
Fluoroquinolones as Tyrosinase Inhibitors; Enzyme Kinetics and Molecular Docking Studies to Explore Their Mechanism of Action

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
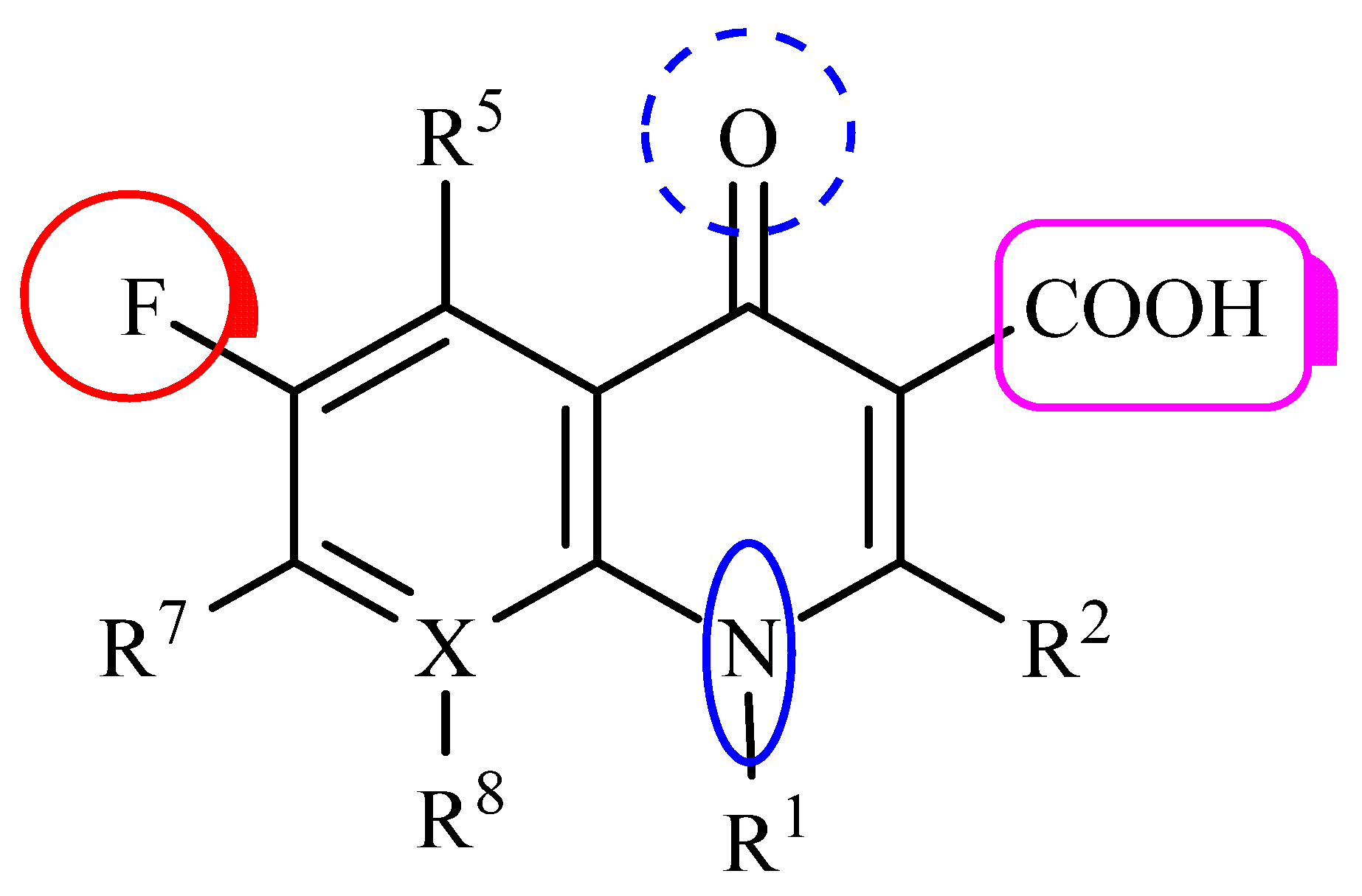
1. Introduction
2. Materials and Methods
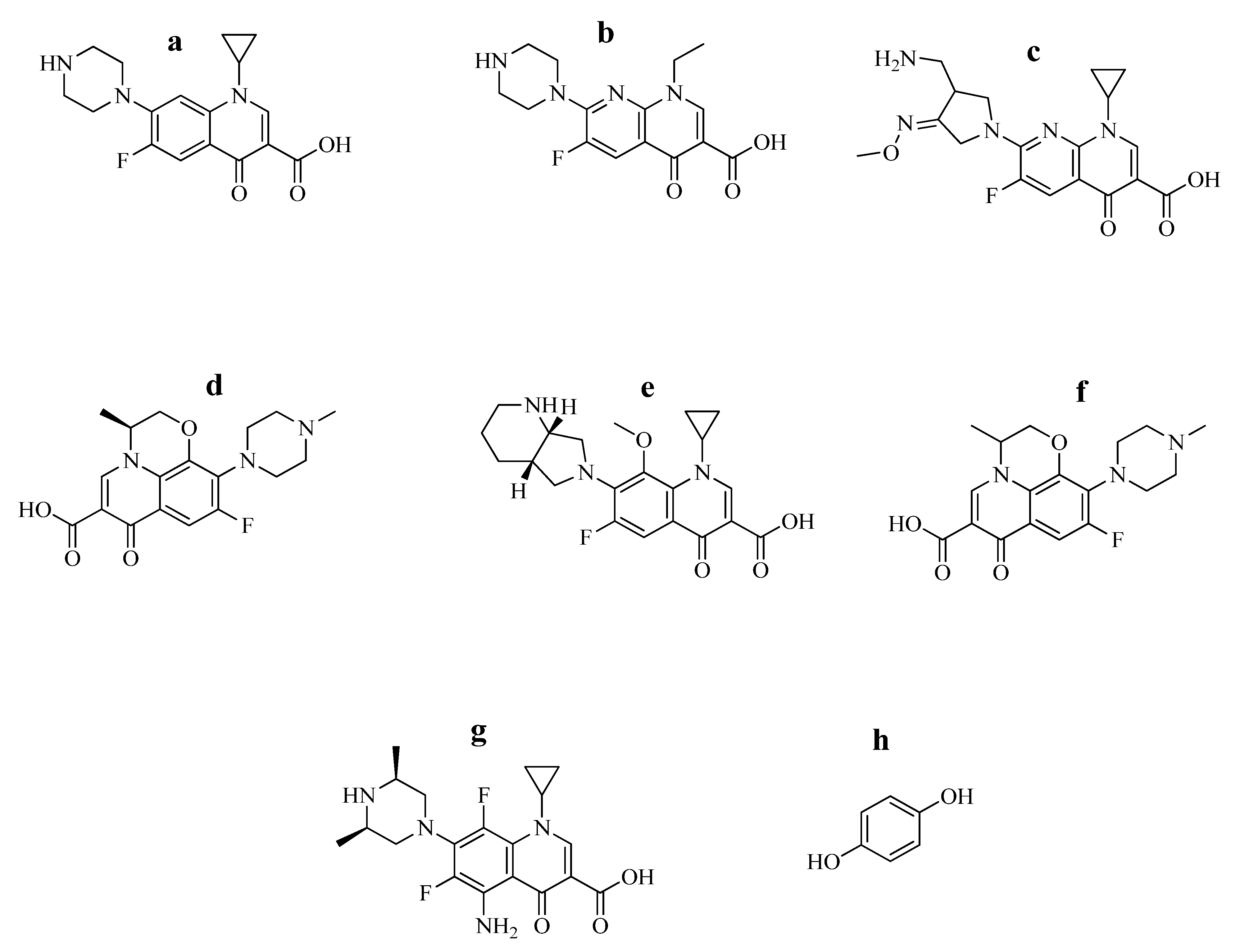
2.1. Chemicals
2.2. In Vitro Anti-Tyrosinase Activity
2.3. Kinetic Analysis of Tyrosinase Inhibition Activity
2.4. Molecular Docking Studies
2.4.1. Repossession of Mushroom Tyrosinase from PDB
2.4.2. Preparation of Ligands and Molecular Docking Simulation
3. Results and Discussion
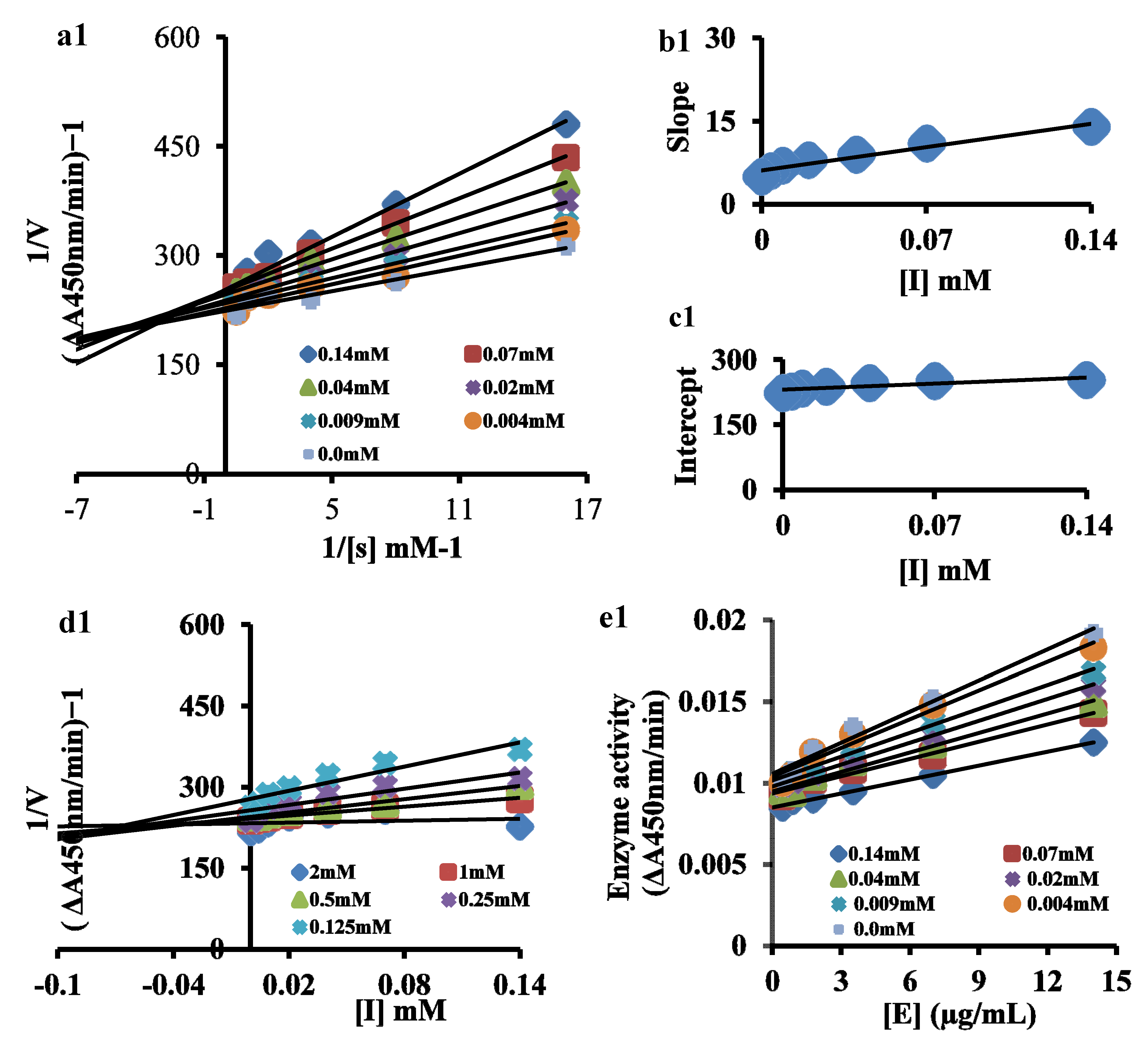
3.1. Mechanism of Enzyme Kinetics
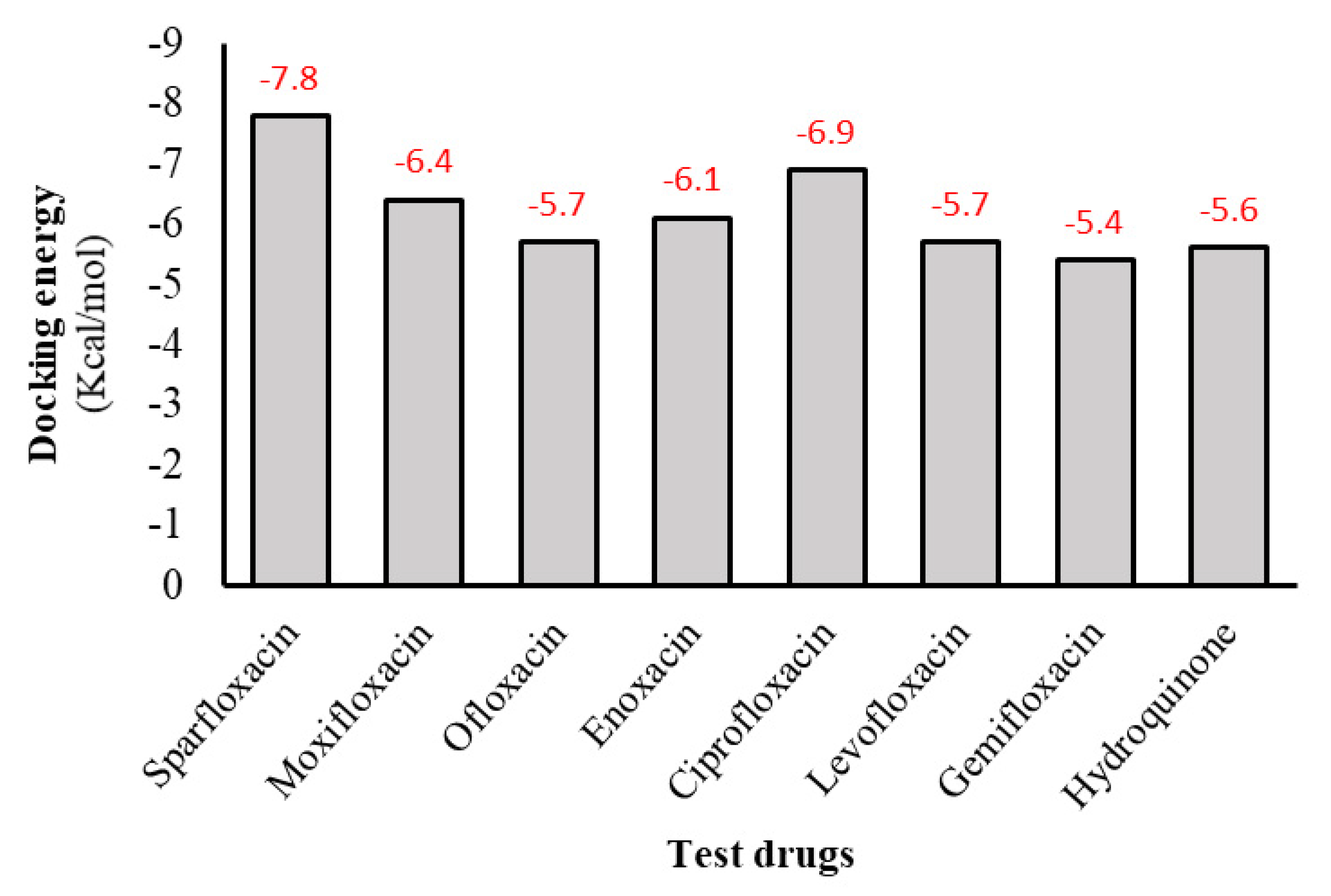
3.2. The Inhibitory Effect of Drugs on Diphenolase Activity of Tyrosinase
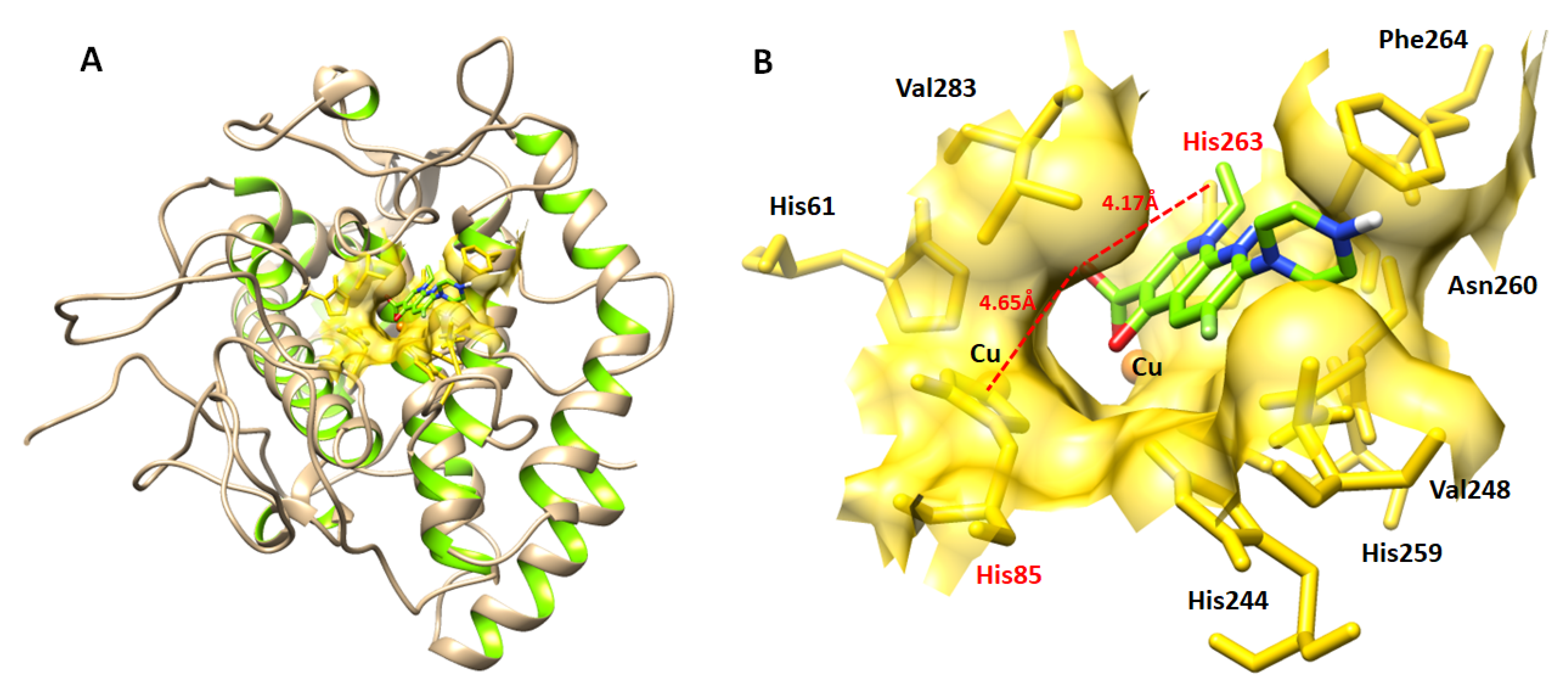
3.3. Molecular Docking and Structural Assessment of Mushroom Tyrosinase
3.4. Binding Analyses of Test Drugs and Tyrosinase Enzyme
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Appelbaum, P.C.; Hunter, P.A. The fluoroquinolone antibacterials: Past, present and future perspectives. Int. J. Antimicrobial. Agents 2000, 16, 5–15. [Google Scholar] [CrossRef]
- Patrick, G.L. An Introduction to Medicinal Chemistry; Oxford University Press: Oxford, UK, 2013; pp. 379–435. [Google Scholar]
- Dalhoff, A. Global fluoroquinolone resistance epidemiology and implications for clinical use. Interdiscip. Perspect. Infect. Dis. 2012, 2012, 976273. [Google Scholar] [CrossRef] [PubMed]
- Linder, J.A.; Huang, E.S.; Steinman, M.A.; Gonzales, R.; Stafford, R. Fluoroquinolone prescribing in the United States: 1995 to 2002. Am. J. Med. 2005, 118, 259–268. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, M.V.; Saraiva, M.F.; de Souza, M.V.; da Costa, C.F.; Vicente, F.R.; Lourenço, M.C. Synthesis and antitubercular activity of lipophilic moxifloxacin and gatifloxacin derivatives. Bioorganic Med. Chem. Lett. 2007, 17, 5661–5664. [Google Scholar] [CrossRef]
- Anquetin, G.; Greiner, J.; Mahmoudi, N.; Santillana-Hayat, M.; Gozalbes, R.; Farhati, K.; Derouin, F.; Aubry, A.; Cambau, E.; Vierling, P. Design, synthesis and activity against Toxoplasma gondii, Plasmodium spp., and Mycobacterium tuberculosis of new 6-fluoroquinolones. Eur. J. Med. Chem. 2006, 41, 1478–1493. [Google Scholar] [CrossRef]
- Redgrave, L.S.; Sutton, S.B.; Webber, M.A.; Piddock, L.J. Fluoroquinolone resistance: Mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol. 2014, 22, 438–445. [Google Scholar] [CrossRef]
- Pham, T.D.; Ziora, Z.M.; Blaskovich, M.A. Quinolone antibiotics. Medchemcomm 2019, 10, 1719–1739. [Google Scholar] [CrossRef]
- Oliveira, H.S.; Gonçalo, M.; Figueiredo, A.C. Photosensitivity to lomefloxacin. A clinical and photobiological study. Photodermatol. Photoimmunol. Photomed. Figueiredo. Photodermatol. Photoimmunol. Photomed. 2000, 16, 116–120. [Google Scholar] [CrossRef]
- De Guidi, G.; Bracchitta, G.; Catalfo, A. Photosensitization reactions of fluoroquinolones and their biological consequences. Photochem. Photobio. 2011, 87, 1214–1229. [Google Scholar] [CrossRef]
- Viola, G.; Facciolo, L.; Canton, M.; Vedaldi, D.; Dall’Acqua, F.; Aloisi, G.G.; Amelia, M.; Barbafina, A.; Elisei, F.; Latterini, L. Photophysical and Phototoxic Properties of the Antibacterial Fluoroquinolones Levofloxacin and Moxifloxacin. Chem. Biodivers. 2004, 1, 782–801. [Google Scholar] [CrossRef]
- Dwivedi, A.; Mujtaba, S.F.; Yadav, N.; Kushwaha, H.N.; Amar, S.K.; Singh, S.K.; Pant, M.C.; Ray, R.S. Cellular and molecular mechanism of ofloxacin induced apoptotic cell death under ambient UV-A and sunlight exposure. Free Radic. Res. 2014, 48, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Ray, R.S.; Farooq, M.; Pant, A.B.; Hans, R.K. Photosensitizing Potential of Ciprofloxacin at Ambient Level of UV Radiation. Photochem. Photobiol. 2007, 83, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Owen, K. Comparative grepafloxacin phototoxicity in mouse skin. J. Antimicrob. Chemother. 1998, 42, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Takahashi, Y.; Kawaguchi, H.; Utsunomiya, S.; Miura, N.; Izumi, H.; Tanimoto, A. A dermal phototoxicity study following intravenous infusion administration of ciprofloxacin hydrochloride in the novel microminipigs. Toxicol. Pathol. 2013, 41, 109–113. [Google Scholar] [CrossRef]
- Scholar, E.M. Fluoroquinolines: Past, present and future of a novel group of antibacterial agents. Am. J. Pharm. Educ. 2002, 66, 164–171. [Google Scholar]
- Ferguson, J.; Dawe, R. Phototoxicity in quinolones: Comparison of ciprofloxacin and grepafloxacin. J. Antimicrob. Chemother. 1997, 4, 93–98. [Google Scholar] [CrossRef][Green Version]
- Cooksey, C.J.; Garratt, P.J.; Land, E.J.; Pavel, S.; Ramsden, C.A.; Riley, P.A.; Smit, N.P. Evidence of the indirect formation of the catecholic intermediate substrate responsible for the autoactivation kinetics of tyrosinase. J. Biol. Chem. 1997, 272, 26226–26235. [Google Scholar] [CrossRef]
- Rozanowska, M.; Sarna, T.; Land, E.J.; Truscott, T. Free radical scavenging properties of melanin: Interaction of eu- and pheo-melanin models with reducing and oxidising radicals. Free Radic. Biol. Med. 1999, 26, 518–525. [Google Scholar] [CrossRef]
- Simon, J.D.; Peles, D.; Wakamatsu, K.; Ito, S. Current challenges in understanding melanogenesis: Bridging chemistry, biological control, morphology, and function. Pigment Cell Melanoma Res. 2009, 22, 563–579. [Google Scholar] [CrossRef]
- Hu, D.N.; Savage, H.E.; Roberts, J.E. Uveal melanocytes, ocular pigment epithelium, and Müller cells in culture: In vitro toxicology. Int. J. Toxicol. 2002, 21, 465–472. [Google Scholar] [CrossRef]
- Knorle, R.; Schniz, E.; Feuerstein, T.J. Drug accumulation in melanin: An affinity chromatographic study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 1998, 714, 171–179. [Google Scholar] [CrossRef]
- Larsson, B.S. Interaction between Chemicals and Melanin. Pigment Cell Res. 1993, 6, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Beberok, A.; Buszman, E.; Wrześniok, D. Interaction of norfloxacin and sparfloxacin with melanin in relation to phototoxic reactions. Ann. Univ. Mariae. Curie. Skłodowska Sectio. DDD Pharm. 2009, 4, 87–92. [Google Scholar]
- Beberok, A.; Buszman, E.; Zdybel, M.; Pilawa, B.; Wrześniok, D. EPR examination of free radical properties of DOPA–melanin complexes with ciprofloxacin, lomefloxacin, norfloxacin and sparfloxacin. Chem. Phy. Lett. 2010, 497, 115–122. [Google Scholar] [CrossRef]
- Beberok, A.; Buszman, E.; Wrześniok, D.; Otręba, M.; Trzcionka, J. Interaction between ciprofloxacin and melanin: The effect on proliferation and melanization in melanocytes. Eur. J. Pharmacol. 2011, 669, 32–37. [Google Scholar] [CrossRef]
- Rok, J.; Buszman, E.; Beberok, A.; Delijewski, M.; Otręba, M.; Wrześniok, D. Modulation of Melanogenesis and Antioxidant Status of Melanocytes in Response to Phototoxic Action of Doxycycline. Photochem. Photobiol. 2015, 91, 1429–1434. [Google Scholar] [CrossRef]
- Rok, J.; Buszman, E.; Delijewski, M.; Otręba, M.; Beberok, A.; Wrześniok, D. Effect of tetracycline and UV radiation on melanization and antioxidant status of melanocytes. J. Photochem. Photobiol. B Biol. 2015, 148, 168–173. [Google Scholar] [CrossRef]
- Buszman, E.; Beberok, A.; Rózańska, R.; Orzechowska, A. Interaction of chlorpromazine, fluphenazine and trifluoperazine with ocular and synthetic melanin in vitro. Die Pharm. J. Pharma. Sci. 2008, 63, 372–376. [Google Scholar]
- Beberok, A.; Otręba, M.; Wrześniok, D.; Buszman, E. Cytotoxic effect of lomefloxacin in culture of human epidermal melanocytes. Pharmacol. Rep. 2013, 65, 689–699. [Google Scholar] [CrossRef]
- Beberok, A.; Wrześniok, D.; Otręba, M.; Miliński, M.; Buszman, E. Effect of norfloxacin and moxifloxacin on melanin synthesis and antioxidant enzymes activity in normal human melanocytes. Mol. Cell Biochem. 2015, 401, 107–114. [Google Scholar] [CrossRef]
- Beberok, A.; Wrześniok, D.; Otręba, M.; Buszman, E. Impact of sparfloxacin on melanogenesis and antioxidant defense system in normal human melanocytes HEMa-LP—An in vitro study. Pharmacol. Rep. 2015, 67, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, H.; Mizutani, H.; Asahig, K.; Shimizu, M. Melanocyte melanin augments sparfloxacin-induced phototoxicity. J. Dermatol. Sci. 1999, 21, 27–33. [Google Scholar] [CrossRef]
- Ono, C.; Tanaka, M. Binding characteristics of fluoroquinolones to synthetic levodopa melanin. J. Pharm. Pharmacol. 2003, 55, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, Z.; Rafiq, M.; Nadeem, H.; Hassan, M.; Afzal, S.; Waseem, M.; Afzal, K.; Latip, J. Carvacrol derivatives as mushroom tyrosinase inhibitors; synthesis, kinetics mechanism and molecular docking studies. PLoS ONE 2017, 12, e0178069. [Google Scholar] [CrossRef]
- A Structural View of Biology. Available online: www.rcsb.org (accessed on 21 April 2021).
- Pettersen, E.F. TD Goddard: CC Huang: GS Couch: DM Greenblatt: EC Meng: TE Ferrin. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B., III; De Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, D.C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Free Download: BIOVIA Discovery Studio Visualizer. Available online: https://discover.3ds.com/discovery-studio-visualizer-download (accessed on 21 April 2021).
- Single (or Multiple) Model Protein Structure Analysis. Available online: http://vadar.wishartlab.com/ (accessed on 21 April 2021).
- O’Donnell, J.A.; Gelone, S.P. Fluoroquinolones. Infect. Dis. Clin. N. Am. 2000, 14, 489–513. [Google Scholar] [CrossRef]
- Guneysel, O.; Onur, O.; Erdede, M.; Denizbasi, A. Trimethoprim/sulfamethoxazole resistance in urinary tract infections. J. Emerg. Med. 2009, 36, 338–341. [Google Scholar] [CrossRef]
- Blandeau, J.M. Expanded activity and utility of the new fluoroquinolones: A review. Clin. Ther. 1999, 21, 3–40. [Google Scholar] [CrossRef]
- Quintero, B.; Miranda, M.A. Mechanisms of photosensitization induced by drugs: A general survey. Ars Pharm. 2000, 41, 27–46. [Google Scholar]
- Domagala, J.M. Structure-activity and structure-side-effect relationships for the quinolone antibacterials. J. Antimicrob. Chemother. 1994, 33, 685–706. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, E. History of Quinolones and Their Side Effects. Chemotherapy 2001, 47, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Tillotson, G.S. Quinolones: Structure-activity relationships and future predictions. J. Med. Microbiol. 1996, 44, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Higgins, P.G.; Fluit, A.C.; Schmitz, F.J. Fluoroquinolones: Structure and target sites. Curr. Drug Targets 2003, 4, 181–190. [Google Scholar] [CrossRef]
- Cecchetti, V.; Filipponi, E.; Fravolini, A.; Tabarrini, O.; Bonelli, D.; Clementi, M.; Cruciani, G.; Clementi, S. Chemometric methodologies in a quantitative structure− activity relationship study: The antibacterial activity of 6-aminoquinolones. J. Med. Chem. 1997, 40, 1698–1706. [Google Scholar] [CrossRef]
- Nawaz, M.S.; Bodla, R.; Kant, R.; Singh, S.P.; Bhutani, R.; Kapoor, G. Fluoroquinolone as antimicrobial agent: A Review. Int. J. Pharm. Sci. Res. 2017, 2, 57. [Google Scholar]
- Daneshtalab, M.; Ahmed, A. Nonclassical Biological Activities of Quinolone Derivatives. J. Pharm. Pharm. Sci. 2012, 15, 52–72. [Google Scholar] [CrossRef]
- Shandil, R.K.; Jayaram, R.; Kaur, P.; Gaonkar, S.; Suresh, B.L.; Mahesh, B.N.; Jayashree, R.; Nandi, V.; Bharath, S.; Balasubramanian, V. Moxifloxacin, ofloxacin, sparfloxacin, and ciprofloxacin against Mycobacterium tuberculosis: Evaluation of in vitro and pharmacodynamic indices that best predict in vivo efficacy. Antimicrob. Agents Chemother. 2007, 51, 576–582. [Google Scholar] [CrossRef]
- Wang, P.; Martin, B.D.; Parida, S.; Rethwisch, D.G.; Dordick, J.S. Multienzymic synthesis of poly (hydroquinone) for use as a redox polymer. J. Am. Chem. Soc. 1995, 117, 12885–12886. [Google Scholar] [CrossRef]
- Yi, W.; Wu, X.; Cao, R.; Song, H.; Ma, L. Biological evaluations of novel vitamin C esters as mushroom tyrosinase inhibitors and antioxidants. Food Chem. 2009, 117, 381–386. [Google Scholar] [CrossRef]
- Rattanangkool, E.; Kittikhunnatham, P.; Damsud, T.; Wacharasindhu, S.; Phuwapraisirisan, P. Quercitylcinnamates, a new series of antidiabetic bioconjugates possessing α-glucosidase inhibition and antioxidant. Eur. J. Med. Chem. 2013, 66, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Cai, D.; Mou, D.; Yan, Q.; Sun, Y.; Pan, W.; Wan, Y.; Song, H.; Yi, W. Design, synthesis and biological evaluation of hydroxy- or methoxy-substituted 5-benzylidene(thio) barbiturates as novel tyrosinase inhibitors. Bioorganic Med. Chem. 2014, 22, 3279–3284. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Chen, Q.-X.; Wang, Q.; Huang, H.; Song, K.-K. Irreversibly inhibitory kinetics of 3,5-dihydroxyphenyl decanoate on mushroom (Agaricus bisporus) tyrosinase. Bioorganic Med. Chem. 2005, 13, 6206–6211. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Ashraf, Z.; Abbas, Q.; Raza, H.; Seo, S.-Y. Exploration of Novel Human Tyrosinase Inhibitors by Molecular Modeling, Docking and Simulation Studies. Interdiscip. Sci. Comput. Life Sci. 2018, 10, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Vanjare, B.D.; Mahajan, P.G.; Dige, N.C.; Raza, H.; Hassan, M.; Han, Y.; Kim, S.J.; Seo, S.-Y.; Lee, K.H. Novel 1,2,4-triazole analogues as mushroom tyrosinase inhibitors: Synthesis, kinetic mechanism, cytotoxicity and computational studies. Mol. Divers. 2020, 25, 2089–2106. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alyami, B.A.; Alqarni, A.O.; Alqahtani, Y.S.; Mahnashi, M.H.; Javed, Q.; Hassan, M.; Tahir, T.; Ali, A.; Kotwica-Mojzych, K.; Mojzych, M. Fluoroquinolones as Tyrosinase Inhibitors; Enzyme Kinetics and Molecular Docking Studies to Explore Their Mechanism of Action. Appl. Sci. 2022, 12, 4849. https://doi.org/10.3390/app12104849
Alyami BA, Alqarni AO, Alqahtani YS, Mahnashi MH, Javed Q, Hassan M, Tahir T, Ali A, Kotwica-Mojzych K, Mojzych M. Fluoroquinolones as Tyrosinase Inhibitors; Enzyme Kinetics and Molecular Docking Studies to Explore Their Mechanism of Action. Applied Sciences. 2022; 12(10):4849. https://doi.org/10.3390/app12104849
Chicago/Turabian StyleAlyami, Bandar A., Ali O. Alqarni, Yahya S. Alqahtani, Mater H. Mahnashi, Qamar Javed, Mubashir Hassan, Tehreem Tahir, Anser Ali, Katarzyna Kotwica-Mojzych, and Mariusz Mojzych. 2022. "Fluoroquinolones as Tyrosinase Inhibitors; Enzyme Kinetics and Molecular Docking Studies to Explore Their Mechanism of Action" Applied Sciences 12, no. 10: 4849. https://doi.org/10.3390/app12104849
APA StyleAlyami, B. A., Alqarni, A. O., Alqahtani, Y. S., Mahnashi, M. H., Javed, Q., Hassan, M., Tahir, T., Ali, A., Kotwica-Mojzych, K., & Mojzych, M. (2022). Fluoroquinolones as Tyrosinase Inhibitors; Enzyme Kinetics and Molecular Docking Studies to Explore Their Mechanism of Action. Applied Sciences, 12(10), 4849. https://doi.org/10.3390/app12104849