Ozone Depletion in Tropospheric Volcanic Plumes: From Halogen-Poor to Halogen-Rich Emissions

Abstract
1. Introduction
2. Challenges to In-Situ Measurement of Ozone in the Near-Source Volcano Plume
2.1. Interferences of Volcanic Gases on the Ozone Measurement
2.2. Intermittent Plume Exposure and Sensor Response Times
3. Materials and Methods
4. Results
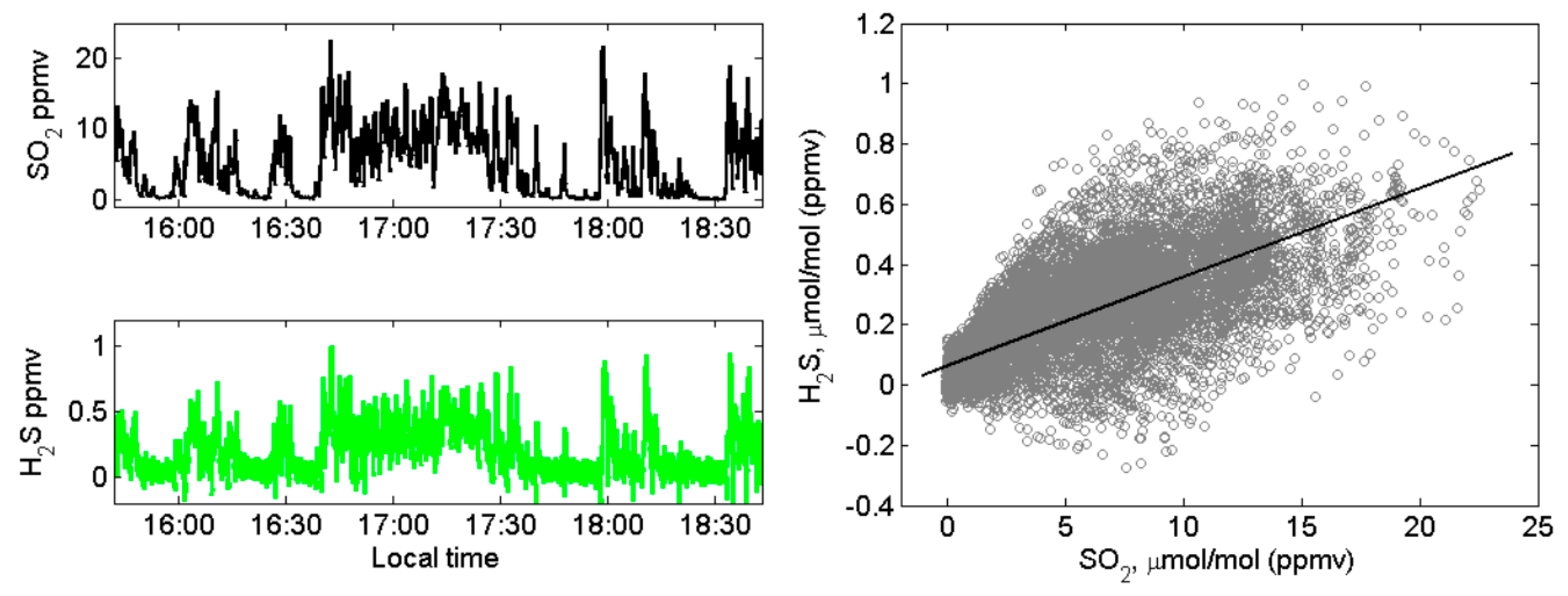
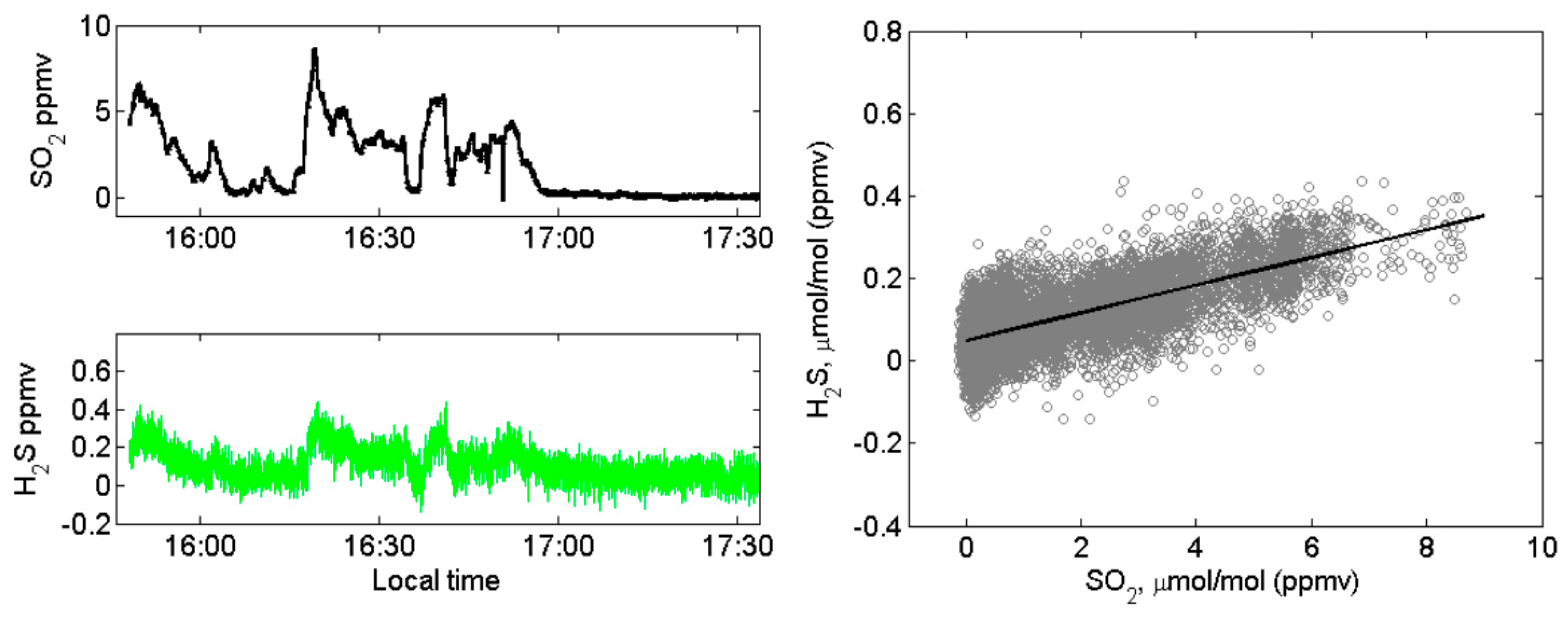
4.1. Volcanic SO2, H2S, and Ozone Measured in the Halema‘uma‘u Crater Rim Emission Plume
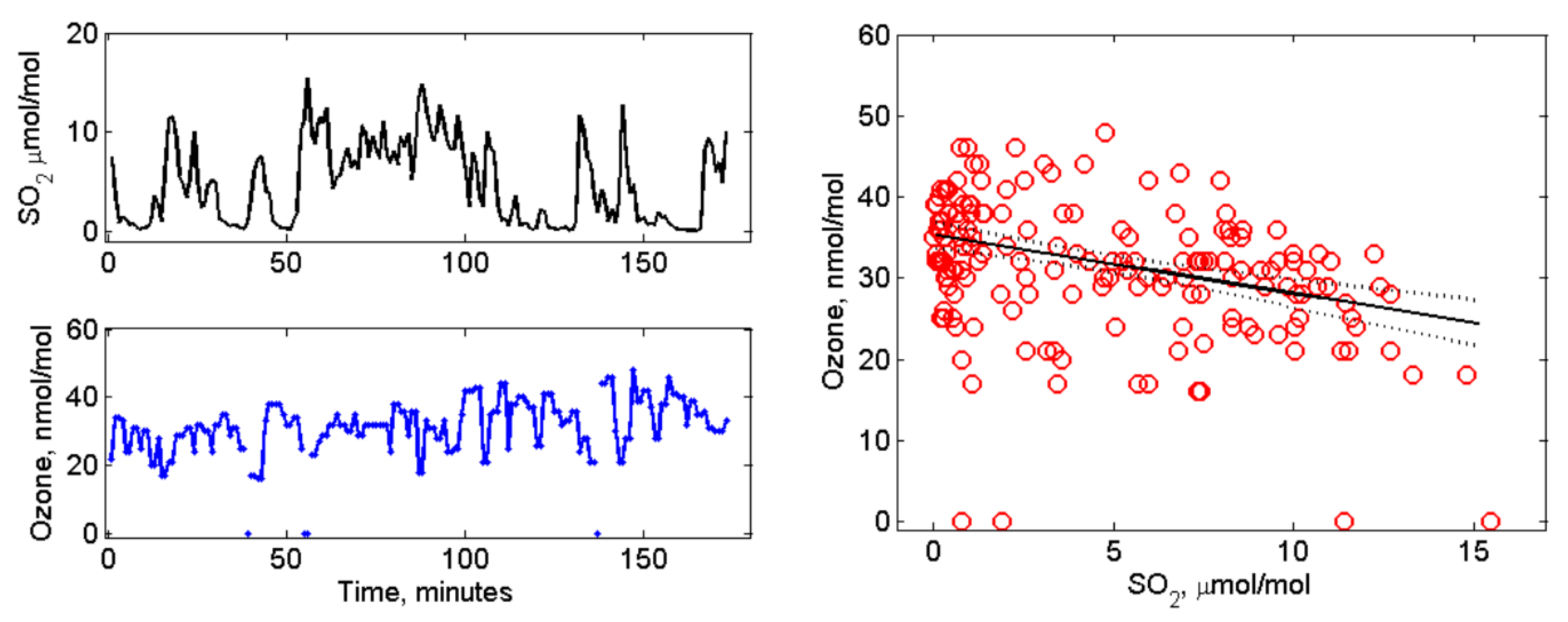
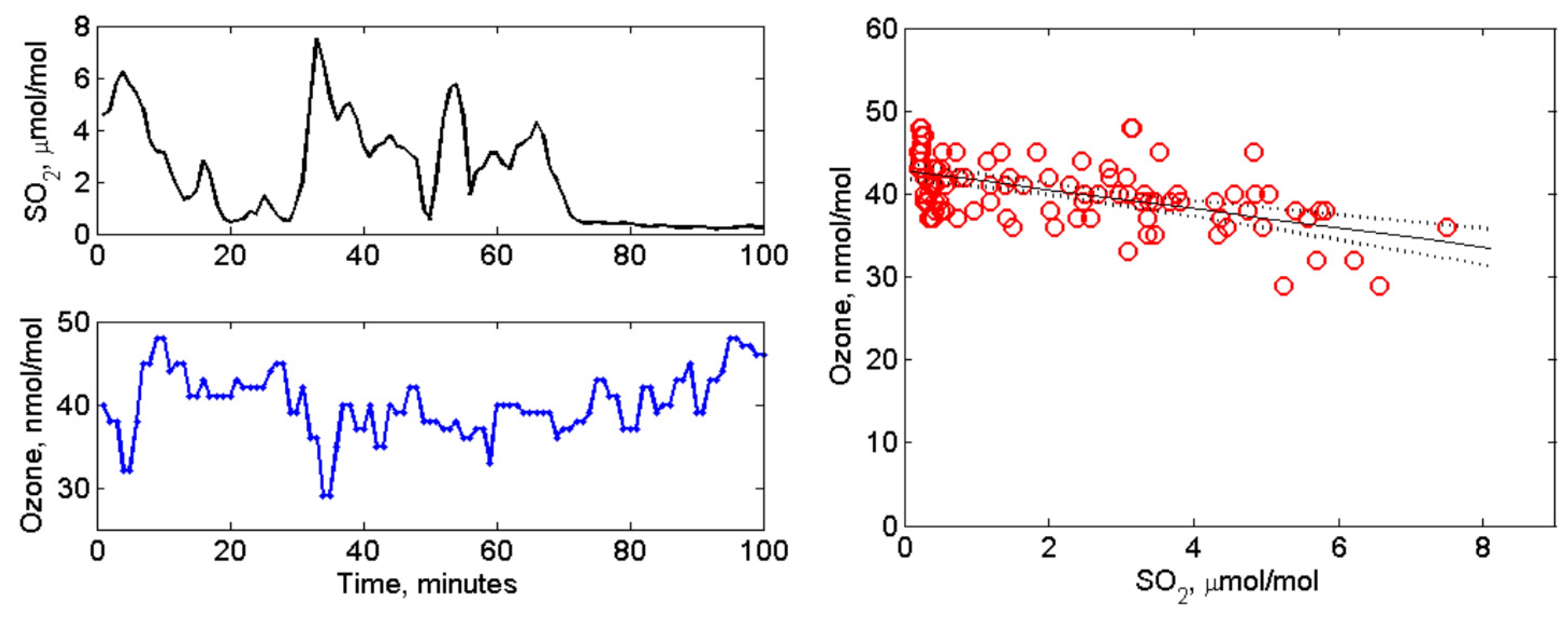
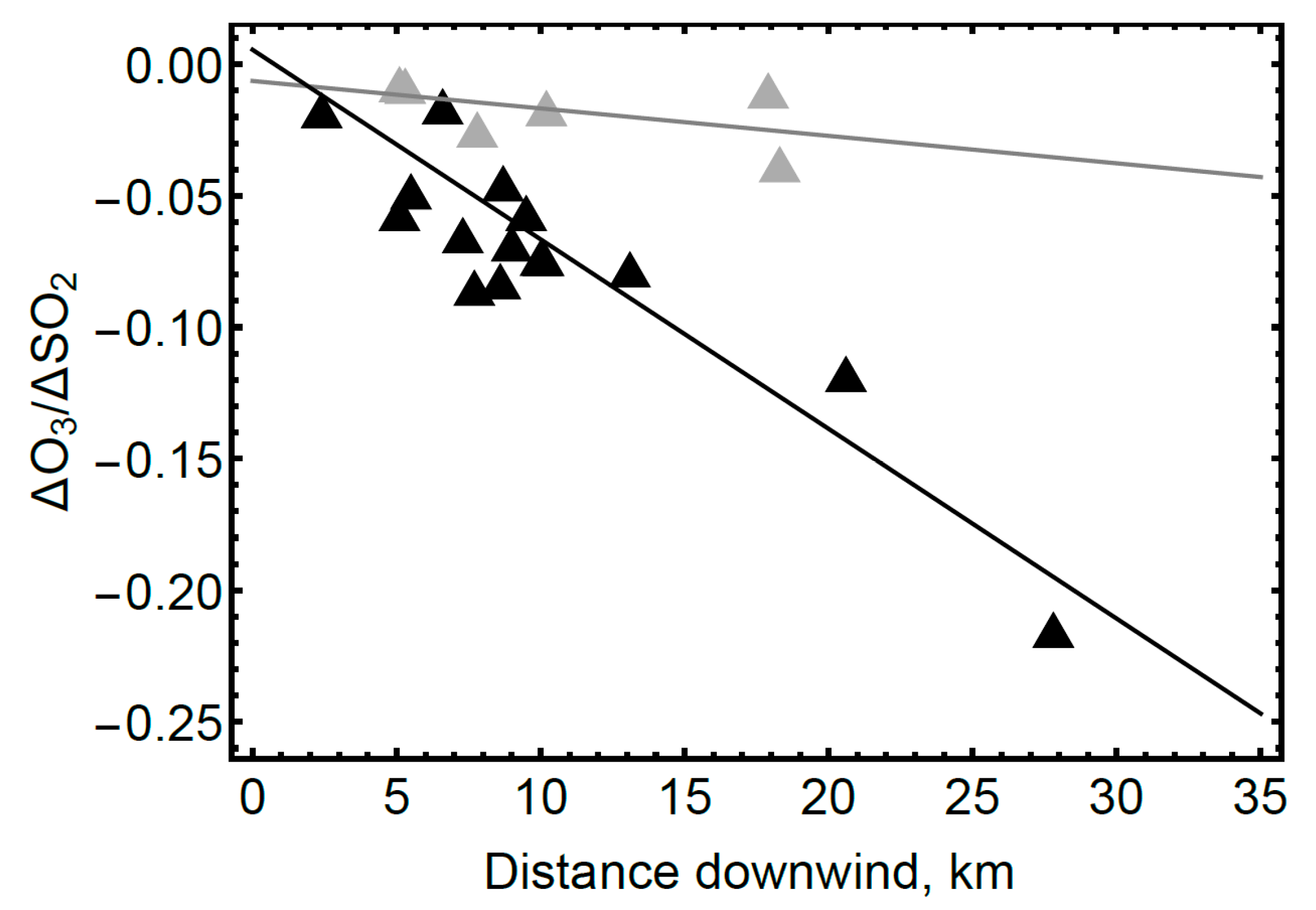
4.2. Volcanic SO2, H2S, and Ozone Measured in the Near-Downwind Plume from Pu‘u ‘Ō‘ō
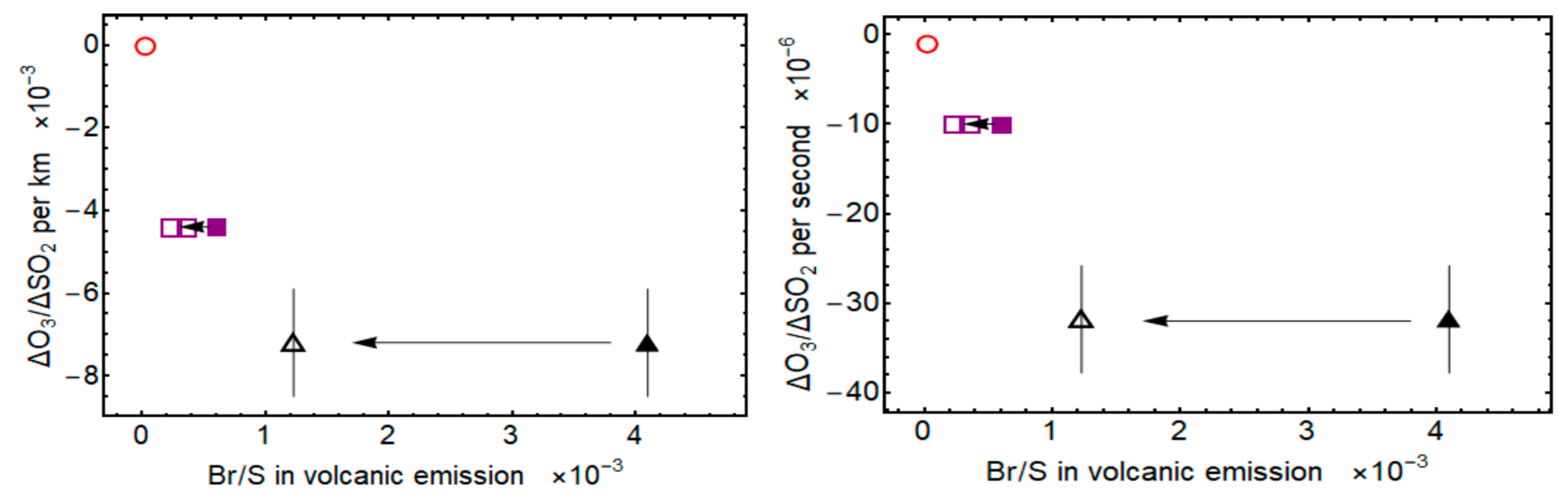
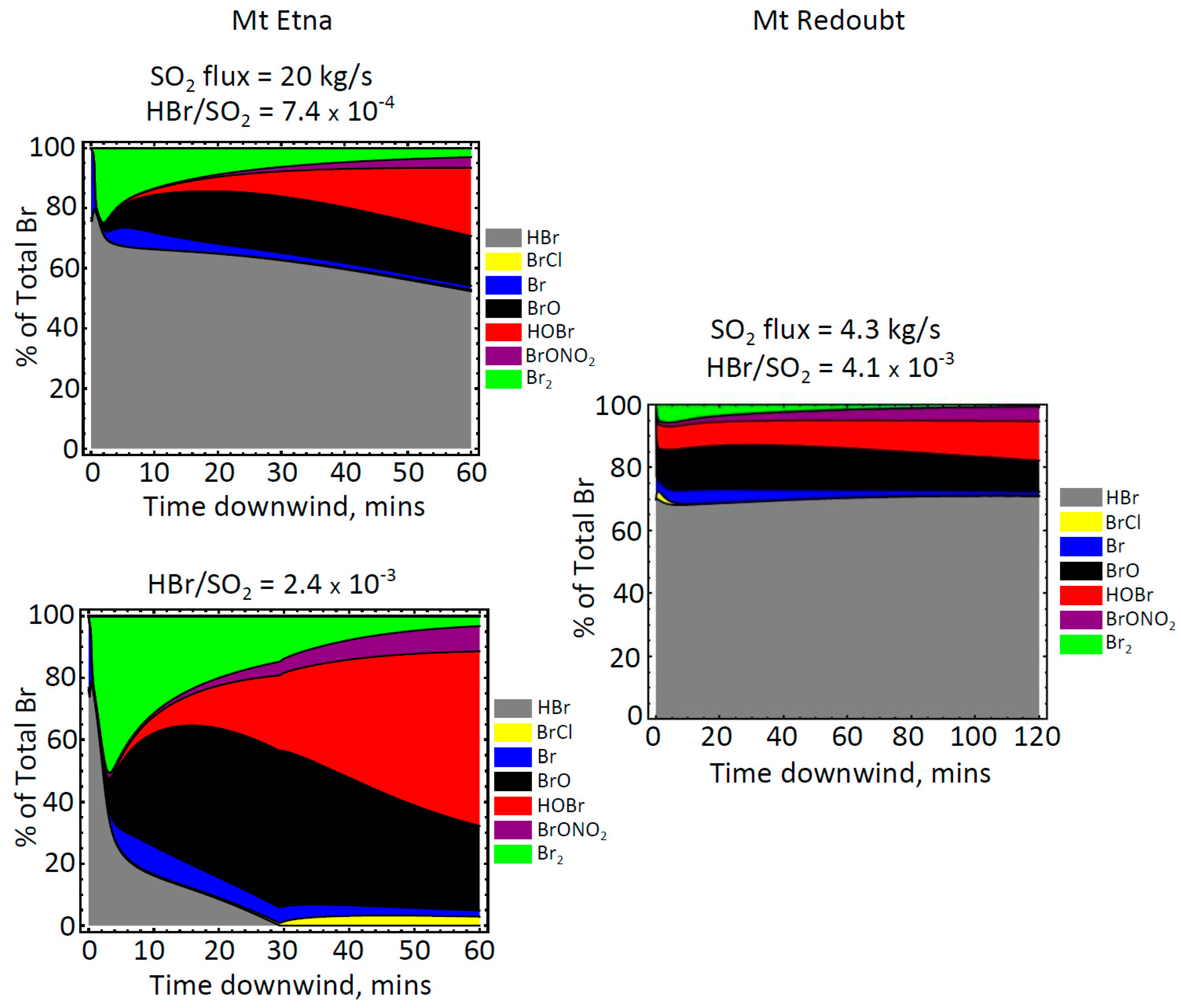
5. Discussion and Conclusion: Ozone Depletion from Halogen-Poor to Halogen-Rich Volcanic Plumes
Acknowledgments
Conflicts of Interest
References
- Carn, S.A.; Fioletov, V.E.; McLinden, C.A.; Li, C.; Krotkov, N.A. A decade of global volcanic SO2 emissions measured from space. Sci. Rep. 2017, 44095. [Google Scholar] [CrossRef] [PubMed]
- Stoffell, M.; Khodri, M.; Corona, C.; Guillet, S.; Poulain, V.; Bekki, S.; Guiot, J.; Luckman, B.H.; Oppenheimer, C.; Lebas, N.; et al. Estimates of volcanic-induced cooling in the Northern Hemisphere over the past 1500 years. Nat. Geosci. 2015, 8, 784–788. [Google Scholar] [CrossRef]
- Sellitto, P.; Zanetel, C.; di Sarra, A.; Salerno, G.; Tapparo, A.; Meloni, D.; Pace, G.; Caltabiano, T.; Briole, P.; Legras, B. The impact of Mount Etna sulfur emissions on the atmospheric composition and aerosol properties in the central Mediterranean: A statistical analysis over the period 2000–2013 based on observations and Lagrangian modelling. Atmos. Environ. 2017, 148, 77–88. [Google Scholar] [CrossRef]
- Berthet, G.; Jégou, F.; Catoire, V.; Krysztofiak, G.; Renard, J.B.; Bourassa, A.E.; Degenstein, D.A.; Brogniez, C.; Dorf, M.; Kreycy, S.; et al. Impact of a moderate volcanic eruption on chemistry in the lower stratosphere: Balloon-borne observations and model calculations. Atmos. Chem. Phys. 2017, 17, 2229–2253. [Google Scholar] [CrossRef]
- Ivy, D.J.; Solomon, S.; Kinnison, D.; Mills, M.J.; Schmidt, A.; Neely, R.R., III. The influence of the Calbuco eruption on the 2015 Antarctic ozone hole in a fully coupled chemistry-climate model. Geophys. Res. Lett. 2017, 44, 2556–2561. [Google Scholar] [CrossRef]
- Aiuppa, A.; Baker, D.R.; Webster, J.D. Halogens in volcanic systems. Chem. Geol. 2009, 263, 1–18. [Google Scholar] [CrossRef]
- Carn, S.; Clarisse, L.; Prata, A.J. Multi-decadal satellite measurements of global volcanic degassing. J. Volcanol. Geotherm. Res. 2016, 311, 99–134. [Google Scholar] [CrossRef]
- Theys, N.; Van Roozendael, M.; Dils, B.; Hendrick, F.; Hao, N.; De Mazière, M. First satellite detection of volcanic bromine monoxide emission after the Kasatochi eruption. Geophys. Res. Lett. 2009, 36. [Google Scholar] [CrossRef]
- Theys, N.; De Smedt, I.; Van Roozendael, M.; Froidevaux, L.; Clarisse, L.; Hendrick, F. First satellite detection of volcanic OClO after the eruption of Puyehue-Cordón Caulle. Geophys. Res. Lett. 2014, 41, 667–672. [Google Scholar] [CrossRef]
- Schönhardt, A.; Richter, A.; Theys, N.; Burrows, J.P. Space-based observation of volcanic iodine monoxide. Atmos. Chem. Phys. 2017, 17, 4857–4870. [Google Scholar] [CrossRef]
- Rose, W.I.; Millard, G.A.; Mather, T.A.; Hunton, D.E.; Anderson, B.; Oppenheimer, C.; Thornton, B.F.; Gerlach, T.; Viggiano, A.A.; Kondo, Y.; et al. Atmospheric chemistry of a 33–34 hour old volcanic cloud from Hekla Volcano (Iceland): Insights from direct sampling and the application of chemical box modeling. J. Geophys. Res. 2006, 111, D20206. [Google Scholar] [CrossRef]
- Millard, G.A.; Mather, T.A.; Pyle, D.M.; Rose, W.I.; Thornton, B. Halogen emissions from a small volcanic eruption: Modeling the peak concentrations, dispersion, and volcanically induced ozone loss in the stratosphere. Geophys. Res. Lett. 2006, 33, L19815. [Google Scholar] [CrossRef]
- Lurton, T.; Jégou, F.; Berthet, G.; Renard, J.-B.; Clarisse, L.; Schmidt, A.; Brogniez, C.; Roberts, T. Model simulations of the chemical and aerosol microphysical evolution of the Sarychev Peak 2009 eruption cloud compared to in-situ and satellite observations. Atmos. Chem. Phys. Discuss. 2017. [Google Scholar] [CrossRef]
- Bobrowski, N.; Honninger, G.; Galle, B.; Plat, U. Detection of bromine monoxide in a volcanic plume. Nature 2003, 423, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Boichu, M.; Oppenheimer, C.; Roberts, T.J.; Tsanev, V.; Kyle, P. On bromine, nitrogen oxides and ozone depletion in the tropospheric plume of Erebus volcano (Antarctica). Atmos. Environ. 2011, 45, 3856–3866. [Google Scholar] [CrossRef]
- Lübcke, P.; Bobrowski, N.; Arellano, S.; Galle, B.; Garzón, G.; Vogel, L.; Platt, U. BrO/SO2 molar ratios from scanning DOAS measurements in the NOVAC network. Solid Earth 2014, 5, 409–424. [Google Scholar] [CrossRef]
- Hörmann, C.; Sihler, H.; Bobrowski, N.; Beirle, S.; de Vries, M.P.; Platt, U.; Wagner, T. Systematic investigation of bromine monoxide in volcanic plumes from space by using the GOME-2 instrument. Atmos. Chem. Phys. 2013, 13, 4749–4781. [Google Scholar] [CrossRef]
- Bobrowski, N.; von Glasow, R.; Aiuppa, A.; Inguaggiato, S.; Louban, I.; Ibrahim, O.W.; Platt, U. Reactive halogen chemistry in volcanic plumes. J. Geophys. Res. 2007, 112, D06311. [Google Scholar] [CrossRef]
- General, S.; Bobrowski, N.; Pöhler, D.; Weber, K.; Fischer, C.; Platt, U. Airborne I-DOAS measurements at Mt. Etna: BrO and OClO evolution in the plume. J. Volcanol. Geotherm. Res. 2015, 300, 175–186. [Google Scholar] [CrossRef]
- Gliß, J.; Bobrowski, N.; Vogel, L.; Pöhler, D.; Platt, U. OClO and BrO observations in the volcanic plume of Mt. Etna—Implications on the chemistry of chlorine and bromine species in volcanic plumes. Atmos. Chem. Phys. 2015, 15, 5659–5681. [Google Scholar] [CrossRef]
- Donovan, A.; Tsanev, V.; Oppenheimer, C.; Edmonds, M. Reactive halogens (BrO and OClO) detected in the plume of Soufrière Hills Volcano during an eruption hiatus. Geochem. Geophys. Geosyst. 2014, 15, 3346–3363. [Google Scholar] [CrossRef]
- Oppenheimer, C.; Tsanev, V.I.; Braban, C.F.; Cox, R.A.; Adams, J.W.; Aiuppa, A.; Bobrowski, N.; Delmelle, P.; Barclay, J.; McGonigle, A.J. BrO fomation in volcanic plumes. Geochim. Cosmochim. Acta 2006, 70, 2935–2941. [Google Scholar] [CrossRef]
- Kelly, P.J.; Kern, C.; Roberts, T.J.; Lopez, T.; Werner, C.; Aiuppa, A. Rapid chemical evolution of tropospheric volcanic emissions from Redoubt Volcano, Alaska, based on observations of ozone and halogen-containing gases. J. Volcanol. Geotherm. Res. 2013, 259, 317–333. [Google Scholar] [CrossRef]
- Surl, L.; Donohoue, D.; Aiuppa, A.; Bobrowski, N.; von Glasow, R. Quantification of the depletion of ozone in the plume of Mount Etna. Atmos. Chem. Phys. 2015, 15, 2613–2628. [Google Scholar] [CrossRef]
- Roberts, T.J.; Braban, C.F.; Martin, R.S.; Oppenheimer, C.; Adams, J.W.; Cox, R.A.; Jones, R.L.; Griffiths, P.T. Modelling reactive halogen formation and ozone depletion in volcanic plumes. Chem. Geol. 2009, 263, 151–163. [Google Scholar] [CrossRef]
- Von Glasow, R. Atmospheric chemistry in volcanic plumes. Proc. Natl. Acad. Sci. USA 2010, 107, 6594–6599. [Google Scholar] [CrossRef] [PubMed]
- Jourdain, L.; Roberts, T.J.; Pirre, M.; Josse, B. Modeling the reactive halogen plume from Ambrym volcano and its impact on the troposphere with the CCATT-BRAMS mesoscale model. Atmos. Chem. Phys. 2016, 16, 12099–12125. [Google Scholar] [CrossRef]
- Von Glasow, R.; Bobrowski, N.; Kern, C. The effects of volcanic eruptions on atmospheric chemistry. Chem. Geol. 2009, 263, 131–142. [Google Scholar] [CrossRef]
- Roberts, T.J.; Martin, R.S.; Jourdain, L. Reactive halogen chemistry in Mt Etna’s volcanic plume: The influence of total Br, high temperature processing, aerosol loading and plume-air mixing (volcanic emissions flux). Atmos. Chem. Phys. 2014, 14, 11201–11219. [Google Scholar] [CrossRef]
- Roberts, T.J.; Vignelles, D.; Liuzzo, M.; Giudice, G.; Aiuppa, A.; Coltelli, M.; Salerno, G.; Chartier, M.; Couté, B.; Berthet, G.; et al. The primary volcanic aerosol emission from Mt Etna: Size-resolved particles with SO2 and role in plume reactive halogen chemistry. Geochim. Cosmochim. Acta 2018, 222, 74–93. [Google Scholar] [CrossRef]
- Flentje, H.; Claude, H.; Elste, T.; Gilge, S.; Köhler, U.; Plass-Dülmer, C.; Steinbrecht, W.; Thomas, W.; Werner, A.; Fricke, W. The Eyjafjallajökull eruption in April 2010—Detection of volcanic plume using in-situ measurements, ozone sondes and lidar-ceilometer profiles. Atmos. Chem. Phys. 2010, 10, 10085–10092. [Google Scholar] [CrossRef]
- Stith, J.L.; Hobbs, P.V.; Radke, L.F. Airborne particle and gas measurements in the emissions from six volcanoes. J. Geophys. Res. 1978, 83, 4009–4017. [Google Scholar] [CrossRef]
- Fruchter, J.S.; Robertson, D.E.; Evans, J.C.; Oslen, K.B.; Lepel, E.A.; Laul, J.C.; Abel, K.H.; Sanders, R.W.; Jackson, P.O.; Wogman, N.S.; et al. Mount St. Helens ash from the 18 May 1980 eruption: Chemical, physical, mineralogical and biological properties. Science 1980, 209, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, P.V.; Tuell, J.P.; Hegg, D.A.; Radke, L.F.; Eltgroth, M.W. Particles and gases in the emissions from the 1980–1981 volcanic eruptions of Mt. St. Helens. J. Geophys. Res. 1982, 87, 11062–11086. [Google Scholar] [CrossRef]
- Vance, A.; McGonigle, A.J.S.; Aiuppa, A.; Stith, J.L.; Turnbull, K.; von Glasow, R. Ozone depletion in tropospheric volcanic plumes. Geophys. Res. Lett. 2010, 37, L22802. [Google Scholar] [CrossRef]
- Schumann, U.; Weinzierl, B.; Reitebuch, O.; Schlager, H.; Miniki, A.; Forster, C.; Baumann, R.; Saile, T.; Graf, K.; Mannstein, H.; et al. Airborne observations of the Eyjafjalla volcano ash cloud over Europe during air space closure in April and May 2010. Atmos. Chem. Phys. 2011, 11, 2245–2279. [Google Scholar] [CrossRef]
- Oppenheimer, C.; Kyle, P.; Eisele, F.; Crawford, J.; Huey, G.; Tanner, D.; Saewung, K.; Mauldin, L.; Blake, D.; Beyersdorf, A.; et al. Atmospheric chemistry of an Antarctic volcanic plume. J. Geophys. Res. 2010, 115, D04303. [Google Scholar] [CrossRef]
- Aiuppa, A.; Federico, C.; Giudice, G.; Gurrieri, S. Chemical mapping of a fumarolic field: La Fossa Crater, Vulcano Island (Aeolian Islands, Italy). Geophys. Res. Lett. 2005, 32, L13309. [Google Scholar] [CrossRef]
- Gerlach, T.M. Volcanic sources of tropospheric ozone-depleting trace gases. Geochem. Geophys. Geosyst. 2004, 5, Q09007. [Google Scholar] [CrossRef]
- Mather, T.A.; Witt, M.L.I.; Pyle, D.M.; Quayle, B.M.; Aiuppa, A.; Bagnato, E.; Martin, R.S.; Sims, K.W.W.; Edmonds, M.; Sutton, A.J.; et al. Halogens and trace metal emissions from the ongoing 2008 summit eruption of Kīlauea volcano, Hawai`I. Geochim. Cosmochim. Acta 2012, 83, 292–323. [Google Scholar] [CrossRef]
- Aeroqual Limited. Datasheets for Aeroqual Series 500 Portable Ozone Instrument. Available online: https://www.aeroqual.com/product/series-500-portable-ozone-gas-monitor (accessed on 24 January 2018).
- Roberts, T.J.; Saffell, J.R.; Dawson, D.H.; Oppenheimer, C.; Lurton, T. Electrochemical sensors applied to pollution monitoring: Measurement error and gas ratio bias—A volcano plume case study. J. Volcanol. Geotherm. Res. 2014, 281, 85–96. [Google Scholar] [CrossRef]
- Roberts, T.J.; Braban, C.; Oppenheimer, C.; Martin, R.S.; Saffell, J.R.; Dawson, D.; Freshwater, R.A.; Griffiths, P.T.; Jones, R.L. Electrochemical sensing of volcanic plumes. Chem. Geol. 2012, 332–333, 74–91. [Google Scholar] [CrossRef]
- Alphasense Sensor Technology Company. Datasheets for Alphasense SO2-AF and H2S-A1 Sensors. Available online: http://www.alphasense.com/index.php/safety/downloads/ (accessed on 24 January 2018).
- Rüdiger, J.; Bobrowski, N.; Liotta, M.; Hoffman, T. Development and application of a sampling method for the determination of reactive halogen species in volcanic gas emissions. Anal. Bioanal. Chem. 2017, 409, 5975–5985. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Gas | Mixing Ratio (ppmv) | Sensor Reading (ppmv) | Sensitivity (%) |
|---|---|---|---|
| Ammonia (NH3) | 25 | −0.02 | −0.08 |
| Butane | 1000 | 0 | 0 |
| Carbon monoxide (CO) | 100 | 0 | 0 |
| Carbon dioxide (CO2) | 1000 | 0 | 0 |
| Chlorine (Cl2) | 0.5 | 0.2 | −40 |
| Ethanol | 20 | −0.02 | −0.1 |
| Ethyl acetate | 100 | −0.02 | −0.02 |
| Heptane | 100 | 0 | 0 |
| Hydrogen sulfide (H2S) | 4 | −0.1 | −2.5 |
| Isopropanol | 20 | 0 | 0 |
| Methane (CH4) | 5000 | 0 | 0 |
| Nitrogen dioxide (NO2) | 0.5 | 0.1 | 20 |
| Ozone (O3) | 0.3 | 0.3 | 100 |
| Perchloroethylene | 50 | 0 | 0 |
| Propane | 5000 | 0 | 0 |
| Sulfur dioxide (SO2) | 10 | 0 | 0 |
| Toluene | 50 | 0 | 0 |
| Volcano | Br/S (mmol/mol) | Distance (km) | Wind Speed * m/s | Time (min) | ΔO3/ΔSO2 Per km (km−1) × 10−3 | ΔO3/ΔSO2 Per Second (s−1) × 10−5 | Reference |
|---|---|---|---|---|---|---|---|
| Mt Redoubt 2009 eruption | 4.1 | 2.4–27.8 | 4.4 | 9–105 | −7.2 ± 1.3 | −3.2 ± 0.6 | [23] (Figure 5 this study) |
| Mt Etna passive degassing 2012 | 0.613 | 0.15–0.40 | ~2.2 | 1–4 | (−4.4 ± 0.3) | −1.02 ± 0.07 | [24] |
| Kīlauea plumes 2007–2008 | 0.029 ± 0.025 | ~10 | ~5 | ~30 | Up to −0.03 | (Up to 0.02) | This study (ΔO3/ΔSO2) [40] (2012) (Br/S) |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roberts, T.J. Ozone Depletion in Tropospheric Volcanic Plumes: From Halogen-Poor to Halogen-Rich Emissions. Geosciences 2018, 8, 68. https://doi.org/10.3390/geosciences8020068
Roberts TJ. Ozone Depletion in Tropospheric Volcanic Plumes: From Halogen-Poor to Halogen-Rich Emissions. Geosciences. 2018; 8(2):68. https://doi.org/10.3390/geosciences8020068
Chicago/Turabian StyleRoberts, Tjarda J. 2018. "Ozone Depletion in Tropospheric Volcanic Plumes: From Halogen-Poor to Halogen-Rich Emissions" Geosciences 8, no. 2: 68. https://doi.org/10.3390/geosciences8020068
APA StyleRoberts, T. J. (2018). Ozone Depletion in Tropospheric Volcanic Plumes: From Halogen-Poor to Halogen-Rich Emissions. Geosciences, 8(2), 68. https://doi.org/10.3390/geosciences8020068