Role of Transglutaminase 2 in Migration of Tumor Cells and How Mouse Models Fit

{kind=link}
Abstract
1. Introduction
1.1. Migration of the Tumor Cells
1.2. Metastasis in Mice
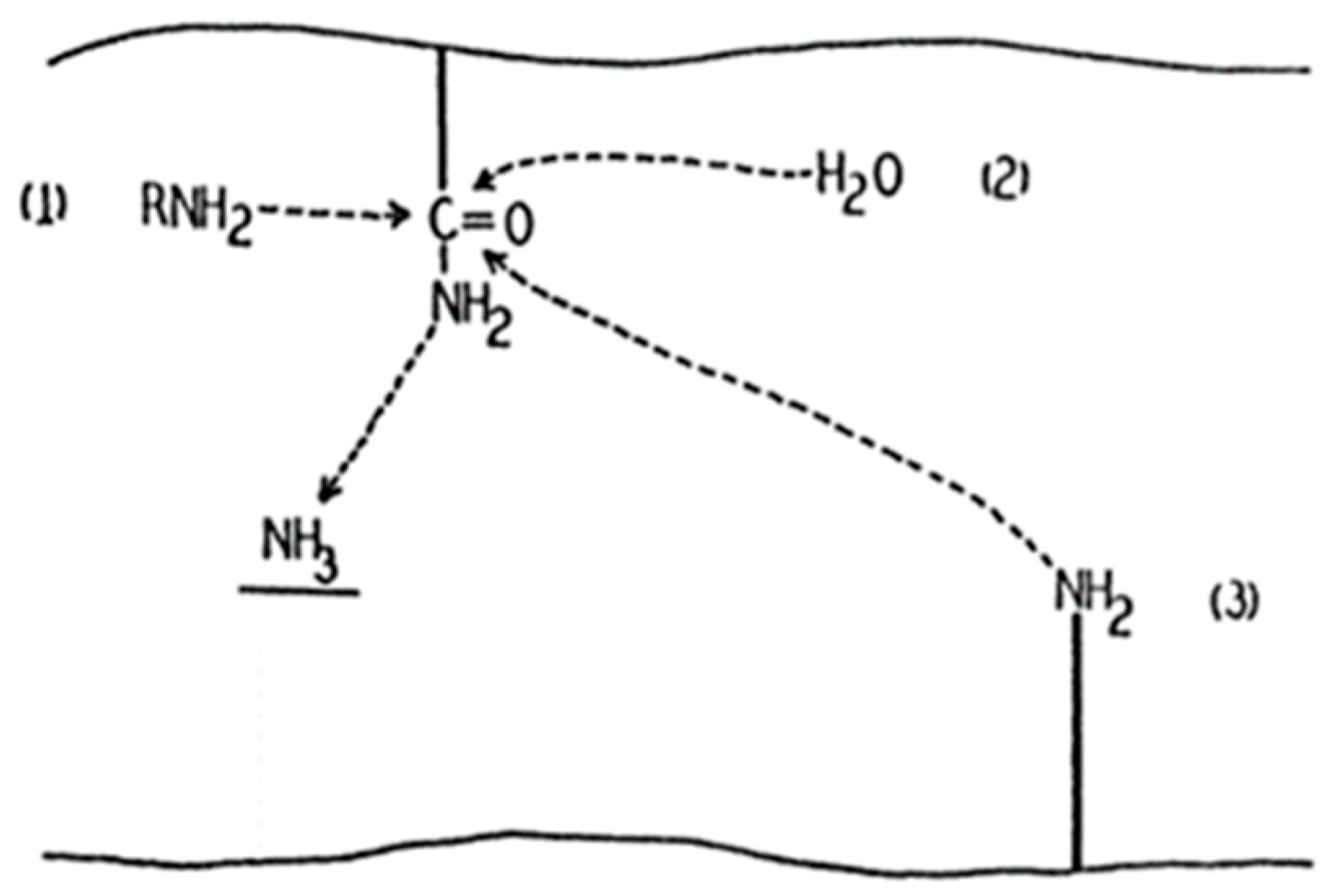
2. Transglutaminase 2
2.1. Transglutaminase 2 in Cancer
2.2. Transglutaminase 2 in Mouse Models
3. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Heppner, G.H.; Miller, B.E. Tumor heterogeneity: Biological implications and therapeutic consequences. Cancer Metastasis Rev. 1983, 2, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [CrossRef]
- Ewing, J. Neoplastic Diseases, a Text-Book on Tumors, James Ewing … with 479 Illustrations; W.B. Saunders Company: Philadelphia, PA, USA; London, UK, 1919; Available online: http://archive.org/details/neoplasticcdiseas00ewinrich (accessed on 27 July 2018).
- Saxena, M.; Christofori, G. Rebuilding cancer metastasis in the mouse. Mol. Oncol. 2013, 7, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Massagué, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J. The pathogenesis of cancer metastasis: The “seed and soil” hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, A.Y. Plasticity of tumor cell migration: Acquisition of new properties or return to the past? Biochemistry 2014, 79, 947–963. [Google Scholar] [CrossRef] [PubMed]
- Hay, E.D. Organization and fine structure of epithelium and mesenchyme in the developing chick embryo. In Epithelial–Mesenchymal Interactions; Billingham, R.E., Ed.; Williams & Wilkins: Baltimore, MD, USA, 1968; pp. 31–55. [Google Scholar]
- Trelstad, R.L.; Hay, E.D.; Revel, J.D. Cell contact during early morphogenesis in the chick embryo. Dev. Biol. 1967, 16, 78–106. [Google Scholar] [CrossRef]
- Friedl, P.; Wolf, K. Plasticity of cell migration: A multiscale tuning model. J. Exp. Med. 2010, 207. [Google Scholar] [CrossRef]
- Lorentzen, A.; Bamber, J.; Sadok, A.; Elson-Schwab, I.; Marshall, C.J. An ezrin-rich, rigid uropod-like structure directs movement of amoeboid blebbing cells. J. Cell Sci. 2011, 124 Pt 8, 1256–1267. [Google Scholar] [CrossRef]
- Cantelli, G.; Orgaz, J.L.; Rodriguez-Hernandez, I.; Karagiannis, P.; Maiques, O.; Matias-Guiu, X.; Nestle, F.O.; Marti, R.M.; Karagiannis, S.N.; Sanz-Moreno, V. TGF-β-Induced Transcription Sustains Amoeboid Melanoma Migration and Dissemination. Curr. Biol. 2015, 25, 2899–2914. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial–mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Barriere, G.; Fici, P.; Gallerani, G.; Fabbri, F.; Rigaud, M. Epithelial Mesenchymal Transition: A double-edged sword. Clin. Transl. Med. 2015, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P.; Fusenig, N.E. “Trans-differentiation” from epidermal to mesenchymal/myogenic phenotype is associated with a drastic change in cell-cell and cell-matrix adhesion molecules. J. Cell Biol. 1993, 120, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial–mesenchymal transition: New insights in signaling, development, and disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Savagner, P. The epithelial–mesenchymal transition (EMT) phenomenon. Ann. Oncol. 2010, 21, vii89–vii92. [Google Scholar] [CrossRef] [PubMed]
- Kopantzev, E.P.; Monastyrskaya, G.S.; Vinogradova, T.V.; Zinovyeva, M.V.; Kostina, M.B.; Filyukova, O.B.; Tonevitsky, A.G.; Sukhikh, G.T.; Sverdlova, E.D. Differences in gene expression levels between early and later stages of human lung development are opposite to those between normal lung tissue and non-small lung cell carcinoma. Lung Cancer 2008, 62, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Samatov, T.R.; Tonevitsky, A.G.; Schumacher, U. Epithelial-mesenchymal transition: Focus on metastatic cascade, alternative splicing, non-coding RNAs and modulating compounds. Mol. Cancer 2013, 12, 107. [Google Scholar] [CrossRef] [PubMed]
- Rørth, P. Collective cell migration. Annu. Rev. Cell Dev. Biol. 2009, 25, 407–429. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Enomoto, A.; Asai, N.; Kato, T.; Takahashi, M. Collective invasion of cancer: Perspectives from pathology and development. Pathol. Int. 2016, 66, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Soulié, P.; Carrozzino, F.; Pepper, M.S.; Strongin, A.Y.; Poupon, M.-F.; Montesano, R. Membrane-type-1 matrix metalloproteinase confers tumorigenicity on nonmalignant epithelial cells. Oncogene 2005, 24, 1689–1697. [Google Scholar] [CrossRef] [PubMed]
- Matise, L.A.; Palmer, T.D.; Ashby, W.J.; Nashabi, A.; Chytil, A.; Aakre, M.; Pickup, M.W.; Gorska, A.E.; Zijlstra, A.; Moses, H.L. Lack of transforming growth factor-β signaling promotes collective cancer cell invasion through tumor-stromal crosstalk. Breast Cancer Res. 2012, 14, R98. [Google Scholar] [CrossRef] [PubMed]
- Pouliot, N.; Pearson, H.B.; Burrows, A. Investigating Metastasis Using In Vitro Platforms Landes Bioscience. 2013. Available online: https://www.ncbi.nlm.nih.gov/books/NBK100379/ (accessed on 27 July 2018).
- Pekkonen, P.; Alve, S.; Balistreri, G.; Gramolelli, S.; Tatti-Bugaeva, O.; Paatero, I.; Niiranen, O.; Tuohinto, K.; Perälä, N.; Taiwo, A.; et al. Lymphatic endothelium stimulates melanoma metastasis and invasion via MMP14-dependent Notch3 and β1-integrin activation. eLife 2018, 7, e32490. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, A.; Żaczek, A.J. The Landscape of Circulating Tumor Cell Research in the Context of Epithelial–Mesenchymal Transition. Pathobiology 2017, 84, 264–283. [Google Scholar] [CrossRef] [PubMed]
- Pandya, P.; Orgaz, J.L.; Sanz-Moreno, V. Modes of invasion during tumour dissemination. Mol. Oncol. 2017, 11, 5–27. [Google Scholar] [CrossRef] [PubMed]
- Pinner, S.; Sahai, E. Imaging amoeboid cancer cell motility in vivo. J. Microsc. 2008, 231, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Sabeh, F.; Shimizu-Hirota, R.; Weiss, S.J. Protease-dependent versus -independent cancer cell invasion programs: Three-dimensional amoeboid movement revisited. J. Cell Biol. 2009, 185, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Hegerfeldt, Y.; Tusch, M.; Bröcker, E.-B.; Friedl, P. Collective cell movement in primary melanoma explants: Plasticity of cell–cell interaction, beta1-integrin function, and migration strategies. Cancer Res. 2002, 62, 2125–2130. [Google Scholar] [PubMed]
- Hay, E.D.; Zuk, A. Transformations between epithelium and mesenchyme: Normal, pathological, and experimentally induced. Am. J. Kidney Dis. 1995, 26, 678–690. [Google Scholar] [CrossRef]
- Apolant, H. Die epithelialen Geschwülste der Maus. Arbeiten ad Koniglchn Inst FExptTher zu Frankfurt aM 1906, 1, 7–68. [Google Scholar]
- Cardiff, R.D. The Pathology of EMT in Mouse Mammary Tumorigenesis. J. Mammary Gland Biol. Neoplasia 2010, 15, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Landesman-Bollag, E.; Romieu-Mourez, R.; Song, D.H.; Sonenshein, G.E.; Cardiff, R.D.; Seldin, D.C. Protein kinase CK2 in mammary gland tumorigenesis. Oncogene 2001, 20, 3247–3257. [Google Scholar] [CrossRef] [PubMed]
- Herschkowitz, J.I.; Zhao, W.; Zhang, M.; Usary, J.; Murrow, G.; Edwards, D.; Knezevic, J.; Greene, S.B.; Darr, D.; Troester, M.A.; et al. Comparative oncogenomics identifies breast tumors enriched in functional tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2012, 109, 2778–2783. [Google Scholar] [CrossRef] [PubMed]
- Moody, S.E.; Perez, D.; Pan, T.; Sarkisian, C.J.; Portocarrero, C.P.; Sterner, C.J.; Notorfrancesco, K.L.; Cardiff, R.D.; Chodosh, L.A. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell 2005, 8, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Moody, S.E.; Sarkisian, C.J.; Hahn, K.T.; Gunther, E.J.; Pickup, S.; Dugan, K.D.; Innocent, N.; Cardiff, R.D.; Schnall, M.D.; Chodosh, L.A. Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell 2002, 2, 451–461. [Google Scholar] [CrossRef]
- Jerry, D.J.; Kittrell, F.S.; Kuperwasser, C.; Laucirica, R.; Dickinson, E.S.; Bonilla, P.J.; Butel, J.S.; Medina, D. A mammary-specific model demonstrates the role of the p53 tumor suppressor gene in tumor development. Oncogene 2000, 19, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.J.; Moreno-Bueno, G.; Sarrio, D.; Locascio, A.; Cano, A.; Palacios, J.; Nieto, M.A. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene 2002, 21, 3241–3246. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.K.; Wilgenbus, P.; Dahl, U.; Semb, H.; Christofori, G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 1998, 392, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.A.; Liu, S.Y.; Tepper, C.G.; Karrim, J.; McGoldrick, E.T.; Rosner, A.; Munn, R.J.; Young, L.J.T.; Borowsky, A.D.; Cardiff, R.D.; et al. Molecular analysis of metastasis in a polyomavirus middle T mouse model: The role of osteopontin. Breast Cancer Res. 2004, 6, R157–R169. [Google Scholar] [CrossRef] [PubMed]
- Damonte, P.; Gregg, J.P.; Borowsky, A.D.; Keister, B.A.; Cardiff, R.D. EMT tumorigenesis in the mouse mammary gland. Lab. Investig. 2007, 87, 1218–1226. [Google Scholar] [CrossRef] [PubMed]
- Cardiff, R.D.; Munn, R.J.; Galvez, J.J. Chapter 24—The Tumor Pathology of Genetically Engineered Mice: A New Approach to Molecular Pathology. In The Mouse in Biomedical Research, 2nd ed.; Fox, J.G., Davisson, M.T., Quimby, F.W., Barthold, S.W., Newcomer, C.E., Smith, A.L., Eds.; Academic Press: Burlington, AN, Canada, 2007; pp. 581–622. Available online: http://www.sciencedirect.com/science/article/pii/B9780123694546500522 (accessed on 27 July 2018).
- Sugino, T.; Kusakabe, T.; Hoshi, N.; Yamaguchi, T.; Kawaguchi, T.; Goodison, S.; Masayuki Sekimata, M.; Homma, Y.; Suzuki, T. An Invasion-Independent Pathway of Blood-Borne Metastasis. Am. J. Pathol. 2002, 160, 1973–1980. [Google Scholar] [CrossRef]
- Lou, Y.; Preobrazhenska, O.; auf dem Keller, U.; Sutcliffe, M.; Barclay, L.; McDonald, P.C.; Roskelley, C.; Overall, C.M.; Dedhar, S. Epithelial-mesenchymal transition (EMT) is not sufficient for spontaneous murine breast cancer metastasis. Dev. Dyn. 2008, 237, 2755–2768. [Google Scholar] [CrossRef] [PubMed]
- Schokrpur, S.; Hu, J.; Moughon, D.L.; Liu, P.; Lin, L.C.; Hermann, K.; Mangul, S.; Guan, W.; Pellegrini, M.; Xu, H.; et al. CRISPR-Mediated VHL Knockout Generates an Improved Model for Metastatic Renal Cell Carcinoma. Sci. Rep. 2016, 6, 29032. [Google Scholar] [CrossRef] [PubMed]
- Manning, C.S.; Hooper, S.; Sahai, E.A. Intravital imaging of SRF and Notch signalling identifies a key role for EZH2 in invasive melanoma cells. Oncogene 2015, 34, 4320–4332. [Google Scholar] [CrossRef] [PubMed]
- Seifert, S.; Sontheimer, H. Bradykinin enhances invasion of malignant glioma into the brain parenchyma by inducing cells to undergo amoeboid migration. J. Physiol. 2014, 592, 5109–5127. [Google Scholar] [CrossRef] [PubMed]
- Menhofer, M.H.; Kubisch, R.; Schreiner, L.; Zorn, M.; Foerster, F.; Mueller, R.; Raedler, J.O.; Wagner, E.; Vollmar, A.M.; Zahler, S. The actin targeting compound Chondramide inhibits breast cancer metastasis via reduction of cellular contractility. PLoS ONE 2014, 9, e112542. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.L.; Kaartinen, M.T.; Nurminskaya, M.; Belkin, A.M.; Colak, G.; Johnson, G.V.W.; Mehta, K. Transglutaminase regulation of cell function. Physiol. Rev. 2014, 94, 383–417. [Google Scholar] [CrossRef] [PubMed]
- Waelsch, H. The amine incorporating system (Transglutaminase). In Monoamines et Systeme Nerveux Central; Ajuriagerra, J., Ed.; Georg et Companie, S.A.: Geneva, Switzerland; Masson et Companie: Paris, France, 1962. [Google Scholar]
- Scarpellini, A.; Germack, R.; Lortat-Jacob, H.; Muramatsu, T.; Billett, E.; Johnson, T.; Verderio, E.A. Heparan sulfate proteoglycans are receptors for the cell-surface trafficking and biological activity of transglutaminase-2. J. Biol. Chem. 2009, 284, 18411–18423. [Google Scholar] [CrossRef] [PubMed]
- Telci, D.; Griffin, M. Tissue transglutaminase (TG2)—A wound response enzyme. Front. Biosci. 2006, 11, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Mehta, K. Tissue transglutaminase: An enzyme with a split personality. Int. J. Biochem. Cell Biol. 1999, 31, 817–836. [Google Scholar] [CrossRef]
- Lai, T.-S.; Lin, C.-J.; Wu, Y.-T.; Wu, C.-J. Tissue transglutaminase (TG2) and mitochondrial function and dysfunction. Front. Biosci. 2017, 22, 1114–1137. [Google Scholar] [CrossRef]
- Gundemir, S.; Colak, G.; Tucholski, J.; Johnson, G.V.W. Transglutaminase 2: A molecular Swiss army knife. Biochim. Biophys. Acta 2012, 1823, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Haroon, Z.A.; Hettasch, J.M.; Lai, T.S.; Dewhirst, M.W.; Greenberg, C.S. Tissue transglutaminase is expressed, active, and directly involved in rat dermal wound healing and angiogenesis. FASEB J. 1999, 13, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Heath, D.J.; Christian, P.; Griffin, M. Involvement of tissue transglutaminase in the stabilisation of biomaterial/tissue interfaces important in medical devices. Biomaterials 2002, 23, 1519–1526. [Google Scholar] [CrossRef]
- Nurminskaya, M.V.; Belkin, A.M. Cellular functions of tissue transglutaminase. Int. Rev. Cell Mol. Biol. 2012, 294, 1–97. [Google Scholar] [PubMed]
- Guilluy, C.; Eddahibi, S.; Agard, C.; Guignabert, C.; Izikki, M.; Tu, L.; Savale, L.; Humbert, M.; Fadel, E.; Adnot, S.; et al. RhoA and Rho kinase activation in human pulmonary hypertension: Role of 5-HT signaling. Am. J. Respir. Crit. Care Med. 2009, 179, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Watts, S.W.; Priestley, J.R.C.; Thompson, J.M. Serotonylation of Vascular Proteins Important to Contraction. PLoS ONE 2009, 4, e5682. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Dudek, N.L.; Patel, T.B.; Muma, N.A. Transglutaminase-catalyzed transamidation: A novel mechanism for Rac1 activation by 5-hydroxytryptamine2A receptor stimulation. J. Pharmacol. Exp. Ther. 2008, 326, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, W.; Ehnis, T.; Bauer, M.; Donner, P.; Volta, U.; Riecken, E.O.; Schuppan, D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 1997, 3, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Molberg, Ø.; Parrot, I.; Hausch, F.; Filiz, F.; Gray, G.M.; Sollid, L.M.; Khosla, C. Structural basis for gluten intolerance in celiac sprue. Science 2002, 297, 2275–2279. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Siegel, M.; Choi, K.; Khosla, C.; Miller, C.R.; Jackson, E.N.; Piwnica-Worms, D.; Rich, K.M. Transglutaminase 2 inhibitor, KCC009, disrupts fibronectin assembly in the extracellular matrix and sensitizes orthotopic glioblastomas to chemotherapy. Oncogene 2007, 26, 2563–2573. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Wang, H.; Manavathi, B.; Fok, J.Y.; Mann, A.P.; Kumar, R.; Mehta, K. Increased expression of tissue transglutaminase in pancreatic ductal adenocarcinoma and its implications in drug resistance and metastasis. Cancer Res. 2006, 66, 10525–10533. [Google Scholar] [CrossRef] [PubMed]
- Erdem, M.; Erdem, S.; Sanli, O.; Sak, H.; Kilicaslan, I.; Sahin, F.; Telci, D. Up-regulation of TGM2 with ITGB1 and SDC4 is important in the development and metastasis of renal cell carcinoma. Urol. Oncol. 2014, 32, 25.e13–25.e20. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.; Fok, J.; Miller, F.R.; Koul, D.; Sahin, A.A. Prognostic significance of tissue transglutaminase in drug resistant and metastatic breast cancer. Clin. Cancer Res. 2004, 10, 8068–8076. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Kim, H.K.; Lee, J.H.; Choi, Y.B.; Park, S.Y.; Yang, S.H.; Kim, S.Y.; Hong, K.M. Transglutaminase 2 as a cisplatin resistance marker in non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2010, 136, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Fok, J.Y.; Ekmekcioglu, S.; Mehta, K. Implications of tissue transglutaminase expression in malignant melanoma. Mol. Cancer Ther. 2006, 5, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Satpathy, M.; Cao, L.; Pincheira, R.; Emerson, R.; Bigsby, R.; Nakshatri, H.; Matei, D. Enhanced peritoneal ovarian tumor dissemination by tissue transglutaminase. Cancer Res. 2007, 67, 7194–7202. [Google Scholar] [CrossRef] [PubMed]
- Iacobuzio-Donahue, C.A.; Ashfaq, R.; Maitra, A.; Adsay, N.V.; Shen-Ong, G.L.; Berg, K.; Hollingsworth, M.A.; Cameron, J.L.; Yeo, C.J.; Kern, S.E.; et al. Highly expressed genes in pancreatic ductal adenocarcinomas: A comprehensive characterization and comparison of the transcription profiles obtained from three major technologies. Cancer Res. 2003, 63, 8614–8622. [Google Scholar] [PubMed]
- Mehta, K.; Kumar, A.; Kim, H.I. Transglutaminase 2: A multi-tasking protein in the complex circuitry of inflammation and cancer. Biochem. Pharmacol. 2010, 80, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Kim, W.-J.; Demircan, B.; Dyer, L.M.; Bray, K.J.; Skehan, R.R.; Massoll, N.A.; Brown, K.D. The transglutaminase 2 gene (TGM2), a potential molecular marker for chemotherapeutic drug sensitivity, is epigenetically silenced in breast cancer. Carcinogenesis 2008, 29, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.; Han, A. Tissue Transglutaminase (TG2)-Induced Inflammation in Initiation, Progression, and Pathogenesis of Pancreatic Cancer. Cancers 2011, 3, 897–912. [Google Scholar] [CrossRef] [PubMed]
- Mohan, K.; Pinto, D.; Issekutz, T.B. Identification of tissue transglutaminase as a novel molecule involved in human CD8+ T cell transendothelial migration. J. Immunol. 2003, 171, 3179–3186. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Collighan, R.J.; Gross, S.R.; Danen, E.H.J.; Orend, G.; Telci, D.; Griffin, M. RGD-independent cell adhesion via a tissue transglutaminase-fibronectin matrix promotes fibronectin fibril deposition and requires syndecan-4/2 α5β1 integrin co-signaling. J. Biol. Chem. 2010, 285, 40212–40229. [Google Scholar] [CrossRef] [PubMed]
- Telci, D.; Wang, Z.; Li, X.; Verderio, E.A.M.; Humphries, M.J.; Baccarini, M.; Basaga, H.; Griffin, M. Fibronectin-Tissue Transglutaminase Matrix Rescues RGD-impaired Cell Adhesion through Syndecan-4 and β1 Integrin Co-signaling. J. Biol. Chem. 2008, 283, 20937–20947. [Google Scholar] [CrossRef] [PubMed]
- Mangala, L.S.; Fok, J.Y.; Zorrilla-Calancha, I.R.; Verma, A.; Mehta, K. Tissue transglutaminase expression promotes cell attachment, invasion and survival in breast cancer cells. Oncogene 2007, 26, 2459–2470. [Google Scholar] [CrossRef] [PubMed]
- Kotsakis, P.; Griffin, M. Tissue transglutaminase in tumour progression: Friend or foe? Amino Acids 2007, 33, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Akimov, S.S.; Krylov, D.; Fleischman, L.F.; Belkin, A.M. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J. Cell Biol. 2000, 148, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Verderio, E.A.M.; Telci, D.; Okoye, A.; Melino, G.; Griffin, M. A Novel RGD-independent Cell Adhesion Pathway Mediated by Fibronectin-bound Tissue Transglutaminase Rescues Cells from Anoikis. J. Biol. Chem. 2003, 278, 42604–42614. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.F.; Mangala, L.S.; Mehta, K. Implications of increased tissue transglutaminase (TG2) expression in drug-resistant breast cancer (MCF-7) cells. Oncogene 2006, 25, 3049–3058. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.H.; Womble, J.R. Transglutaminase may mediate certain physiological effects of endogenous amines and of amine-containing therapeutic agents. Life Sci. 1982, 30, 1499–1508. [Google Scholar] [CrossRef]
- Kumar, S.; Mehta, K. Tissue transglutaminase constitutively activates HIF-1α promoter and nuclear factor-κB via a non-canonical pathway. PLoS ONE 2012, 7, e49321. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Xu, J.; Brady, S.; Gao, H.; Yu, D.; Reuben, J.; Mehta, K. Tissue Transglutaminase Promotes Drug Resistance and Invasion by Inducing Mesenchymal Transition in Mammary Epithelial Cells. PLoS ONE 2010, 5, e13390. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Shao, M.; Schilder, J.; Guise, T.; Mohammad, K.S.; Matei, D. Tissue transglutaminase links TGF-β, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene 2012, 31, 2521–2534. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.A.; Kotsakis, P.; Johnson, T.S.; Chau, D.Y.S.; Ali, S.; Melino, G.; Griffin, M. Matrix changes induced by transglutaminase 2 lead to inhibition of angiogenesis and tumor growth. Cell Death Differ. 2006, 13, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, G.; Lentini, A.; Beninati, S.; Piacentini, M.; Rodolfo, C. In vivo evaluation of type 2 transglutaminase contribution to the metastasis formation in melanoma. Amino Acids 2009, 36, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Iismaa, S.E.; Mearns, B.M.; Lorand, L.; Graham, R.M. Transglutaminases and disease: Lessons from genetically engineered mouse models and inherited disorders. Physiol. Rev. 2009, 89, 991–1023. [Google Scholar] [CrossRef] [PubMed]
- Ikenaga, N.; Peng, Z.-W.; Vaid, K.A.; Liu, S.B.; Yoshida, S.; Sverdlov, D.Y.; Mikels-Vigdal, A.; Smith, V.; Schuppan, D.; Popov, Y.V. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut 2017, 66, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Iismaa, S.E.; Aplin, M.; Holman, S.; Yiu, T.W.; Jackson, K.; Burchfield, J.G.; Mitchell, C.J.; O’Reilly, L.; Davenport, A.; Cantley, J.; et al. Glucose Homeostasis in Mice Is Transglutaminase 2 Independent. PLoS ONE 2013, 8, e63346. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-J.; Jung, S.-H.; Kim, S.-H.; Kim, M.-S.; Lee, S.; Hwang, J.; Kim, S.Y.; Kim, Y.M.; Ha, K.S. Essential Role of Transglutaminase 2 in Vascular Endothelial Growth Factor-Induced Vascular Leakage in the Retina of Diabetic Mice. Diabetes 2016, 65, 2414–2428. [Google Scholar] [CrossRef] [PubMed]
- Szondy, Z.; Mastroberardino, P.G.; Váradi, J.; Farrace, M.G.; Nagy, N.; Bak, I.; Viti, I.; Wieckowski, M.R.; Melino, G.; Rizzuto, R.; et al. Tissue transglutaminase (TG2) protects cardiomyocytes against ischemia/reperfusion injury by regulating ATP synthesis. Cell Death Differ. 2006, 13, 1827–1829. [Google Scholar] [CrossRef] [PubMed]
- Balajthy, Z.; Csomós, K.; Vámosi, G.; Szántó, A.; Lanotte, M.; Fésüs, L. Tissue-transglutaminase contributes to neutrophil granulocyte differentiation and functions. Blood 2006, 108, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Rossin, F.; D’Eletto, M.; Falasca, L.; Sepe, S.; Cocco, S.; Fimia, G.M.; Campanella, M.; Mastroberardino, P.G.; Farrace, M.G.; Piacentini, M. Transglutaminase 2 ablation leads to mitophagy impairment associated with a metabolic shift towards aerobic glycolysis. Cell Death Differ. 2015, 22, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Szondy, Z.; Sarang, Z.; Molnar, P.; Nemeth, T.; Piacentini, M.; Mastroberardino, P.G.; Falasca, L.; Aeschlimann, D.; Kovacs, J.; Kiss, I.; et al. Transglutaminase 2−/− mice reveal a phagocytosis-associated crosstalk between macrophages and apoptotic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 7812–7817. [Google Scholar] [CrossRef] [PubMed]
- Van Herck, J.L.; Schrijvers, D.M.; De Meyer, G.R.Y.; Martinet, W.; Van Hove, C.E.; Bult, H.; Vrints, C.J.; Herman, A.G. Transglutaminase 2 deficiency decreases plaque fibrosis and increases plaque inflammation in apolipoprotein-E-deficient mice. J. Vasc. Res. 2010, 47, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Arbiser, J.L.; Raab, G.; Rohan, R.M.; Paul, S.; Hirschi, K.; Flynn, E.; Price, E.R.; Fisher, D.E.; Cohen, C.; Klagsbrun, M. Isolation of mouse stromal cells associated with a human tumor using differential diphtheria toxin sensitivity. Am. J. Pathol. 1999, 155, 723–729. [Google Scholar] [CrossRef]
- Blasco, M.A.; Lee, H.W.; Hande, M.P.; Samper, E.; Lansdorp, P.M.; DePinho, R.A.; Greider, C.W. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 1997, 91, 25–34. [Google Scholar] [CrossRef]
- Gonzalez, F.J.; Kimura, S. Understanding the role of xenobiotic-metabolism in chemical carcinogenesis using gene knockout mice. Mutat. Res. 2001, 477, 79–87. [Google Scholar] [CrossRef]
- Adelman, R.; Saul, R.L.; Ames, B.N. Oxidative damage to DNA: Relation to species metabolic rate and life span. Proc. Natl. Acad. Sci. USA 1988, 85, 2706–2708. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R. Neoplastic transformation: The contrasting stability of human and mouse cells. Cancer Surv. 1996, 28, 103–115. [Google Scholar] [PubMed]
- Rangarajan, A.; Weinberg, R.A. Comparative biology of mouse versus human cells: Modelling human cancer in mice. Nat. Rev. Cancer 2003, 3, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.E.; Yu, J.S.; Quigley, D.A.; To, M.D.; Jen, K.-Y.; Huang, P.Y.; Del Rosario, R.; Balmain, A. Inflammation and Hras signaling control epithelial–mesenchymal transition during skin tumor progression. Genes Dev. 2013, 27, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Perez, M.; Lee, E.-S.; Kojima, S.; Griffin, M. The functional relationship between transglutaminase 2 and transforming growth factor β1 in the regulation of angiogenesis and endothelial–mesenchymal transition. Cell Death Dis. 2017, 8, e3032. [Google Scholar] [CrossRef] [PubMed]
- Kerr, C.; Szmacinski, H.; Fisher, M.L.; Nance, B.; Lakowicz, J.R.; Akbar, A.; Keillor, J.W.; Wong, T.L.; Godoy-Ruiz, R.; Toth, E.A.; et al. Transamidase site-targeted agents alter the conformation of the transglutaminase cancer stem cell survival protein to reduce GTP binding activity and cancer stem cell survival. Oncogene 2017, 36, 2981–2990. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.L.; Keillor, J.W.; Xu, W.; Eckert, R.L.; Kerr, C. Transglutaminase Is Required for Epidermal Squamous Cell Carcinoma Stem Cell Survival. Mol. Cancer Res. 2015, 13, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Paupe, V.; Prudent, J. New insights into the role of mitochondrial calcium homeostasis in cell migration. Biochem. Biophys. Res. Commun. 2018, 500, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Sileno, S.; D’Oria, V.; Stucchi, R.; Alessio, M.; Petrini, S.; Bonetto, V.; Maechler, P.; Bertuzzi, F.; Grasso, V.; Paolella, K.; et al. A possible role of transglutaminase 2 in the nucleus of INS-1E and of cells of human pancreatic islets. J. Proteomics 2014, 96, 314–327. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bihorac, A. Role of Transglutaminase 2 in Migration of Tumor Cells and How Mouse Models Fit. Med. Sci. 2018, 6, 70. https://doi.org/10.3390/medsci6030070
Bihorac A. Role of Transglutaminase 2 in Migration of Tumor Cells and How Mouse Models Fit. Medical Sciences. 2018; 6(3):70. https://doi.org/10.3390/medsci6030070
Chicago/Turabian StyleBihorac, Ajna. 2018. "Role of Transglutaminase 2 in Migration of Tumor Cells and How Mouse Models Fit" Medical Sciences 6, no. 3: 70. https://doi.org/10.3390/medsci6030070
APA StyleBihorac, A. (2018). Role of Transglutaminase 2 in Migration of Tumor Cells and How Mouse Models Fit. Medical Sciences, 6(3), 70. https://doi.org/10.3390/medsci6030070