parent, L.R.; Bakalis, E.; Ramírez-Hernández, A.; Kammeyer, J.K.; Park, C.; De Pablo, J.; Zerbetto, F.; Patterson, J.P.; Gianneschi, N.C. Directly observing micelle fusion and growth in solution by liquid-cell transmission electron microscopy. J. Am. Chem. Soc. 2017, 139, 17140–17151. [Google Scholar] [CrossRef] [PubMed]
- Alberstein, R.G.; Tezcan, F.A. Observations of the birth of crystals. Nature 2018, 556, 41–42. [Google Scholar] [CrossRef] [PubMed]
- Demichelis, R.; Schuitemaker, A.; Garcia, N.A.; Koziara, K.B.; De La Pierre, M.; Raiteri, P.; Gale, J.D. Simulation of crystallization of biominerals. Annu. Rev. Mater. Res. 2018, 48, 327–352. [Google Scholar] [CrossRef]
- Whitelam, S. Control of pathways and yields of protein crystallization through the interplay of nonspecific and specific attractions. Phys. Rev. Lett. 2010, 105, 088102. [Google Scholar] [CrossRef] [PubMed]
- Sosso, G.C.; Chen, J.; Cox, S.J.; Fitzner, M.; Pedevilla, P.; Zen, A.; Michaelides, A. Crystal nucleation in liquids: Open questions and future challenges in molecular dynamics simulations. Chem. Rev. 2016, 116, 7078–7116. [Google Scholar] [CrossRef] [PubMed]
- Toxvaerd, S. Molecular dynamics simulation of heterogeneous nucleation at a structureless solid surface. J. Chem. Phys. 2002, 117, 10303–10310. [Google Scholar] [CrossRef]
- Cacciuto, A.; Auer, S.; Frenkel, D. Onset of heterogeneous crystal nucleation in colloidal suspensions. Nature 2004, 428, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Meng, S. Atomistic nature of NaCl nucleation at the solid-liquid interface. J. Chem. Phys. 2007, 126, 044708. [Google Scholar] [CrossRef] [PubMed]
- Webb, E.B.; Grest, G.S.; Heine, D.R. Precursor film controlled wetting of Pb on Cu. Phys. Rev. Lett. 2003, 91, 236102. [Google Scholar] [CrossRef] [PubMed]
- Gránásy, L.; Pusztai, T.; Saylor, D.; Warren, J.A. Phase field theory of heterogeneous crystal nucleation. Phys. Rev. Lett. 2007, 98, 035703. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S.; Shimosaka, A.; Shirakawa, Y.; Hidaka, J. Molecular dynamics simulations of the formation for NaCl cluster at the interface between the supersaturated solution and the substrate. J. Nanopart. Res. 2010, 12, 831–839. [Google Scholar] [CrossRef]
- Dashtian, H.; Wang, H.; Sahimi, M. Nucleation of salt crystals in clay minerals: Molecular dynamics simulation. J. Phys. Chem. Lett. 2017, 8, 3166–3172. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Patey, G.N. How crystals nucleate and grow in aqueous NaCl solution. J. Phys. Chem. Lett. 2013, 4, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Lanaro, G.; Patey, G.N. Molecular dynamics simulation of NaCl dissolution. J. Phys. Chem. B 2015, 119, 4275–4283. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Liang, Y.; Sakka, T.; Matsuoka, T. Molecular dynamics study of salt–solution interface: Solubility and surface charge of salt in water. J. Chem. Phys. 2014, 140, 144705. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Patey, G.N. Evidence that crystal nucleation in aqueous NaCl solution occurs by the two-step mechanism. Chem. Phys. Lett. 2013, 587, 25–29. [Google Scholar] [CrossRef]
- Macedonio, F.; Drioli, E. Hydrophobic membranes for salts recovery from desalination plants. Desalin. Water Treat. 2010, 18, 224–234. [Google Scholar] [CrossRef]
- Materials Studio 7.0; Dassault Systèmes BIOVIA: San Diego, CA, USA, 2013.
- Sun, H. COMPASS: An ab initio force-field optimized for condensed-phase applications overview with details on alkane and benzene compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Theodorou, D.N.; Suter, U.W. Detailed molecular structure of a vinyl polymer glass. Macromolecules 1985, 18, 1467–1478. [Google Scholar] [CrossRef]
- Meirovitch, H. Computer simulation of self-avoiding walks: Testing the scanning method. J. Chem. Phys. 1983, 79, 502–508. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Byutner, O.G.; Smith, G.D. Quantum chemistry based force field for simulations of poly(vinylidene fluoride). Macromolecules 2000, 33, 4264–4270. [Google Scholar] [CrossRef]
- Erdtman, E.; Satyanarayana, K.C.; Bolton, K. Simulation of α- and β-PVDF melting mechanisms. Polymer 2012, 53, 2919–2926. [Google Scholar] [CrossRef]
- Berendsen, H.; Grigera, J.; Straatsma, T. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Stephen, H.; Stephen, T.; Silcock, H.L. Solubilities of Inorganic and Organic Compounds; Stephen, H., Stephen Macmillan, T., Eds.; A Pergamon Press: New York, NY, USA, 1963. [Google Scholar]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. JACS 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press: New York, NY, USA, 1987; Volume 1989, ISBN 9780198556459. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Aqvist, J. Ion-water interaction potentials derived from free energy perturbation simulations. J. Phys. Chem. 1990, 94, 8021–8024. [Google Scholar] [CrossRef]
- Chandrasekhar, J.; Spellmeyer, D.C.; Jorgensen, W.L. Energy component analysis for dilute aqueous solutions of lithium (1+), sodium (1+), fluoride (1−), and chloride (1−) ions. JACS 1984, 106, 903–910. [Google Scholar] [CrossRef]
- Lachet, V.; Teuler, J.M.; Rousseau, B. Classical force field for hydrofluorocarbon molecular simulations. Application to the study of gas solubility in poly(vinylidene fluoride). J. Phys. Chem. A 2015, 119, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.; Verma, P.K.; Cranswick, L.M.D.; Jones, R.L.; Clark, S.M.; Buhre, S. Halite-sylvite thermoelasticity. Am. Mineral. 2004, 89, 204–210. [Google Scholar] [CrossRef]
- Zhao, J.; Miao, H.; Duan, L.; Kang, Q.; He, L. The mass transfer process and the growth rate of NaCl crystal growth by evaporation based on temporal phase evaluation. Opt. Lasers Eng. 2012, 50, 540–546. [Google Scholar] [CrossRef]
- An, S.; Li, J.; Li, Y.; Li, S.; Wang, Q.; Liu, B. Two-step crystal growth mechanism during crystallization of an undercooled Ni50Al50 alloy. Sci. Rep. 2016, 6, 31062. [Google Scholar] [CrossRef] [PubMed]
- Lupi, L.; Peters, B.; Molinero, V. Pre-ordering of interfacial water in the pathway of heterogeneous ice nucleation does not lead to a two-step crystallization mechanism. J. Chem. Phys. 2016, 145, 211910. [Google Scholar] [CrossRef] [PubMed]
- Vekilov, P.G. Two-step mechanism for the nucleation of crystals from solution. J. Cryst. Growth 2005, 275, 65–76. [Google Scholar] [CrossRef]
- Ten Wolde, P.R.; Frenkel, D. Homogeneous nucleation and the ostwald step rule. Phys. Chem. Chem. Phys. 1999, 1, 2191–2196. [Google Scholar] [CrossRef]
- Sauter, A.; Roosen-Runge, F.; Zhang, F.; Lotze, G.; Feoktystov, A.; Jacobs, R.M.J.; Schreiber, F. On the question of two-step nucleation in protein crystallization. Faraday Discuss. 2015, 179, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Ten Wolde, P.R.; Ruiz-Montero, M.J.; Frenkel, D. Numerical evidence for BCC ordering at the surface of a critical FCC nucleus. Phys. Rev. Lett. 1995, 75, 2714–2717. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Li, X.; Zhang, Y.; Wang, Z.; Gugliuzza, A.; Militano, F.; Drioli, E.; Macedonio, F. Testing of three different PVDF membranes in membrane assisted-crystallization process: Influence of membrane structural-properties on process performance. Desalination 2018, 440, 68–77. [Google Scholar] [CrossRef]
- Wang, J.; Sun, Z.; Lu, G.; Yu, J. Molecular dynamics simulations of the local structures and transport coefficients of molten alkali chlorides. J. Phys. Chem. B 2014, 118, 10196–10206. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, I.F.; Bunker, B.A.; Cygan, R.T. Molecular dynamics modeling of ion adsorption to the basal surfaces of kaolinite. J. Phys. Chem. C 2007, 111, 6753–6762. [Google Scholar] [CrossRef]
- Rowley, C.N.; Roux, B. The solvation structure of Na+ and K+ in liquid water determined from high level ab initio molecular dynamics simulations. J. Chem. Theory Comput. 2012, 8, 3526–3535. [Google Scholar] [CrossRef] [PubMed]
- Skipper, N.T.; Neilson, G.W. X-ray and neutron diffraction studies on concentrated aqueous solutions of sodium nitrate and silver nitrate. J. Phys. Condens. Matter 1989, 1, 4141–4154. [Google Scholar] [CrossRef]
- Mähler, J.; Persson, I. A study of the hydration of the alkali metal ions in aqueous solution. Inorg. Chem. 2012, 51, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Macedonio, F.; Politano, A.; Drioli, E.; Gugliuzza, A. Bi2Se3-assisted membrane crystallization. Mater. Horiz. 2018, 5, 912–919. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fiber Type | Outer Diameter (mm) | Inner Diameter (mm) | Thickness (mm) | Pore Size (μm) | Porosity (%) | Contact Angle (°) |
|---|---|---|---|---|---|---|
| PVDF | 1.75 | 0.94 | 0.40 | 0.47 | 80.8 | 91 |
| PP | 1.80 | 1.56 | 0.12 | 0.20 | 70.0 | 115 |
| Bonds | (nm) | (kJ/mol nm2) | ||||
|---|---|---|---|---|---|---|
| CH–H | 0.1085 | 274,135.68 | ||||
| CF–F | 0.1357 | 417,814.24 | ||||
| CF–CH | 0.1534 | 258,487.52 | ||||
| Angles | (deg) | (kJ/mol rad2) | ||||
| F–CF–F | 105.27 | 1004.1600 | ||||
| F–CF–CH | 107.74 | 753.1200 | ||||
| CH–CF–CH | 118.24 | 671.9504 | ||||
| H–CH–H | 109.27 | 322.1680 | ||||
| H–CH–CF | 108.45 | 358.9872 | ||||
| CF–CH–CF | 118.24 | 671.9504 | ||||
| Torsions (kJ/mol) | ||||||
| CF–CH–CF–CH | 1.65268 | 3.01248 | −1.58992 | −0.85772 | 1.77820 | −0.10460 |
| F–CF–CH–CF | 1.48532 | 1.44348 | −1.58992 | 0.58576 | 0.60668 | −0.10460 |
| Atom Type | σ (Å) | ε (kJ/mol) | Charge (e) |
|---|---|---|---|
| Ow | 3.166 | 0.6501 | −0.847600 |
| Hw | 0.0 | 0.0 | +0.423800 |
| Na+ | 3.330 | 0.0116 | +1 |
| Cl− | 4.417 | 0.4928 | −1 |
| CPVDF, H | 3.500 | 0.2763 | −0.650250 |
| CPVDF, F | 3.500 | 0.2763 | +0.765000 |
| HPVDF | 2.500 | 0.1256 | +0.225875 |
| FPVDF | 2.983 | 0.2512 | −0.283250 |
| PVDF | PP | |
|---|---|---|
| Minimum time for detecting and recovering crystals [min] | 360 | from 165 to 283 a) |
| Crystals growth rate [mm/min] a) | from 6.5 × 10−5 to 2.2 × 10−4 | from 2.5 × 10−5 to 5.7 × 10−5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, J.-H.; Perrotta, M.L.; Gugliuzza, A.; Macedonio, F.; Giorno, L.; Drioli, E.; Tung, K.-L.; Tocci, E. Membrane-Assisted Crystallization: A Molecular View of NaCl Nucleation and Growth. Appl. Sci. 2018, 8, 2145. https://doi.org/10.3390/app8112145
Tsai J-H, Perrotta ML, Gugliuzza A, Macedonio F, Giorno L, Drioli E, Tung K-L, Tocci E. Membrane-Assisted Crystallization: A Molecular View of NaCl Nucleation and Growth. Applied Sciences. 2018; 8(11):2145. https://doi.org/10.3390/app8112145
Chicago/Turabian StyleTsai, Jheng-Han, Maria Luisa Perrotta, Annarosa Gugliuzza, Francesca Macedonio, Lidietta Giorno, Enrico Drioli, Kuo-Lun Tung, and Elena Tocci. 2018. "Membrane-Assisted Crystallization: A Molecular View of NaCl Nucleation and Growth" Applied Sciences 8, no. 11: 2145. https://doi.org/10.3390/app8112145
APA StyleTsai, J.-H., Perrotta, M. L., Gugliuzza, A., Macedonio, F., Giorno, L., Drioli, E., Tung, K.-L., & Tocci, E. (2018). Membrane-Assisted Crystallization: A Molecular View of NaCl Nucleation and Growth. Applied Sciences, 8(11), 2145. https://doi.org/10.3390/app8112145