
Mechanistic Insight into SARS-CoV-2 Mpro Inhibition by Organoselenides: The Ebselen Case Study


 ,
,  , and
, and 
Abstract
Featured Application
Abstract
1. Introduction
2. Materials and Methods
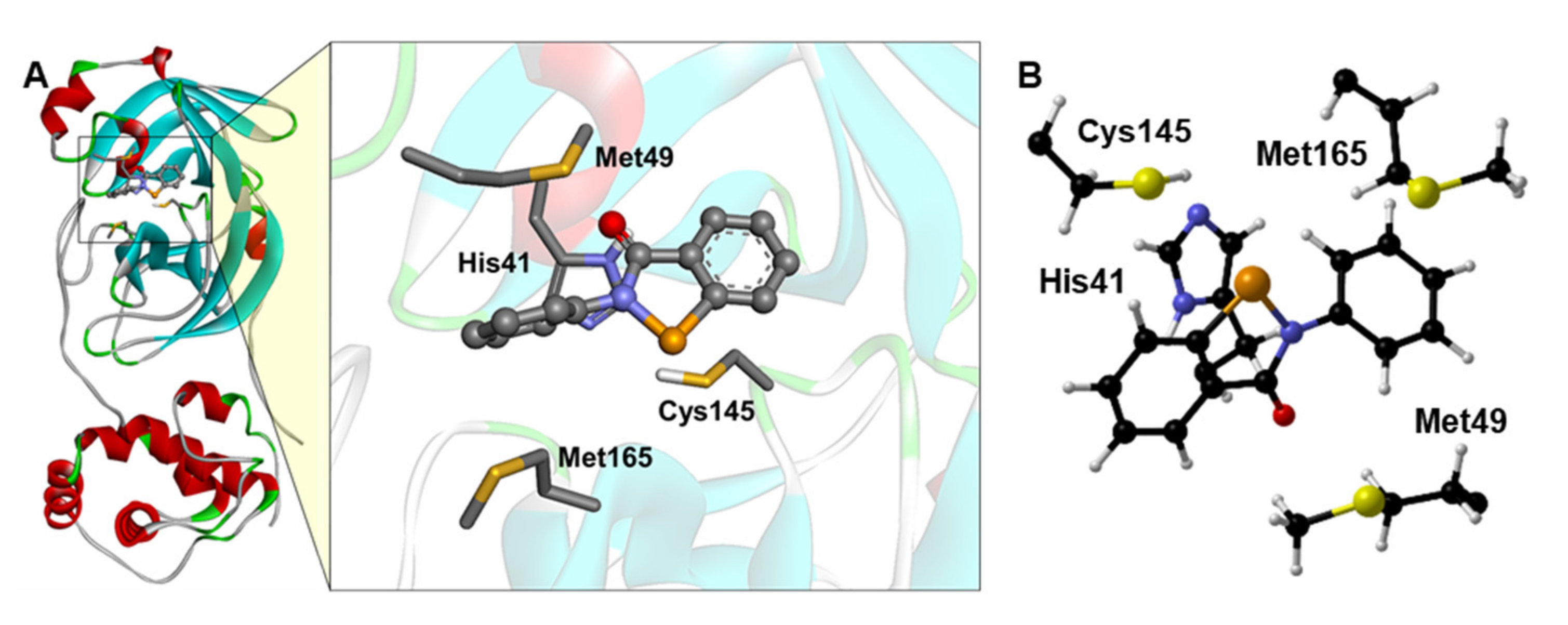
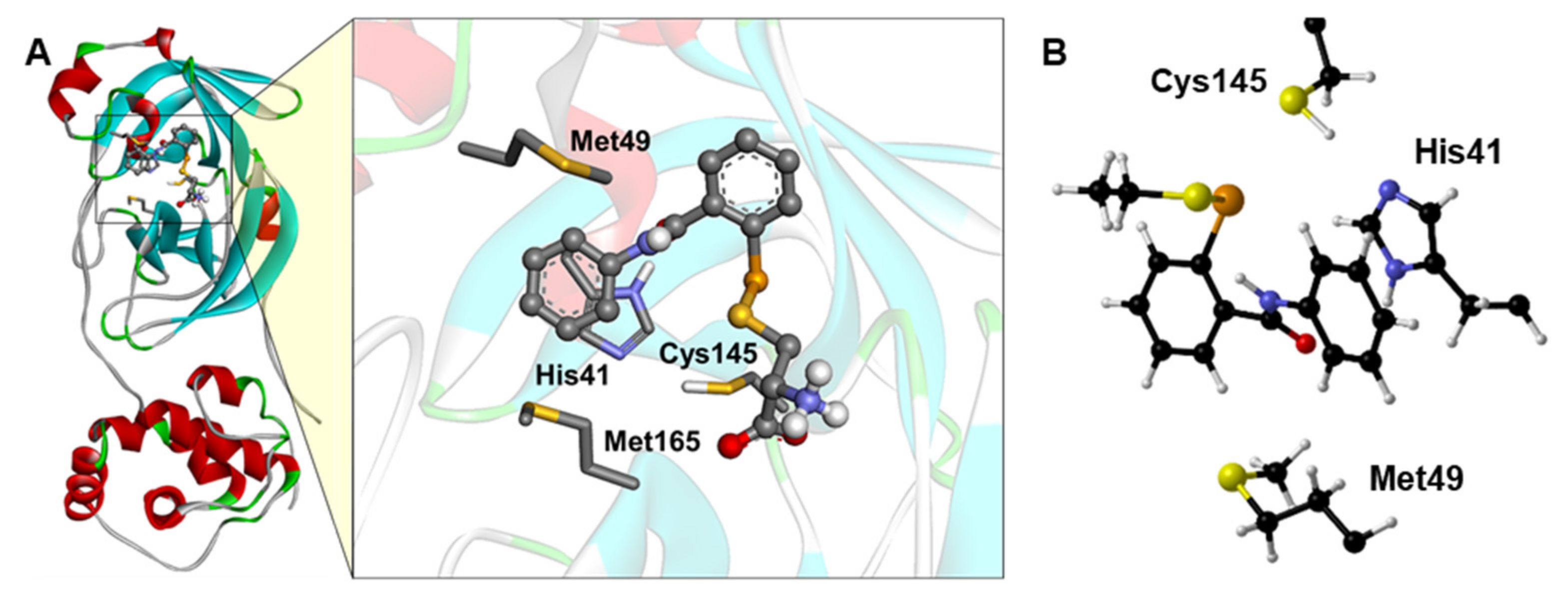
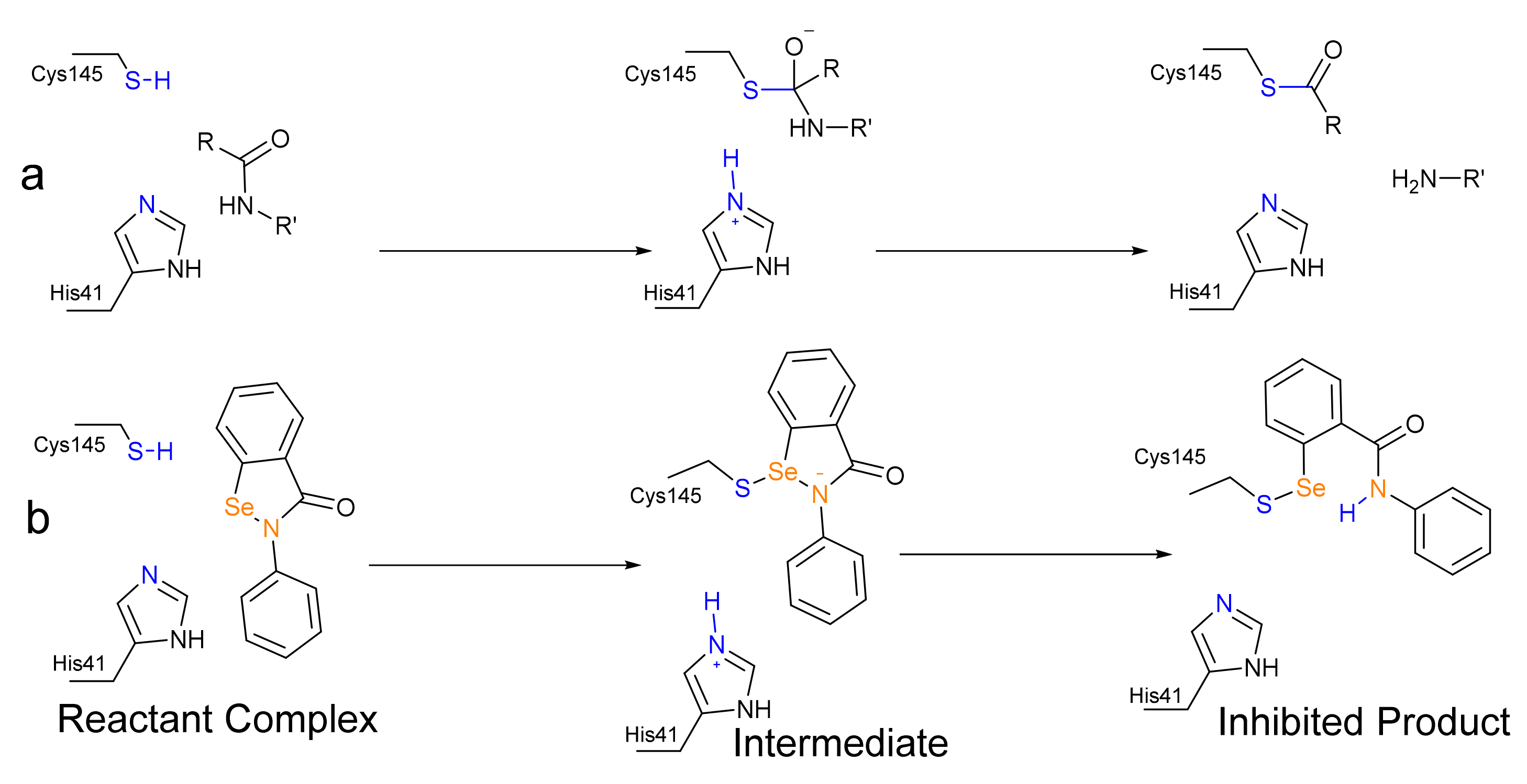
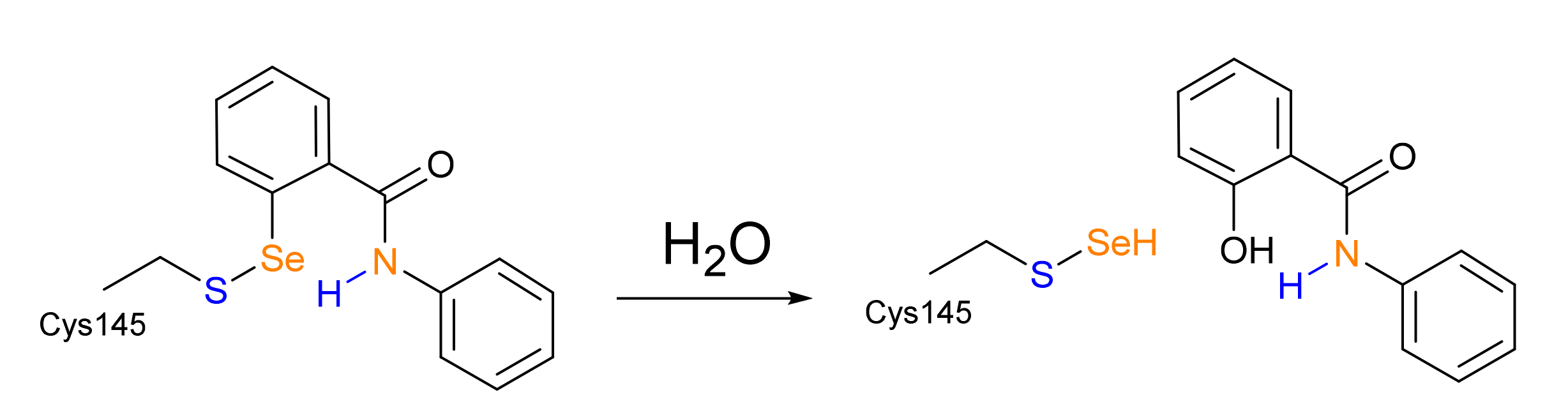
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Francés-Monerris, A.; Hognon, C.; Miclot, T.; García-Iriepa, C.; Iriepa, I.; Terenzi, A.; Grandemange, S.; Barone, G.; Marazzi, M.; Monari, A. Molecular Basis of SARS-CoV-2 Infection and Rational Design of Potential Antiviral Agents: Modeling and Simulation Approaches. J. Proteome Res. 2020, 19, 4291–4315. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, L.V. Overview of Targets and Potential Drugs of SARS-CoV-2 According to the Viral Replication. J. Proteome Res. 2021, 20, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Cannalire, R.; Cerchia, C.; Beccari, A.R.; Di Leva, F.S.; Summa, V. Targeting SARS-CoV-2 Proteases and Polymerase for COVID-19 Treatment: State of the Art and Future Opportunities. J. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef] [PubMed]
- Świderek, K.; Moliner, V. Revealing the molecular mechanisms of proteolysis of SARS-CoV-2 Mproby QM/MM computational methods. Chem. Sci. 2020, 11, 10626–10630. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Favrot, L.; Grzegorzewicz, A.E.; Lajiness, D.H.; Marvin, R.K.; Boucau, J.; Isailovic, D.; Jackson, M.; Ronning, D.R. Mechanism of inhibition of Mycobacterium tuberculosis antigen 85 by ebselen. Nat. Commun. 2013, 4, 1–10. [Google Scholar] [CrossRef]
- Chiou, J.; Wan, S.; Chan, K.-F.; So, P.-K.; He, D.; Chan, E.W.-C.; Chan, T.-H.; Wong, K.-Y.; Tao, J.; Chen, S. Ebselen as a potent covalent inhibitor of New Delhi metallo-β-lactamase (NDM-1). Chem. Commun. 2015, 51, 9543–9546. [Google Scholar] [CrossRef]
- Sies, H.; Parnham, M.J. Potential therapeutic use of ebselen for COVID-19 and other respiratory viral infections. Free Radic. Biol. Med. 2020, 156, 107–112. [Google Scholar] [CrossRef]
- Kil, J.; Lobarinas, E.; Spankovich, C.; Griffiths, S.K.; Antonelli, P.J.; Lynch, E.D.; Le Prell, C. Safety and efficacy of ebselen for the prevention of noise-induced hearing loss: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2017, 390, 969–979. [Google Scholar] [CrossRef]
- Parnham, M.J.; Sies, H. The early research and development of ebselen. Biochem. Pharmacol. 2013, 86, 1248–1253. [Google Scholar] [CrossRef]
- Thenin-Houssier, S.; de Vera, I.M.S.; Pedro-Rosa, L.; Brady, A.; Richard, A.; Konnick, B.; Opp, S.; Buffone, C.; Fuhrmann, J.; Kota, S.; et al. Ebselen, a Small-Molecule Capsid Inhibitor of HIV-1 Replication. Antimicrob. Agents Chemother. 2016, 60, 2195–2208. [Google Scholar] [CrossRef] [PubMed]
- Zmudzinski, M.; Rut, W.; Olech, K.; Granda, J.; Giurg, M.; Burda-Grabowska, M.; Zhang, L.; Sun, X.; Lv, Z.; Nayak, D.; et al. Ebselen Derivatives Are Very Potent Dual Inhibitors of SARS-CoV-2 Proteases—PLpro and Mpro in in Vitro Studies. bioRxiv 2020. [Google Scholar] [CrossRef]
- Weglarz-Tomczak, E.; Tomczak, J.M.; Talma, M.; Burda-Grabowska, M.; Giurg, M.; Brul, S. Identification of ebselen and its analogues as potent covalent inhibitors of papain-like protease from SARS-CoV-2. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Sancineto, L.; Mangiavacchi, F.; Dąbrowska, A.; Pacuła, A.; Obieziurska-Fabisiak, M.; Scimmi, C.; Lei, Y.; Kong, J.; Zhao, Y.; Machado, K.d.S.; et al. Organoselenium Mild Electrophiles in the Inhibition of Mpro and SARSCoV-2 Replication. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Ma, C.; Hu, Y.; Townsend, J.A.; Lagarias, P.I.; Marty, M.T.; Kolocouris, A.; Wang, J. Ebselen, Disulfiram, Carmofur, PX-12, Tideglusib, and Shikonin Are Nonspecific Promiscuous SARS-CoV-2 Main Protease Inhibitors. ACS Pharmacol. Transl. Sci. 2020, 3, 1265–1277. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.-Y.; Chen, C.; Su, J.; Li, J.-Q.; Jiang, Z.; Gao, H.; Chigan, J.-Z.; Ding, H.-H.; Zhai, L.; Yang, K.-W. Ebsulfur and Ebselen as highly potent scaffolds for the development of potential SARS-CoV-2 antivirals. Bioorg. Chem. 2021, 112, 104889. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, V.; Weber, P.; Meisch, F.; von Appen, F. Ebselen-binding equilibria between plasma and target proteins. Biochem. Pharmacol. 1996, 52, 15–19. [Google Scholar] [CrossRef]
- Wagner, G.; Schuch, G.; Akerboom, T.P.; Sies, H. Transport of ebselen in plasma and its transfer to binding sites in the hepatocyte. Biochem. Pharmacol. 1994, 48, 1137–1144. [Google Scholar] [CrossRef]
- Amporndanai, K.; Meng, X.; Shang, W.; Jin, Z.; Rogers, M.; Zhao, Y.; Rao, Z.; Liu, Z.-J.; Yang, H.; Zhang, L.; et al. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat. Commun. 2021, 12, 3061. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M.; Himo, F. Recent developments of the quantum chemical cluster approach for modeling enzyme reactions. J. Biol. Inorg. Chem. 2009, 14, 643–651. [Google Scholar] [CrossRef]
- Prabhakar, R.; Vreven, T.; Morokuma, K.; Musaev, D.G. Elucidation of the Mechanism of Selenoprotein Glutathione Peroxidase (GPx)-Catalyzed Hydrogen Peroxide Reduction by Two Glutathione Molecules: A Density Functional Study. Biochemistry 2005, 44, 11864–11871. [Google Scholar] [CrossRef]
- Parks, J.M.; Guo, H.; Momany, C.; Liang, L.; Miller, S.M.; Summers, A.; Smith, J. Mechanism of Hg−C Protonolysis in the Organomercurial Lyase MerB. J. Am. Chem. Soc. 2009, 131, 13278–13285. [Google Scholar] [CrossRef]
- Ngo, P.D.; Mansoorabadi, S.O.; Frey, P.A. Serine Protease Catalysis: A Computational Study of Tetrahedral Intermediates and Inhibitory Adducts. J. Phys. Chem. B 2016, 120, 7353–7359. [Google Scholar] [CrossRef]
- Hu, Q.; Jayasinghe-Arachchige, V.M.; Prabhakar, R. Degradation of a Main Plastic Pollutant Polyethylene Terephthalate by Two Distinct Proteases (Neprilysin and Cutinase-like Enzyme). J. Chem. Inf. Model. 2021, 61, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.W.; Sameera, W.M.C.; Ramozzi, R.; Page, A.J.; Hatanaka, M.; Petrova, G.P.; Harris, T.V.; Li, X.; Ke, Z.; Liu, F.; et al. The ONIOM Method and Its Applications. Chem. Rev. 2015, 115, 5678–5796. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, R.; Vreven, T.; Frisch, M.J.; Morokuma, K.; Musaev, D.G. Is the Protein Surrounding the Active Site Critical for Hydrogen Peroxide Reduction by Selenoprotein Glutathione Peroxidase? An ONIOM Study. J. Phys. Chem. B 2006, 110, 13608–13613. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Gaillard, T. Evaluation of AutoDock and AutoDock Vina on the CASF-2013 Benchmark. J. Chem. Inf. Model. 2018, 58, 1697–1706. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Awoonor-Williams, E.; Abu-Saleh, A.A.-A.A. Covalent and non-covalent binding free energy calculations for peptidomimetic inhibitors of SARS-CoV-2 main protease. Phys. Chem. Chem. Phys. 2021, 23, 6746–6757. [Google Scholar] [CrossRef]
- Suárez, D.; Díaz, N. SARS-CoV-2 Main Protease: A Molecular Dynamics Study. J. Chem. Inf. Model. 2020, 60, 5815–5831. [Google Scholar] [CrossRef]
- Sies, H. Ebselen, a Selenorganic Compound as Glutathione Peroxidase Mimic. Free Radic. Biol. Med. 1993, 14, 313–323. [Google Scholar] [CrossRef]
- Schewe, T. Molecular actions of Ebselen—An antiinflammatory antioxidant. Gen. Pharmacol. Vasc. Syst. 1995, 26, 1153–1169. [Google Scholar] [CrossRef]
- Haenen, G.R.M.M.; De Rooij, B.M.; Vermeulen, N.P.E.; Bast, A. Mechanism of the Reaction of Ebselen with Endogenous Thiols: Dihydrolipoate Is a Better Cofactor than Glutathione in the Peroxidase Activity of Ebselen. Mol. Pharmacol. 1990, 37, 412–422. [Google Scholar]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 1–17. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Sepay, N.; Saha, P.C.; Shahzadi, Z.; Chakraborty, A.; Halder, U.C. A crystallography-based investigation of weak interactions for drug design against COVID-19. Phys. Chem. Chem. Phys. 2021, 23, 7261–7270. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Rev. C.01; Gaussian: Wallingford, CT, USA, 2016. [Google Scholar]
- Dalla Tiezza, M.; Bickelhaupt, F.; Flohé, L.; Maiorino, M.; Ursini, F.; Orian, L. A dual attack on the peroxide bond. The common principle of peroxidatic cysteine or selenocysteine residues. Redox Biol. 2020, 34, 101540. [Google Scholar] [CrossRef]
- Bortoli, M.; Torsello, M.; Bickelhaupt, F.M.; Orian, L. Role of the Chalcogen (S, Se, Te) in the Oxidation Mechanism of the Glutathione Peroxidase Active Site. ChemPhysChem 2017, 18, 2990–2998. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Johnson, E.R. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Raghavachari, K. Perspective on “Density functional thermochemistry. III. The role of exact exchange”. Theor. Chem. Acc. 2000, 103, 361–363. [Google Scholar] [CrossRef]
- Grimme, S. Density functional theory with London dispersion corrections. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 211–228. [Google Scholar] [CrossRef]
- Becke, A.D.; Johnson, E.R. A density-functional model of the dispersion interaction. J. Chem. Phys. 2005, 123, 154101. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Li, L.; Li, C.; Zhang, Z.; Alexov, E. On the Dielectric “Constant” of Proteins: Smooth Dielectric Function for Macromolecular Modeling and Its Implementation in DelPhi. J. Chem. Theory Comput. 2013, 9, 2126–2136. [Google Scholar] [CrossRef]
- Kenyon, R.L. EDITORIAL. Chem. Eng. News Arch. 1968, 46, 363–368. [Google Scholar] [CrossRef][Green Version]
- Ramos-Guzmán, C.A.; Ruiz-Pernía, J.J.; Tuñón, I. Unraveling the SARS-CoV-2 Main Protease Mechanism Using Multiscale Methods. ACS Catal. 2020, 10, 12544–12554. [Google Scholar] [CrossRef]
- Dalla Tiezza, M.; Bickelhaupt, F.M.; Flohé, L.; Orian, L. Proton Transfer and S N 2 Reactions as Steps of Fast Selenol and Thiol Oxidation in Proteins: A Model Molecular Study Based on GPx. ChemPlusChem 2021, 86, 525–532. [Google Scholar] [CrossRef]
- Nogara, P.A.; Omage, F.B.; Bolzan, G.R.; Delgado, C.P.; Aschner, M.; Orian, L.; Rocha, J.B.T. In silico Studies on the Interaction Between Mpro and PLpro from SARS-CoV-2 and Ebselen, its Metabolites and Derivatives. Mol. Inform. 2021, 2100028, 1–13. [Google Scholar] [CrossRef]
- Bortoli, M.; Wolters, L.P.; Orian, L.; Bickelhaupt, F.M. Addition–Elimination or Nucleophilic Substitution? Understanding the Energy Profiles for the Reaction of Chalcogenolates with Dichalcogenides. J. Chem. Theory Comput. 2016, 12, 2752–2761. [Google Scholar] [CrossRef]
- Wiberg, K. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Rohrbach, S.; Murphy, J.A.; Tuttle, T. Computational Study on the Boundary Between the Concerted and Stepwise Mechanism of Bimolecular SNAr Reactions. J. Am. Chem. Soc. 2020, 142, 14871–14876. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D. Improving the Accuracy of Hybrid Meta-GGA Density Functionals by Range Separation. J. Phys. Chem. Lett. 2011, 2, 2810–2817. [Google Scholar] [CrossRef]
- Bortoli, M.; Madabeni, A.; Nogara, P.A.; Omage, F.B.; Ribaudo, G.; Zeppilli, D.; Rocha, J.B.T.; Orian, L. Chalcogen–Nitrogen Bond: Insights into a Key Chemical Motif. Catalysts 2021, 11, 114. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EbS(e)S | CH3S(S/SeCH3)SCH3 2 | |
|---|---|---|
| S1–S2 | 0.190 3 | 0.450 |
| S2–S3 | 0.833 3 | 0.610 |
| S1–Se | 0.325 | 0.535 |
| Se–S3 | 0.703 | 0.537 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madabeni, A.; Nogara, P.A.; Omage, F.B.; Rocha, J.B.T.; Orian, L. Mechanistic Insight into SARS-CoV-2 Mpro Inhibition by Organoselenides: The Ebselen Case Study. Appl. Sci. 2021, 11, 6291. https://doi.org/10.3390/app11146291
Madabeni A, Nogara PA, Omage FB, Rocha JBT, Orian L. Mechanistic Insight into SARS-CoV-2 Mpro Inhibition by Organoselenides: The Ebselen Case Study. Applied Sciences. 2021; 11(14):6291. https://doi.org/10.3390/app11146291
Chicago/Turabian StyleMadabeni, Andrea, Pablo Andrei Nogara, Folorunsho Bright Omage, João Batista Teixeira Rocha, and Laura Orian. 2021. "Mechanistic Insight into SARS-CoV-2 Mpro Inhibition by Organoselenides: The Ebselen Case Study" Applied Sciences 11, no. 14: 6291. https://doi.org/10.3390/app11146291
APA StyleMadabeni, A., Nogara, P. A., Omage, F. B., Rocha, J. B. T., & Orian, L. (2021). Mechanistic Insight into SARS-CoV-2 Mpro Inhibition by Organoselenides: The Ebselen Case Study. Applied Sciences, 11(14), 6291. https://doi.org/10.3390/app11146291