3.1. Optimization of Chemical Conditions for EPD and Co-Crosslinking
As pointed out in
Section 1, only charged particles can migrate under an electrical field in EPD and then be deposited onto the target electrode. Glucose oxidase (GOD) and bovine serum albumin (BSA) have isoelectric points at pH 4.3 and 4.7, respectively [
10], so they are positively charged in an acidic solution while being negatively charged in neutral and alkaline solutions. Since low pH values significantly reduce the GOD activity and induce the denaturation of both proteins, EPD was performed in neutral solutions (i.e., a phosphate buffer at pH 7,
I 0.1 M), so both proteins were expected to be in their negatively charged forms and be attracted by the anode of EPD cell.
Crosslinking [
24,
25] is a conventional method for enzyme immobilization able to produce insoluble polymeric structures of enzyme molecules covalently linked by a crosslinking agent. By using, e.g., glutaraldehyde (GLU) as a bifunctional crosslinker, both aldehyde groups react with primary amino group residues of proteins, initially creating Schiff bases, followed by the formation of stable secondary amine linkages. The kinetics and the degree of crosslinking are usually controlled by the concentration of both glutaraldehyde and enzyme, reaction time, pH, and temperature: usually, higher concentrations of protein and crosslinker result in stronger crosslinking, which adversely affects the catalytic activity of the immobilized structure due to enzyme crowding. For this reason, co-crosslinking [
24,
25], i.e., inert protein-assisted enzyme crosslinking, was preferred; indeed, the use of an inert protein like BSA significantly reduces the crowding of GOD, resulting in a immobilized structure with sufficient catalytic activity. Typical co-crosslinking chemical conditions were already studied and optimized elsewhere, e.g., for GOD [
26], by varying enzyme, BSA, and GLU concentrations as well as buffer type and pH. Anyway, the aim of the present research was to drastically slow down the kinetics of the co-crosslinking reaction in the bath solution to avoid any gelation process in the cell while inducing co-crosslinking exclusively on the electrode surface during the EPD process. With this aim in mind, a carefully study showed that it is more effective to reduce the concentrations of both proteins than the concentration of the crosslinker agent; in fact, reducing the GLU concentration to 0.5 ÷ 1%
v/
v, to avoid any co-crosslinking in the solution, did not lead to any deposition, even if, for example, the protein concentration was higher than 10%
w/
v. Typically, a GOD concentration of 0.5%
w/
v, a BSA concentration of 1%
w/
v, and a GLU concentration of 2.5%
v/
v in phosphate buffer at pH 7,
I 0.1 M, guaranteed the absence of any gelation processes in the cell for as long as several hours but permitted in situ co-crosslinking of GOD and BSA under suitable EPD conditions (vide infra). It is worth noting that a buffer with high ionic strength was preferred due to its influence on the size of the diffusion layer (i.e., the electrical double layer) surrounding the protein molecules; in fact, as
Scheme S1 shows, higher ionic strengths, reducing the size of the diffusion layer, permit fewer retardation effects on the electrophoretic mobilities of proteins and constitute a proper approach for molecules in the co-crosslinking reaction.
3.2. Galvanostatic and Potentiostatic Deposition
In an initial series of experiments, the electrophoretic deposition and in situ co-crosslinking of GOD and BSA onto the electrode surface were investigated at a constant current, which was ensured using the galvanostatic technique. The chemical conditions were those already optimized above, while several constant current densities, in the range of 0.5 ÷ 3 mA/cm
2, were investigated for protein deposition. In this context,
Figure S1 shows the relevant chronopotentiometric curves observed in these experiments, while
Figure 2 shows the potential attained at the deposition electrode for each performed constant-current experiment.
In all cases, notable dioxygen evolution at the deposition electrode was evident, while a foamy coating was observed only at the higher applied current densities; unfortunately, in the latter conditions, the degree of dioxygen bubbling was so strong that it quickly removed any newly formed coating. Indeed, as reported in
Figure 2, for each applied current, the potential at the deposition electrode quickly reached values high enough, even at the lowest current densities, to promote water oxidation and dioxygen bubbling, ruining any protein deposition. These findings strictly correspond with similar side effects observed in metal electroplating [
21] and agree with those already pointed out elsewhere [
17]; it is worth noting that the use of the galvanostatic deposition technique has already been attempted for EPD deposition [
7,
8,
9,
10], but these side effects were unexpected and have never been reported.
Similar results were observed even for potentiostatic (i.e., constant potential) deposition experiments (not shown here), so the constant current and potential deposition approaches were abandoned.
3.3. Galvanodynamic Deposition
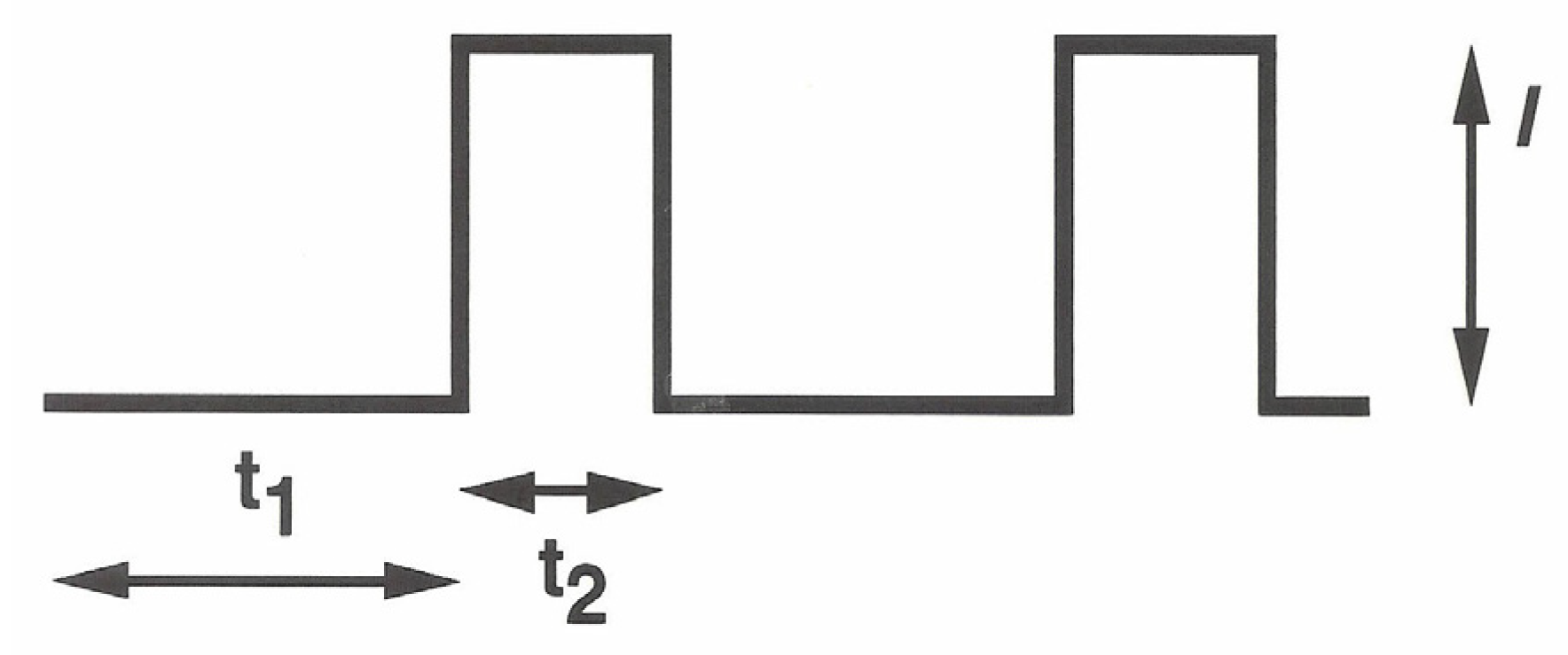
As the previous experiments show, EPD should be efficiently promoted by applying high current (or potential) values at the deposition electrode while minimizing the concurrent and undesired solvent discharge causing gas bubbling on top of it. Operating under the hypothesis that the kinetics of the two concurrent processes were almost different, a galvanodynamic electrophoretic deposition technique was investigated. In this approach, a pulsed current sequence (see
Scheme 1) was applied at the deposition electrode while varying the maximal current
I and the pulse duty cycle by varying
t1 and
t2.
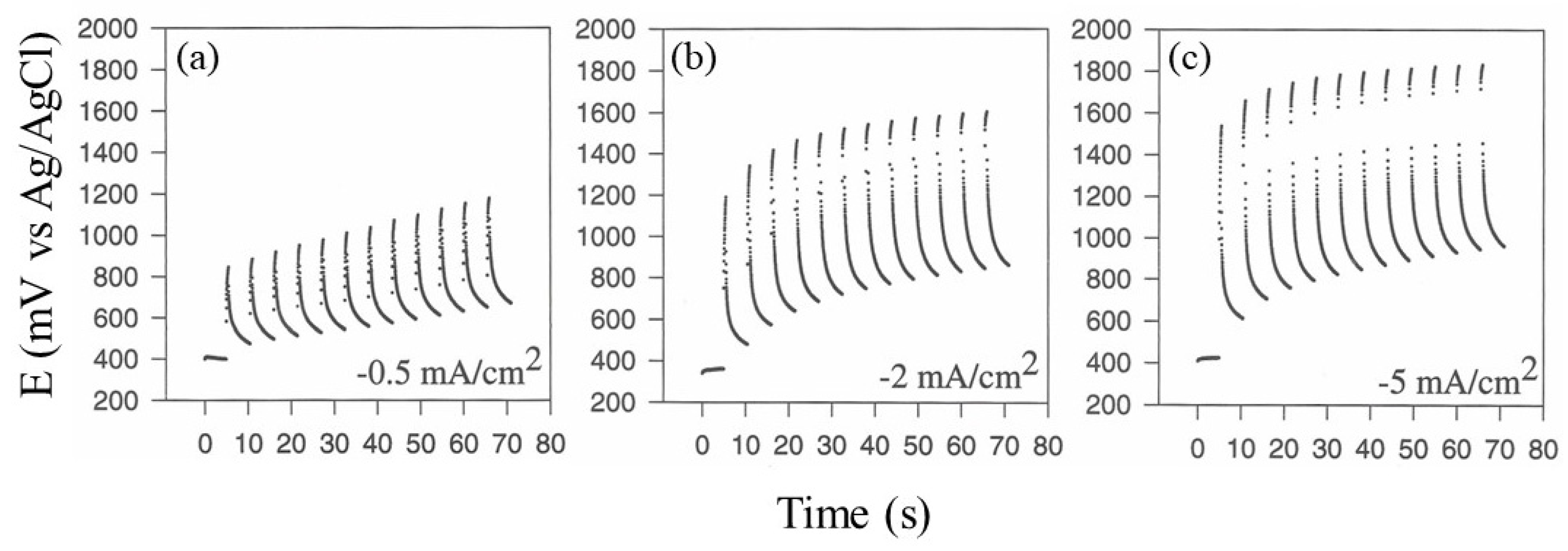
Figure 3 shows typical chronopotentiometric curves at the deposition electrode due to the application of current pulse sequences in a fixed duty cycle while varying
I, as described in the figure. As can be seen, whatever the value of
I, the expected potential increase was observed with the application of current pulse. Notably, a hysteresis effect was also evident, wherein the maximal potential reached by the deposition electrode increased with the increase in the time of the current pulse sequence application: due to the formation of platinum oxides on the electrode surface, this last effect was particularly revealing since it is well known that water discharge (and hence gas evolution at the electrode) is indeed mediated by the formation of metal oxides on the electrode surface. As evident from
Figure 3, increasing
I in the current pulse perturbation at the electrode promotes higher oxide formation on the surface of the electrode and hence dioxygen evolution. In particular, lower
I values ensured lower, if any, water discharge, but electrodeposition was practically absent: these experimental conditions evidently do not promote any significant electrophoretic movements of proteins and hence any in situ co-crosslinking, even when extending the experiment for a very long time. Oppositely, higher
I values promoted an evident and significant degree of protein deposition, but the water discharge was so intense that it destroyed any deposits. A
I value of 2 mA/cm
2 was an optimal current density value and a good compromise between efficient protein deposition and lower gas evolution at the electrode surface.
Regarding the duty cycle optimization of the current pulse sequence and its frequency,
Figure 4 shows typical chronopotentiometric curves at the deposition electrode due to the application of a current pulse sequence at a fixed maximal current
I (2 mA/cm
2) while varying
t1 and
t2, as described in the figure. As can be seen, an increase in the duty cycle of the pulse sequence, i.e., increasing
t2 while keeping
t1 +
t2 constant, was equivalent to an increase in
I, as observed in the previous experiments; in fact, the deposition electrode reached higher potential values (suggesting intense kinetics of platinum oxide formation on its surface), and dioxygen evolution was more evident, destroying any formation of the protein deposit. On the contrary, lower duty cycle values permitted the formation of a compact protein layer even if at a lower rate. Further, increasing the frequency of the current pulse sequence by lowering both
t1 and
t2 was not effective for protein deposition: in fact, due to the inherent inertia of the electrophoresis of big molecules like proteins, electrophoresis could be considered negligible at the higher frequency values. Accordingly, the application of a current pulse sequence at low frequency values and low duty cycles constitutes mandatory conditions for the optimal electrophoresis of both proteins and in situ co-crosslinking: a current pulse sequence with
t1 5 s and
t2 0.5 s yielded the best protein deposit at a current density of 2 mA/cm
2.
In these optimized conditions, after just a few seconds, an opalescent layer was observed on the electrode surface, without any evidence of gas evolution. After 1 min and more, a yellowish proteic layer was clearly visible; it did not have any gas bubbles entrapped on it and was quite resistant to being washed with water when the electrode was removed from the bath solution.
More insights into the growth of protein deposition and in situ co-crosslinking were gained via an electrochemical quartz crystal microbalance (EQCM) study. Here, a platinum-plated quartz disk electrode was used as the deposition electrode in the electrochemical cell: by measuring the frequency change of the quartz crystal resonator, the growth of the proteic deposit could be followed and eventually quantified [
34].
Since EQCM measurements allowed for the measurement of mass changes as low as about 1 ng/cm2 and are sensitive to viscosity changes, preliminary experiments were performed to exclude significant frequency changes due to protein adsorption at the surface of the deposition electrode and/or changes in the viscosity of the solution due to crosslinking in a bath solution. Using a 1 h time window, the frequency changes in a pure buffer reached a maximal value of about 0.1 kHz, while those observed in the presence of both proteins and a crosslinker but without any current pulse application were on the order of 1 kHz; finally, frequency changes due to protein adsorption, induced in the absence of GLU in the solution and without any current pulse application at the deposition electrode, were about 0.1 kHz as well.
These frequency changes were surely lower than those observed for EPD experiments wherein protein deposition and in situ co-crosslinking were expected. In fact, as reported in
Figure 5, the frequency changes due to EPD/co-crosslinking were significantly higher, increasing with the time of protein deposition and starting and stopping with the current pulse sequence application as expected for a process under electrostatic control; further, the EQCM profiles were almost reproducible, as curves (b) and (c) in
Figure 5 show, since they represented a couple of replicate experiments differing only in their deposition time. Notably, at the end of the deposition time, as shown in
Figure 5 for both curves (b) and (c), which depict the cessation of current pulse application, the frequency changes reversed (i.e., the resonance frequency increased). This is likely due to the disruption of the concentration gradient of these proteins, still not crosslinked, near the electrode surface/solution interface, originally produced by their electrophoretic movements towards the deposition electrode; anyway, these frequency changes soon reached a plateau, demonstrating the presence of a deposit on the electrode surface and further detachment from it.
Figure 5 also shows that frequency changes varied almost linearly with the low deposition time (case (b)) and in a hyperbolic fashion at longer durations, with an overall frequency change of about 0.3 kHz/min; using the well-known Sauerbrey equation [
34], this corresponded to about 3 × 10
−7 g/min, so, e.g., in the case (c) of
Figure 5, a proteic deposition of about 10 µg (corresponding to nearly 500 ng/mm
2 and roughly to a one-micron thickness) was observed in less than an hour. Similar EQCM studies [
4,
22] about the EPD of GOD performed using different electrical perturbation techniques and different chemical conditions reported significantly lower amounts of protein deposition: in ultrapure water, the amounts were comparable with the other conditions investigated, demonstrating that efficient EPD in these poor and uncontrolled chemical conditions is not mandatory. Finally, it should be stressed that the Sauerbrey equation, here and elsewhere used and sometimes misused, is strictly valid only for elastic mass measurements [
34], i.e., when the deposit onto the quartz sensor is so rigid that it is mechanically coupled with the vibrating quartz, as in the case of metal deposition or thin film formation; on the contrary, in the present case, as for similarly fabricated biosensors proposed elsewhere, the nature of the enzymatic deposit is quite different from the rigid case, being essentially a dense gel with a viscoelastic nature wherein density, viscosity, and hydration states depend on the chemical conditions used for its formation. Accordingly, care should be taken when considering these mass or thickness figures as the real mass amounts of the proteic deposit.
3.4. Potentiodynamic Deposition
Even if the galvanodynamic approach optimized in this study proved capable of successfully depositing and in situ co-crosslinking GOD and BSA on the electrode surface, further approaches were investigated and studied. As previously pointed out, the concurrent and deleterious processes of water discharge and dioxygen bubbling are catalyzed by platinum oxide formation: since this last process is merely controlled by the potential of the deposition electrode under suitable chemical conditions, a potentiodynamic technique, wherein the potential is under the control of the EPD process, should increase the selectivity of the electrophoretic deposition towards gas evolution, increasing the efficiency of the overall EPD/co-crosslinking process; in fact, the galvanodynamic approach investigated herein, even if optimized, did not permit the true control of the potential reached by the deposition electrode since, as
Figure 3 and
Figure 4 show, its value increased, even if only slightly, during the current sequence application.
Since the preliminary potentiostatic experiments (see
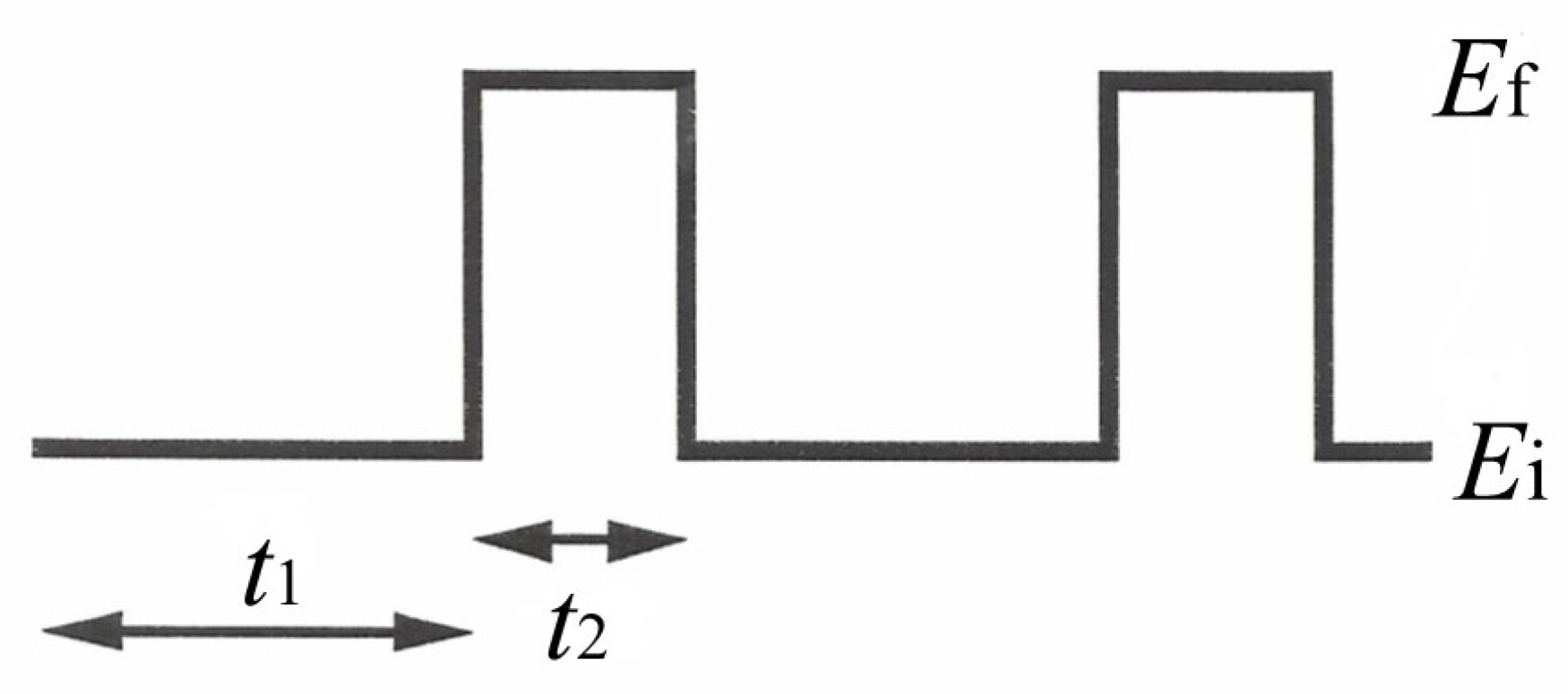
Section 3.2) failed to reach the goal of the present research, a potentiodynamic electrophoretic deposition technique was developed and investigated for protein deposition: here, a pulsed potential sequence (see
Scheme 2) was applied at the deposition electrode while varying the potential step values
Ei and
Ef and the pulse duty cycle by
t1 and
t2.
Apart from
t1 and
t2 optimization, the selection of
Ei and
Ef was of paramount importance for removing any Pt oxide formed during the electrophoretic pulse step and minimizing oxide formation, respectively; accordingly, several electrochemical experiments were preliminarily conducted for this purpose. In this respect,
Figure S2 shows typical cyclic voltammograms of the deposition Pt electrode as a function of scan rate in the media used for EPD/co-crosslinking deposition. Clearly evident are (a) the oxidation of the adsorbed dihydrogen and the subsequent Pt oxide formation due to Pt electrooxidation during the anodic scan (shown from left to right by the lower curves in both panels) and (b) Pt oxide stripping due to Pt electroreduction and the subsequent water reduction generating dihydrogen evolution during the reversal cathodic scan (right to left, the upper curves in both panels). As can be seen, the stripping peak, corresponding to the removal of Pt oxide from the electrode, shifts toward cathodic values, increasing the scan rate, but it was not lower than −0.1 V vs. Ag/AgCl, even for the fastest scan rate; accordingly,
Ei was set at this potential value to assure the complete removal of the Pt oxide on the surface of the deposition electrode.
Figure S2 also shows that the Pt oxide was formed in the range 0.2 ÷ 1 V vs. Ag/AgCl and shifted toward more positive values, increasing the scan rate.
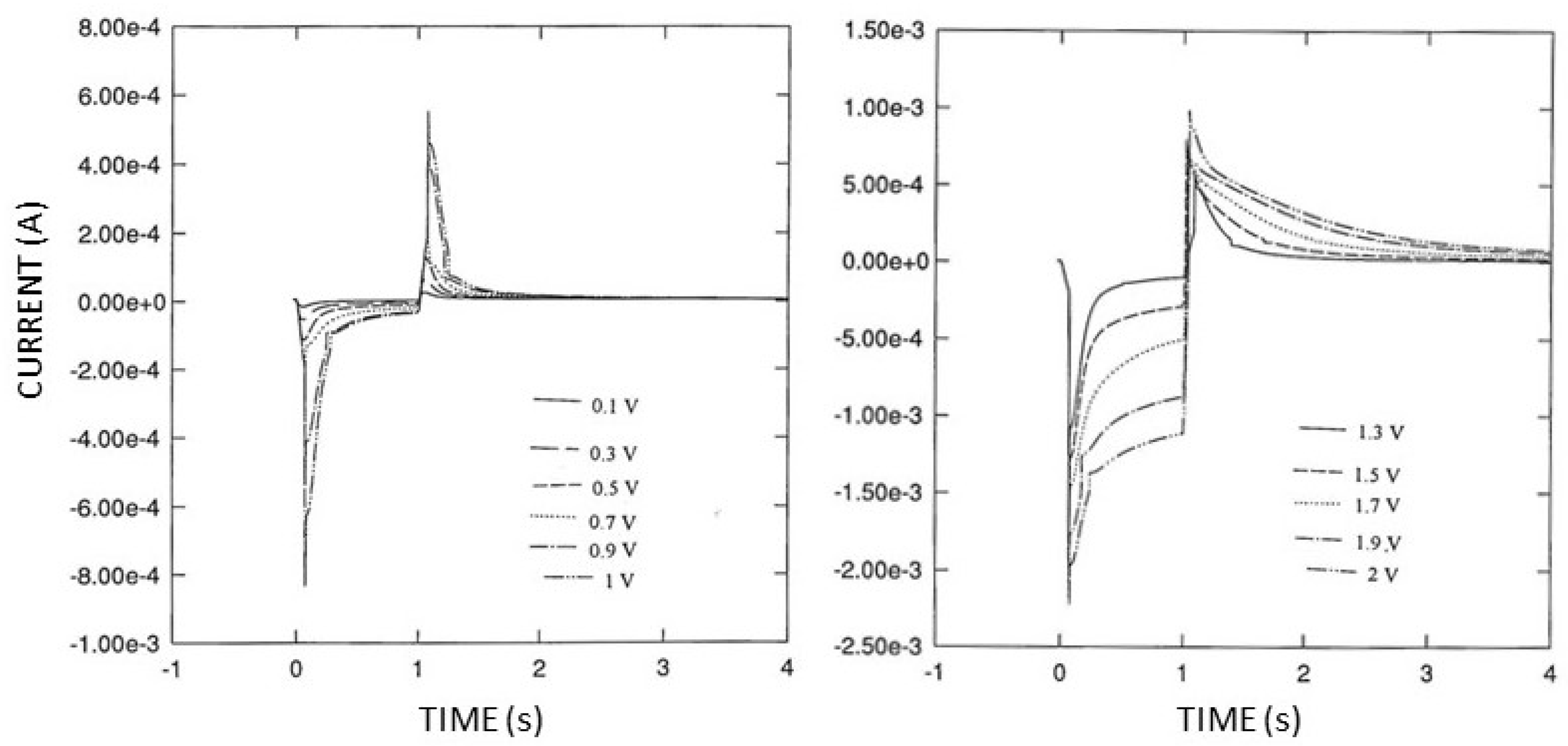
Due to this significant dependence on the scan rate, double potential step experiments (see
Figure 6) were performed; particularly, chronoamperometric curves were acquired after the application of a single potential step of potential starting from
Ef (Pt oxide formation) in the range of 0.1 ÷ 2 V vs. Ag/AgCl to
Ei (oxide stripping) set at −0.1 V; in this case,
t2 was 1 s, and
t1 was 5 s. These experiments were preferred over simple cyclic voltammograms since the conditions were more akin to the potential pulse sequence under study.
The double potential step experiments shown in
Figure 6 clearly evidenced that both the chronoamperometric curves, the positive ones due to Pt oxide stripping and the negative ones due to Pt oxide formation, increased in terms of current values and time extension upon increasing the final potential
Ef. Nevertheless, this concurrent increase was biased for the high values. Considering that the areas under these curves correspond to the charges due to reduction (positive curves) and oxidation processes (negative curves), plots of these charge values, as shown in
Figure 7, better highlight this occurrence: in fact, the net charge due to the oxidation processes increased to a greater extent than that due to the reduction processes upon increasing the final potential starting from values higher than 0.7 V vs. Ag/AgCl. Since this charge hysteresis means that the produced Pt oxide persists onto the electrode surface (thus catalyzing water discharge) even after the reduction-stripping step, care should be taken in choosing the right timing of the potential pulse sequence: in fact,
t1 should be sufficiently higher than
t2, and the latter should be significantly lower to assure the almost complete removal of Pt oxide formed during the application of
Ef to induce the electrophoretic movement of proteins; on the other hand, the value of the latter potential should be sufficiently high to promote efficient electrophoresis in the time scale of the deposition experiment while minimizing Pt oxide formation and hence gas evolution.
Further experiments performed using pulsed voltammetry permitted us to optimize the potential and timing parameters of the potential pulse sequence shown in
Scheme 2. Using an
Ei value of −0.1 V vs. Ag/AgCl, as previously optimized, it was found that an
Ef value of 1.8 V was a good compromise between efficient and quick protein deposition and low dioxygen evolution. Regarding the timing of the potential pulse sequence,
t2 was reduced at 12 ms (a value much lower than that used in galvanodynamic deposition), while
t1 was set to 5 s; indeed,
t1 could be set even higher, up to 20 s, assuring the complete absence of any side effects, but higher
t1 values were not adequate for promoting sufficient electrophoresis/co-crosslinking in the time scale of the experiment in order to produce a deposit that had good mechanical properties and was firmly attached to the deposition electrode.
In these optimized conditions, the growth of the proteic layer somewhat resembled that already described for the galvanodynamic deposition technique. Even using this potentiodynamic approach, after just a few seconds, an opalescent foamy-like layer was evident onto the electrode surface; on the contrary, after less than 1 min and more, a visual inspection showed a nearly “double” proteic layer, with the inner layer much more compact and adherent to the electrode, while the outer layer was foamy and easily detachable via a simple water washing procedure.
Further insights were gained via an EQCM study.
Figure 8 shows a typical EQCM profile of the growth of the proteic layer using this approach. Unlike the previous EQCM experiments (see
Figure 5), after the application of the optimized potential pulse sequence at the deposition electrode, the frequency change abruptly varied in less than 1 min, reaching a plateau in less than 5 min: this feature suggested quick protein deposition of the layer, which, after a few minutes, quickly stopped growing, as confirmed via a visual inspection. Even if the growth rate was much greater (roughly 10
−6 g/min) than that observed for the galvanodynamic deposition technique, the amount of the deposit formed at the plateau was significantly lower, estimated to be about 3.3 µg, with the deposit requiring less than 5 min to be formed. While a fast growth rate is surely desirable, unfortunately, it seems the growth of the proteic layer cannot be changed so that higher amounts are obtained. The reasons for this undesirable behavior are unknown: a severe ohmic drop due to the presence of a compact proteic layer, significantly reducing the electrical field around the electrode, may be tentatively suggested; even if a similar deposit is expected when employing the galvanodynamic approach, on the contrary, there, the increase in potential, as previously evidenced in this technique, should maintain an adequate electrical field to continue promoting satisfactory electrophoresis of proteins towards the electrode.
Finally, a further frequency change was observed after stopping the potential pulse application (see
Figure 8), wherein the growth of the deposit somewhat continued even in the absence of any electrophoretic movement of the proteins. Anyway, this last frequency change, which was significantly lower, exhibited a slower rate and reached a plateau after a few minutes. Notably, this peculiar feature disappeared completely if the potential pulse sequence application was protracted for more than 30 min, even if a plateau had already been reached a while ago. This last piece of evidence suggested the chemical (as opposed to electrophoretic) nature of this frequency change: it is probable that the co-crosslinking kinetics were less intense than the electrophoretic kinetics, so the frequency change observed after the stop time in
Figure 8 was due to residual co-crosslinking still acting after the end of the electrophoretic movement of the proteins. This feature was not evident in the galvanodynamic deposition approaches since the EPD growth was slower and the co-crosslinking was not limiting.
Even if the potentiodynamic approach apparently permitted the quick formation of a proteic deposit of a well-defined mass and thickness, which cannot induce further growth, the preliminary experiments showed that thicker deposits can be grown by varying some electrical parameters, like Ef, instead of increasing the deposition time. In this context, further experiments are ongoing in our laboratory.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}