

An Attempted Correlation Between the Fecal Microbial Community of Chinese Forest Musk Deer (Moschus berezovskii) and Differences in Musk Production and Quality
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Animals and Sample Collection
2.2. Musk Morphological Characteristics Assessment
2.3. DNA Extraction and PCR Amplification
2.4. Illumina Sequencing
2.5. Amplicon Sequence Processing and Analysis
2.6. Statistical Analysis
2.6.1. Diversity Analysis
2.6.2. Fecal Microbiota Health Index (GMHI) and Microbial Dysbiosis Index (MDI) Assessment
2.6.3. Community Difference Analysis
2.6.4. Correlation Heatmap Construction
2.6.5. Prediction of the Functional Profiles of the Microbial Communities
3. Results
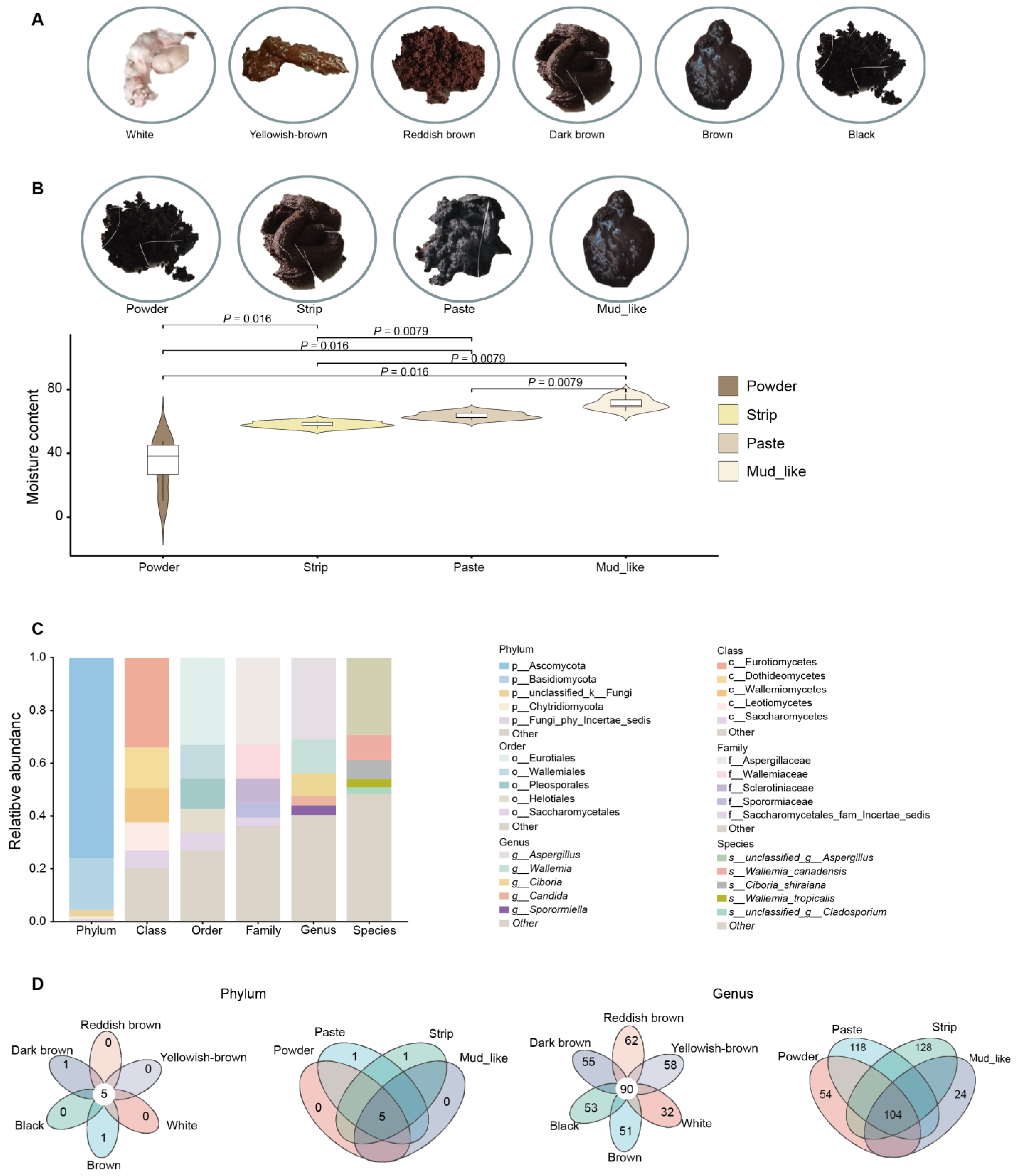
3.1. Characteristics and Grouping of Musk Quality
3.2. Statistics of Sequencing Data
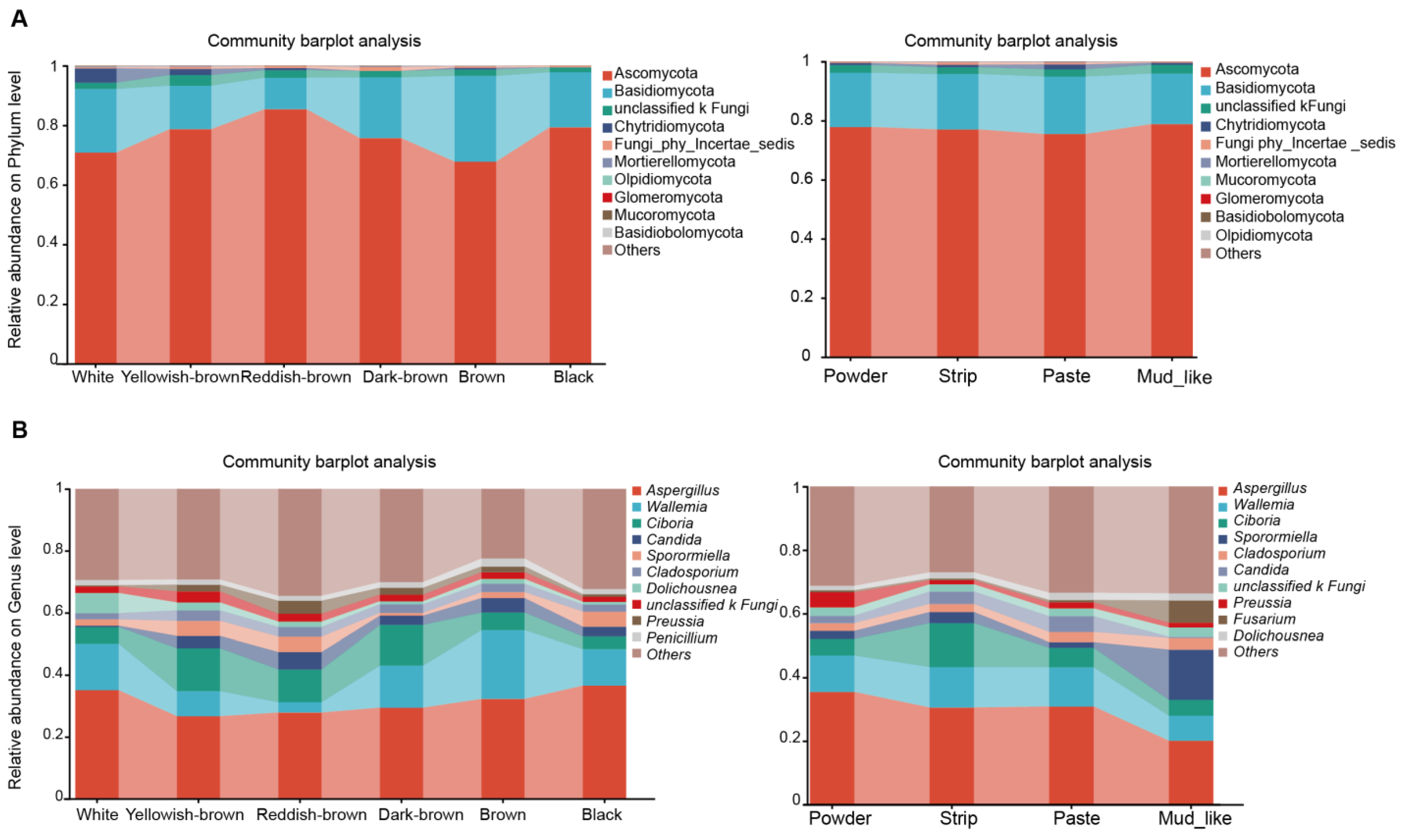
3.3. Fungal Content in Different Musk Color and Moisture Content Groups
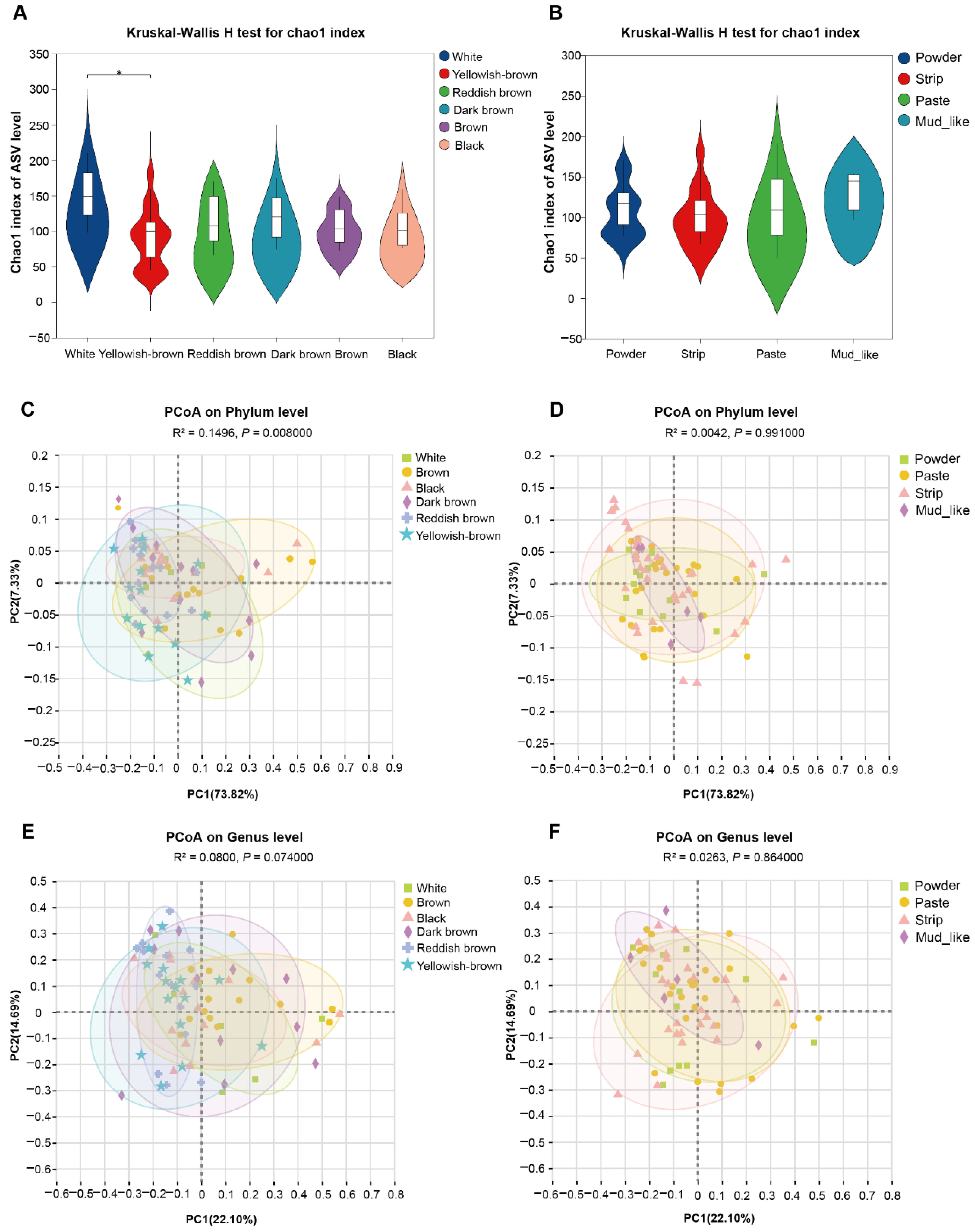
3.4. Diversity of Fecal Fungal Communities in Musk Deer with Different Musk Quality Groups
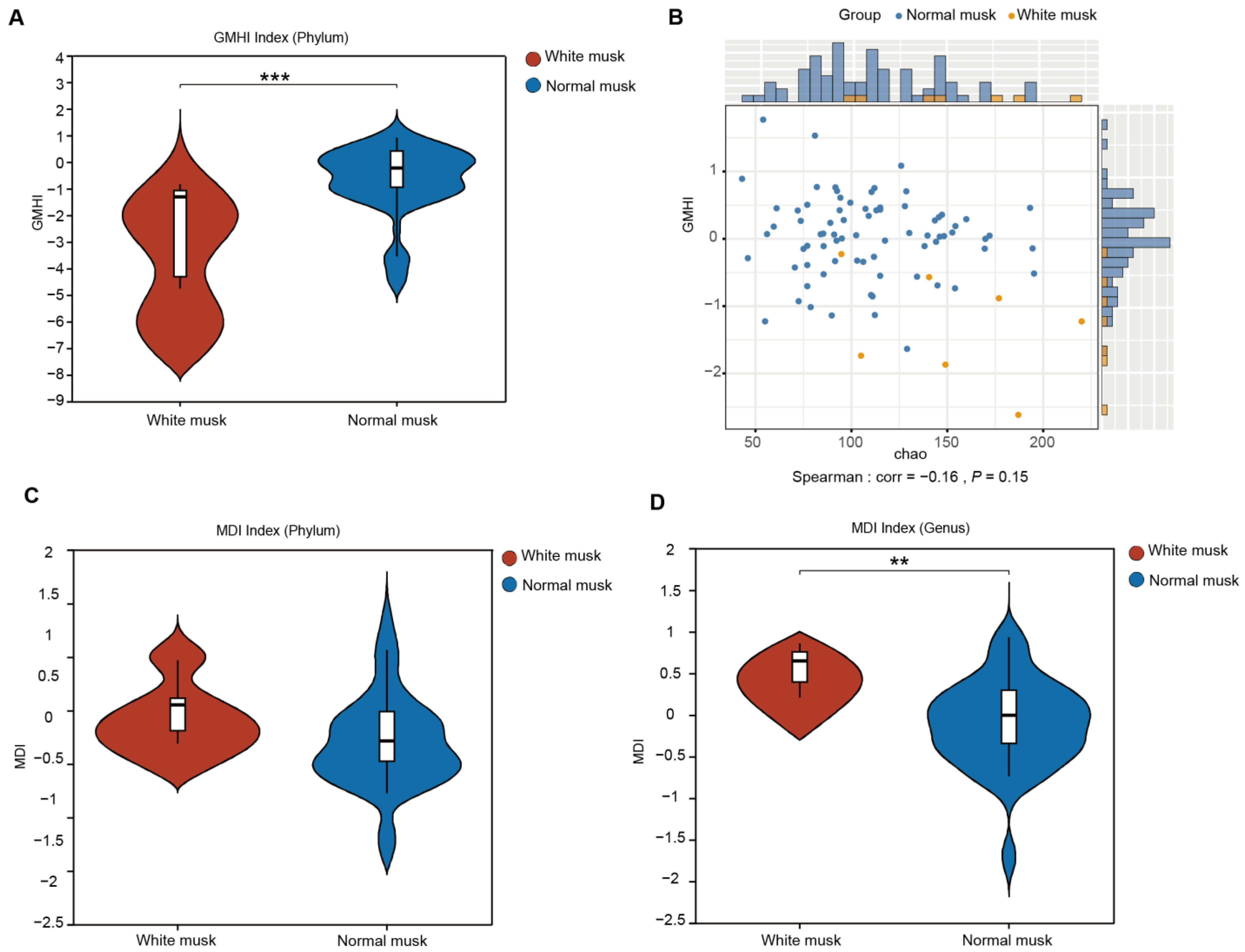
3.5. Forest Musk Deer Producing Normal Musk Have More Stable Fecal Fungal Communities
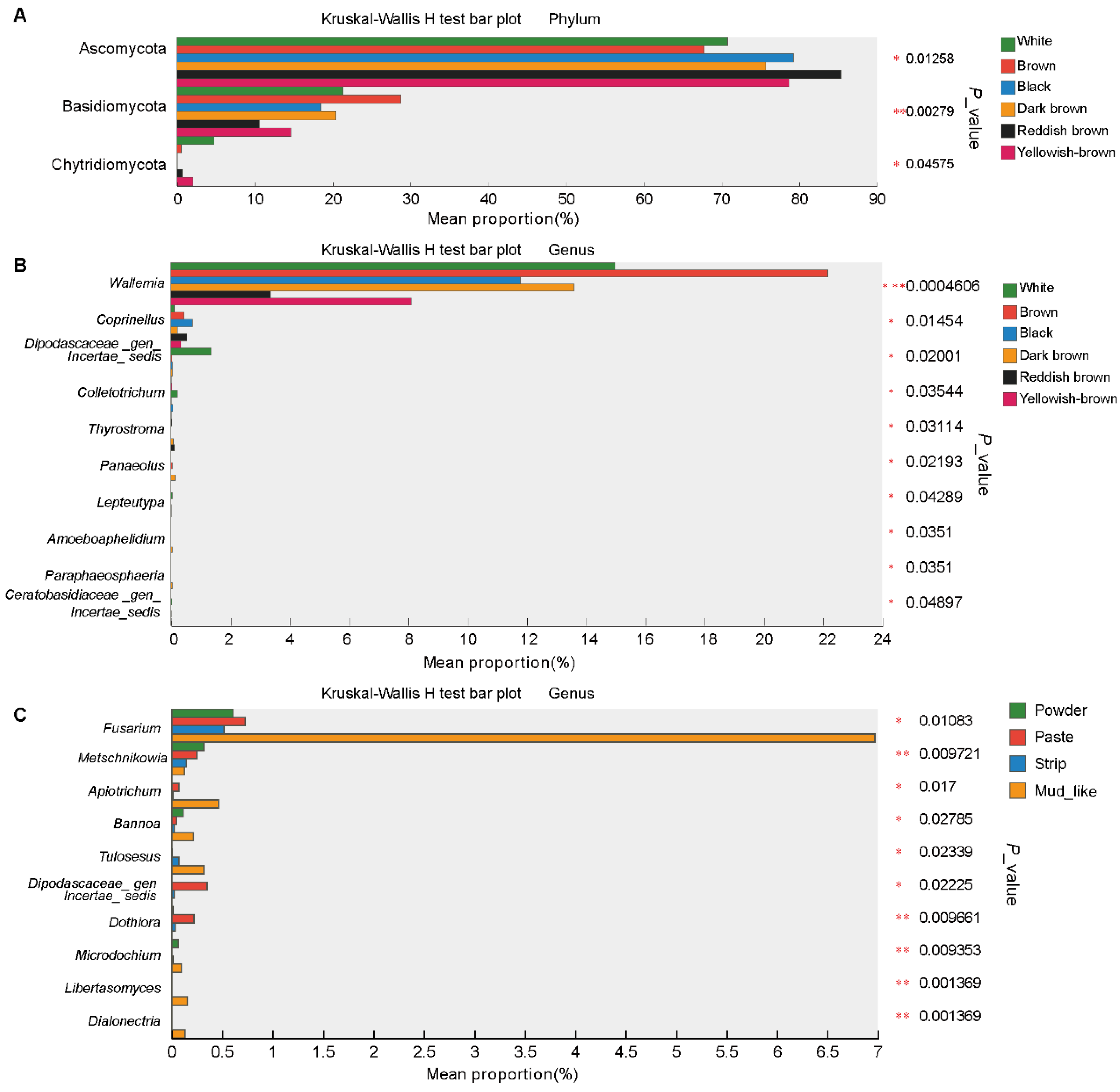
3.6. Differential Fungi Among the Different Musk Colors and Moisture Content Groups
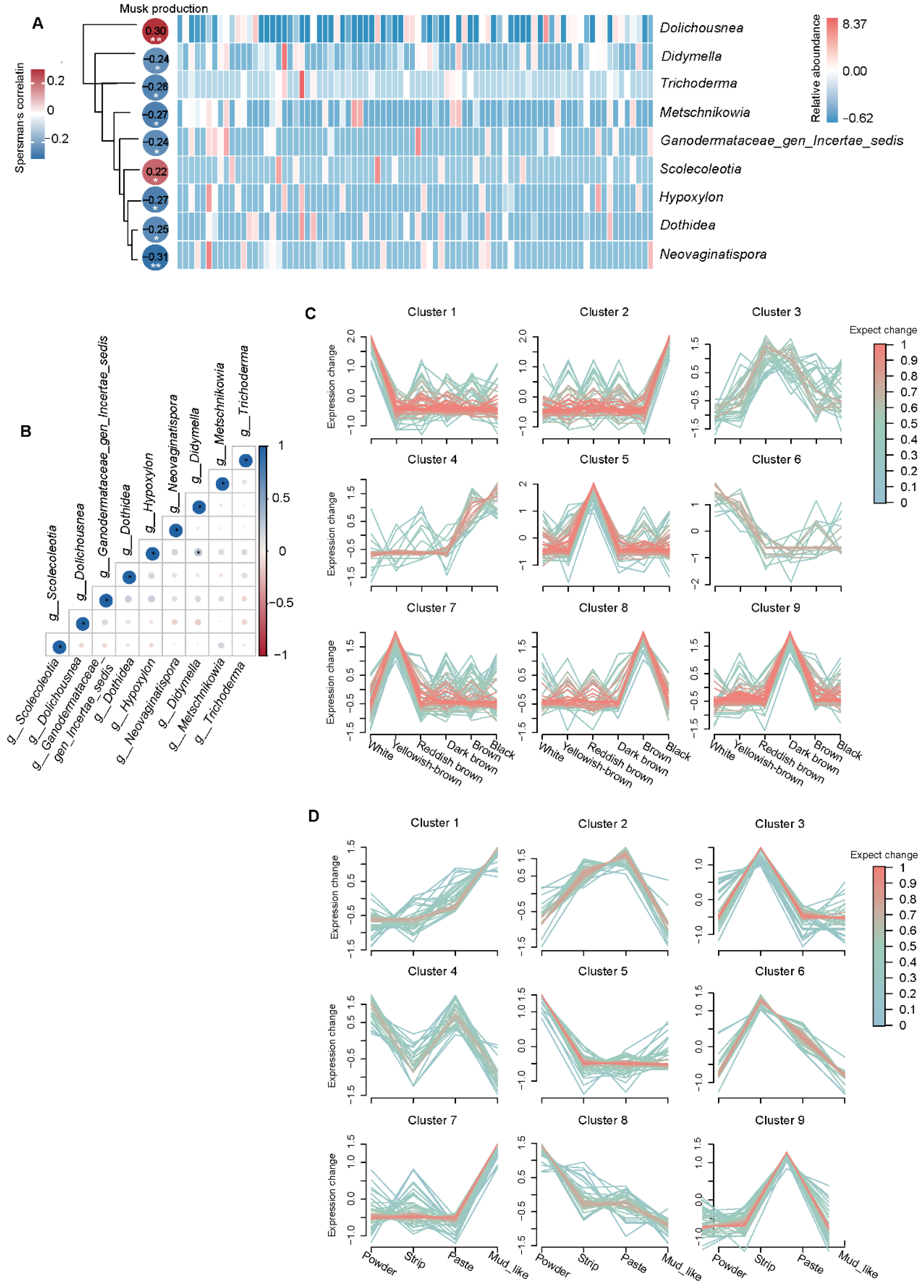
3.7. Relationships Between Musk Production and Fecal Fungi
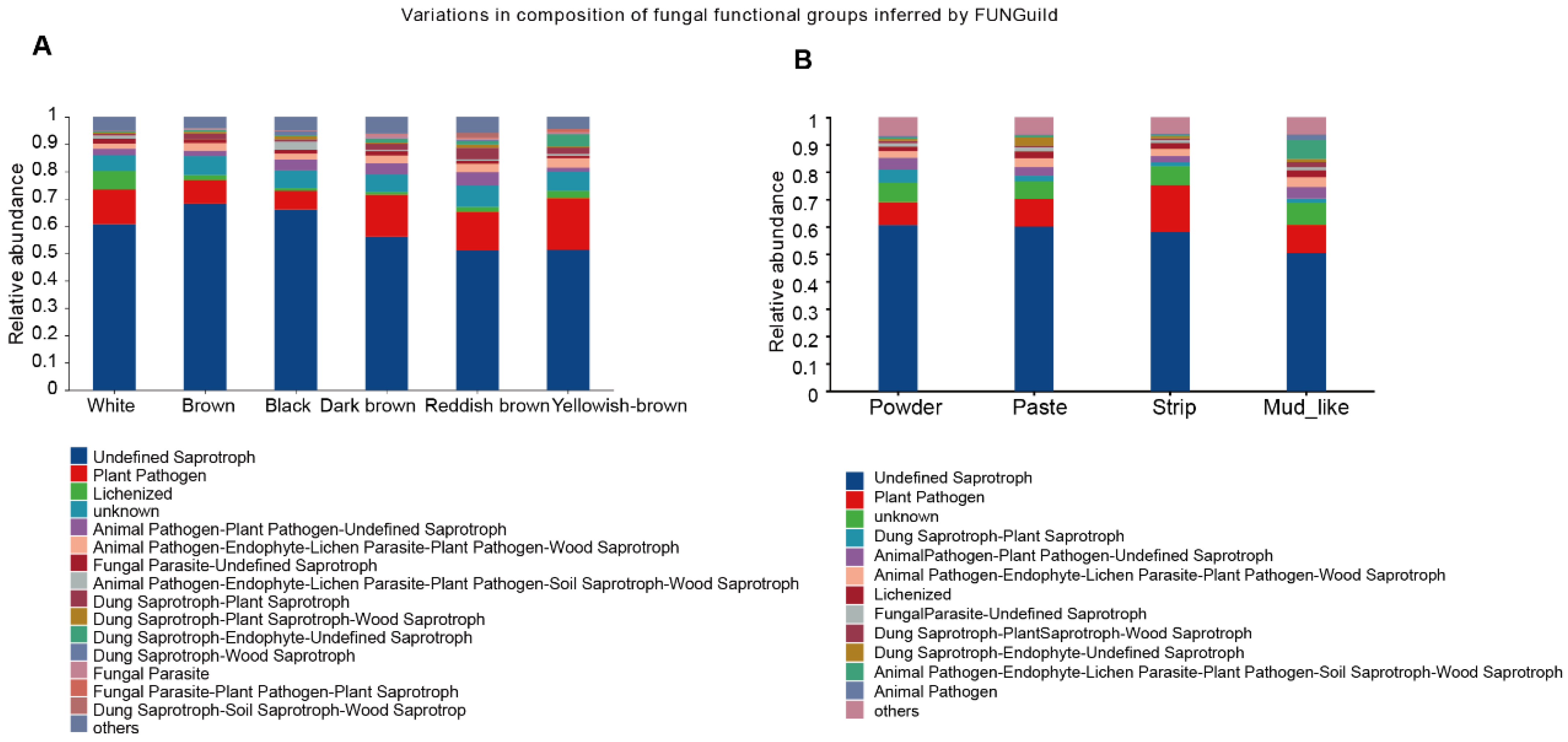
3.8. Predicted Functions of the Fecal Fungal Community of Musk Deer
4. Discussion
4.1. Dominant Fungal Communities in Musk
4.2. Pathogenic Fungi Related to Abnormal Musk
4.3. Beneficial Fungi Facilitating Musk Production
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feng, H.; Feng, C.; Huang, Y.; Tang, J. Structure of mitochondrial DNA control region and genetic diversity of Moschus berezovskii populations in Shaanxi Province. Genet. Mol. Res. 2016, 15, gmr.15027578. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Wang, L.; Cao, F.; Ma, J.; Tang, J.; Feng, C.; Su, Z. Forest musk deer (Moschus berezovskii) in China: Research and protection. J. Vertebr. Biol. 2023, 72, 22067. [Google Scholar] [CrossRef]
- Peng, H.; Liu, S.; Zou, F.; Zeng, B.; Yue, B. Genetic diversity of captive forest musk deer (Moschus berezovskii) inferred from the mitochondrial DNA control region. Anim. Genet. 2009, 40, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Meng, X.; Xia, L.; Feng, Z. Conservation status and causes of decline of musk deer (Moschus spp.) in China. Biol. Conserv. 2003, 109, 333–342. [Google Scholar] [CrossRef]
- Fan, M.; Zhang, M.; Shi, M.; Zhang, T.; Qi, L.; Yu, J.; Li, X.; Lin, S.; Huang, Z.; Yang, S.; et al. Sex hormones play roles in determining musk composition during the early stages of musk secretion by musk deer (Moschus berezovskii). Endocr. J. 2018, 65, 1111–1120. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, T.; Qi, L.; Yang, S.; Xu, S.; Cha, M.; Zhang, M.; Huang, Z.; Yu, J.; Hu, D.; et al. Microbiota Changes in the Musk Gland of Male Forest Musk Deer During Musk Maturation. Front. Microbiol. 2018, 9, 3048. [Google Scholar] [CrossRef]
- Li, D.; Chen, B.; Zhang, L.; Gaur, U.; Ma, T.; Jie, H.; Zhao, G.; Wu, N.; Xu, Z.; Xu, H.; et al. The musk chemical composition and microbiota of Chinese forest musk deer males. Sci. Rep. 2016, 6, 18975. [Google Scholar] [CrossRef]
- Luo, Y.; Yang, M.; Liao, Z.G.; Zhang, J.; Zhao, H.P. Research progresses on quality evaluation methods for musk and its alternative. Chin. J. Pharm. Anal. 2017, 37, 13–19. [Google Scholar]
- Jie, H.; Feng, X.-L.; Zhao, G.-J.; Zeng, D.-J.; Zhang, C.-L.; Chen, Q. Research progress on musk secretion mechanism of forest musk deer. Zhongguo Zhong Yao Za Zhi 2014, 39, 4522–4525. [Google Scholar]
- Zhang, T.; Jin, W.; Yang, S.; Li, Y.; Zhang, M.; Shi, M.; Guo, X.; Li, D.; Zhang, B.; Liu, S.; et al. Study of compositions of musks in different types secreted by forest musk deer (Moschus berezovskii). PLoS ONE 2021, 16, e0245677. [Google Scholar] [CrossRef]
- Li, Y.; Hu, X.; Yang, S.; Zhou, J.; Qi, L.; Sun, X.; Fan, M.; Xu, S.; Cha, M.; Zhang, M.; et al. Comparison Between the Fecal Bacterial Microbiota of Healthy and Diarrheic Captive Musk Deer. Front. Microbiol. 2018, 9, 300. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Cai, R.; Jin, X.; Shafer, A.B.A.; Hu, X.; Yang, S.; Li, Y.; Qi, L.; Liu, S.; Hu, D. Blood transcriptomics of captive forest musk deer (Moschus berezovskii) and possible associations with the immune response to abscesses. Sci. Rep. 2018, 8, 599. [Google Scholar] [CrossRef] [PubMed]
- You, N.; Zhuo, L.; Zhou, J.; Song, Y.; Shi, J. The Role of Intestinal Fungi and Its Metabolites in Chronic Liver Diseases. Gut Liver 2020, 14, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.-W.; Yu, Y.; Wang, P.; Wan, Y.; Gao, Y.; Zhang, C.-X. Advances in the research of intestinal fungi in Crohn’s disease. World J. Gastroenterol. 2024, 30, 4318–4323. [Google Scholar] [CrossRef]
- Jiang, F.; Song, P.; Wang, H.; Zhang, J.; Liu, D.; Cai, Z.; Gao, H.; Chi, X.; Zhang, T. Comparative analysis of gut microbial composition and potential functions in captive forest and alpine musk deer. Appl. Microbiol. Biotechnol. 2022, 106, 1325–1339. [Google Scholar] [CrossRef]
- Xu, Z.; Li, F.; Liu, Q.; Ma, T.; Feng, X.; Zhao, G.; Zeng, D.; Li, D.; Jie, H. Chemical composition and microbiota changes across musk secretion stages of forest musk deer. Front. Microbiol. 2024, 15, 1322316. [Google Scholar] [CrossRef]
- Zhao, G.; Ma, T.; Tang, W.; Li, D.; Mishra, S.K.; Xu, Z.; Wang, Q.; Jie, H. Gut Microbiome of Chinese Forest Musk Deer Examined across Gender and Age. Biomed Res. Int. 2019, 2019, 9291216. [Google Scholar] [CrossRef]
- Bo, X.; Chen, J.; Mu, J.; Dong, X.; Ren, Z.; Liu, J.; Wang, S. Quercetin promotes the secretion of musk by regulating the hormone level and microbial structure of forest musk deer. Integr. Zoöl. 2024, 19, 596–611. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, C.; Wang, Z.; Wang, Q.; Nielsen, J.; Dai, Z. Dual β-oxidation pathway and transcription factor engineering for methyl ketones production in Saccharomyces cerevisiae. Metab. Eng. 2022, 73, 225–234. [Google Scholar] [CrossRef]
- Gu, Y.; Wang, C.; Qin, X.; Zhou, B.; Liu, X.; Liu, T.; Xie, R.; Liu, J.; Wang, B.; Cao, H. Saccharomyces boulardii, a yeast probiotic, inhibits gut motility through upregulating intestinal serotonin transporter and modulating gut microbiota. Pharmacol. Res. 2022, 181, 106291. [Google Scholar] [CrossRef]
- Zhu, M.; Shang, B.; Zhou, F.; Yuan, Y.; Yin, F.; Cao, J.; Huang, J.; Wang, K.; Zeng, X.; Li, M.; et al. Polysaccharides of Aspergillus cristatus attenuate obesity by regulating gut microbiota and gut microbiota-related metabolites. Food Sci. Hum. Wellness 2024, 13, 1513–1530. [Google Scholar] [CrossRef]
- Pellon, A.; Begum, N.; Nasab, S.D.S.; Harzandi, A.; Shoaie, S.; Moyes, D.L. Role of Cellular Metabolism during Candida-Host Interactions. Pathogens 2022, 11, 184. [Google Scholar] [CrossRef] [PubMed]
- Alhussaini, M.S.; Mosle, M.A.; Alghonaim, M.I.; Al-Ghanaye, A.A.; Al-Yahya, A.A.; Hefny, H.M.; Saadabi, A.M. Characterization of Cladosporium Species by Internal Transcribed Spacer-PCR and Microsatellites-PCR. Pak. J. Biol. Sci. 2016, 19, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Mukabayire, O.; Boccolini, D.; Lochouarn, L.; Fontenille, D.; Besansky, N.J. Mitochondrial and ribosomal internal transcribed spacer (ITS2) diversity of the African malaria vector Anopheles funestus. Mol. Ecol. 1999, 8, 289–297. [Google Scholar] [CrossRef]
- Ahasan, S.A.; De Elías-Escribano, A.; Artigas, P.; Alam, M.Z.; Mondal, M.M.H.; Blair, D.; Chowdhury, E.H.; Bargues, M.D.; Mas-Coma, S. Wide variation of heterozygotic genotypes of recent fasciolid hybrids from livestock in Bangladesh assessed by rDNA internal transcribed spacer region sequencing and cloning. One Health 2023, 17, 100614. [Google Scholar] [CrossRef]
- Jagadeesh, D.; Kumar, M.P.; Chandrakanth, R.; Devaki, N. Molecular diversity of internal transcribed spacer among the monoconidial isolates of Magnaporthe oryzae isolated from rice in Southern Karnataka, India. J. Genet. Eng. Biotechnol. 2018, 16, 631–638. [Google Scholar] [CrossRef]
- Buehler, A.; Evanowski, R.; Martin, N.; Boor, K.; Wiedmann, M. Internal transcribed spacer (ITS) sequencing reveals considerable fungal diversity in dairy products. J. Dairy Sci. 2017, 100, 8814–8825. [Google Scholar] [CrossRef]
- Alanagreh, L.; Pegg, C.; Harikumar, A.; Buchheim, M. Assessing intragenomic variation of the internal transcribed spacer two: Adapting the Illumina metagenomics protocol. PLoS ONE 2017, 12, e0181491. [Google Scholar] [CrossRef]
- Jie, H.; Li, F.; Liu, Q.; Zheng, T.; Tan, H.; Feng, X.; Zhao, G.; Zeng, D.; Li, D.; Xu, Z.; et al. Elucidating metabolites and biosynthetic pathways during musk maturation: Insights from forest musk deer. Front. Pharmacol. 2025, 16, 1503138. [Google Scholar] [CrossRef]
- Nishijima, S.; Stankevic, E.; Aasmets, O.; Schmidt, T.S.; Nagata, N.; Keller, M.I.; Ferretti, P.; Juel, H.B.; Fullam, A.; Robbani, S.M.; et al. Fecal microbial load is a major determinant of gut microbiome variation and a confounder for disease associations. Cell 2025, 188, 222–236.e15. [Google Scholar] [CrossRef]
- Wei, G.; Lu, H.; Zhou, Z.; Xie, H.; Wang, A.; Nelson, K.; Zhao, L. The microbial community in the feces of the giant panda (Ailuropoda melanoleuca) as determined by PCR-TGGE profiling and clone library analysis. Microb. Ecol. 2007, 54, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.I.; Miletto, M.; Taylor, J.W.; Bruns, T.D. Dispersal in microbes: Fungi in indoor air are dominated by outdoor air and show dispersal limitation at short distances. ISME J. 2013, 7, 1262–1273. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Gupta, V.K.; Kim, M.; Bakshi, U.; Cunningham, K.Y.; Davis, J.M.; Lazaridis, K.N.; Nelson, H.; Chia, N.; Sung, J. A predictive index for health status using species-level gut microbiome profiling. Nat. Commun. 2020, 11, 4635. [Google Scholar] [CrossRef]
- Gunathilake, M.; Lee, J.; Choi, I.J.; Kim, Y.-I.; Yoon, J.; Sul, W.J.; Kim, J.F.; Kim, J. Alterations in Gastric Microbial Communities Are Associated with Risk of Gastric Cancer in a Korean Population: A Case-Control Study. Cancers 2020, 12, 2619. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Liu, S.; Xiao, Y.; Wang, X.; Guo, D.; Wang, Y.; Wang, Y. Effects of Microhabitat Temperature Variations on the Gut Microbiotas of Free-Living Hibernating Animals. Microbiol. Spectr. 2023, 11, e0043323. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, Y.; Xiao, H.; Liu, X. Research progress of gut mycobiota. Mycosystema 2023, 42, 26–37. [Google Scholar]
- Wang, J.; Zhu, N.; Su, X.; Gao, Y.; Yang, R. Gut-Microbiota-Derived Metabolites Maintain Gut and Systemic Immune Homeostasis. Cells 2023, 12, 793. [Google Scholar] [CrossRef]
- Azad, M.A.K.; Sarker, M.; Li, T.; Yin, J. Probiotic Species in the Modulation of Gut Microbiota: An Overview. Biomed. Res. Int. 2018, 2018, 9478630. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Warmbrunn, M.V.; Nieuwdorp, M.; Clément, K. Metabolism and Metabolic Disorders and the Microbiome: The Intestinal Microbiota Associated with Obesity, Lipid Metabolism, and Metabolic Health—Pathophysiology and Therapeutic Strategies. Gastroenterology 2021, 160, 573–599. [Google Scholar] [CrossRef]
- Schoeler, M.; Caesar, R. Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472. [Google Scholar] [CrossRef]
- Yin, X.-J.; Ji, S.-K.; Duan, C.-H.; Tian, P.-Z.; Ju, S.-S.; Yan, H.; Zhang, Y.-J.; Liu, Y.-Q. Dynamic change of fungal community in the gastrointestinal tract of growing lambs. J. Integr. Agric. 2022, 21, 3314–3328. [Google Scholar] [CrossRef]
- Solomon, K.V.; Haitjema, C.H.; Henske, J.K.; Gilmore, S.P.; Borges-Rivera, D.; Lipzen, A.; Brewer, H.M.; Purvine, S.O.; Wright, A.T.; Theodorou, M.K.; et al. Early-branching gut fungi possess a large, comprehensive array of biomass-degrading enzymes. Science 2016, 351, 1192–1195. [Google Scholar] [CrossRef]
- Gruninger, R.J.; Puniya, A.K.; Callaghan, T.M.; Edwards, J.E.; Youssef, N.; Dagar, S.S.; Fliegerova, K.; Griffith, G.W.; Forster, R.; Tsang, A.; et al. Anaerobic fungi (phylum Neocallimastigomycota): Advances in understanding their taxonomy, life cycle, ecology, role and biotechnological potential. FEMS Microbiol. Ecol. 2014, 90, 1–17. [Google Scholar] [CrossRef]
- Osińska-Jaroszuk, M.; Jarosz-Wilkołazka, A.; Jaroszuk-Ściseł, J.; Szałapata, K.; Nowak, A.; Jaszek, M.; Ozimek, E.; Majewska, M. Extracellular polysaccharides from Ascomycota and Basidiomycota: Production conditions, biochemical characteristics, and biological properties. World J. Microbiol. Biotechnol. 2015, 31, 1823–1844. [Google Scholar] [CrossRef]
- Wang, X.; Liu, J.; Wei, J.; Zhang, Y.; Xu, Y.; Yue, T.; Yuan, Y. Protective Mechanism of Eurotium amstelodami from Fuzhuan Brick Tea against Colitis and Gut-Derived Liver Injury Induced by Dextran Sulfate Sodium in C57BL/6 Mice. Nutrients 2024, 16, 1178. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sun, Y.; Shi, Z.; Li, Z.; Zhao, C.; Lu, T.; Gao, H.; Zhu, F.; Chen, R.; Zhang, J.; et al. Gut Microbiota of Wild and Captive Alpine Musk Deer (Moschus chrysogaster). Front. Microbiol. 2019, 10, 3156. [Google Scholar] [CrossRef] [PubMed]
- Villa-Rivera, M.G.; Conejo-Saucedo, U.; Lara-Marquez, A.; Cano-Camacho, H.; Lopez-Romero, E.; Zavala-Paramo, M.G. The Role of Virulence Factors in the Pathogenicity of Colletotrichum sp. Curr. Protein Pept. Sci. 2017, 18, 1005–1018. [Google Scholar] [CrossRef]
- Li, J.; Sun, K.; Ma, Q.; Chen, J.; Wang, L.; Yang, D.; Chen, X.; Li, X. Colletotrichum gloeosporioides- Contaminated Tea Infusion Blocks Lipids Reduction and Induces Kidney Damage in Mice. Front. Microbiol. 2017, 8, 2089. [Google Scholar] [CrossRef]
- Shivaprakash, M.R.; Appannanavar, S.B.; Dhaliwal, M.; Gupta, A.; Gupta, S.; Gupta, A.; Chakrabarti, A. Colletotrichum truncatum: An unusual pathogen causing mycotic keratitis and endophthalmitis. J. Clin. Microbiol. 2011, 49, 2894–2898. [Google Scholar] [CrossRef]
- Schelstraete, W.; Devreese, M.; Croubels, S. Comparative toxicokinetics of Fusarium mycotoxins in pigs and humans. Food Chem. Toxicol. 2020, 137, 111140. [Google Scholar] [CrossRef]
- Yang, Z.-K.; Huang, X.-L.; Peng, L. Transcriptome analysis reveals gene expression changes of the basidiomycetous yeast Apiotrichum mycotoxinivorans in response to ochratoxin A exposure. Ecotoxicol. Environ. Saf. 2022, 246, 114146. [Google Scholar] [CrossRef]
- Crous, P.; Wingfield, M.; Richardson, D.; Leroux, J.; Strasberg, D.; Edwards, J.; Roets, F.; Hubka, V.; Taylor, P.; Heykoop, M.; et al. Fungal Planet description sheets: 400–468. Persoonia 2016, 36, 316–458. [Google Scholar] [CrossRef]
- Thomas, P.A.; Kaliamurthy, J. Mycotic keratitis: Epidemiology, diagnosis and management. Clin. Microbiol. Infect. 2013, 19, 210–220. [Google Scholar] [CrossRef]
- Nucci, M.; Anaissie, E. Fusarium infections in immunocompromised patients. Clin. Microbiol. Rev. 2007, 20, 695–704. [Google Scholar] [CrossRef]
- Xiaochun, J.; Naisheng, Z.; Jiaren, Z.; Yueyi, L. Isolation and Identification of Fusariotoxin of F. campto-ceras Causing Foot Rot and Tail Decay in Cattle. Chin. J. Vet. Sci. 1994, 14, 121–126. [Google Scholar]
- de Borja Martínez Muñiz, F.; Redondo, M.M.; Sánchez, C.P.; Rodríguez, J.G. Chronic Lung Infection Caused by Trichosporon mycotoxinivorans and Trichosporon mucoides in an Immunocompetent Cystic Fibrosis Patient. Arch. Bronconeumol. 2016, 52, 400. [Google Scholar] [CrossRef]
- Hatori, H.; Shibata, T.; Nishikawa, M.; Ueda, H.; Hino, M.; Fujii, T. FR171456, a novel cholesterol synthesis inhibitor produced by Sporormiella minima No. 15604. I. Taxonomy, fermentation, isolation, physico-chemical properties. J. Antibiot. 2004, 57, 260–263. [Google Scholar] [CrossRef]
- Carpouron, J.E.; de Hoog, S.; Gentekaki, E.; Hyde, K.D. Emerging Animal-Associated Fungal Diseases. J. Fungi 2022, 8, 611. [Google Scholar] [CrossRef]
- Antonissen, G.; Martel, A.; Pasmans, F.; Ducatelle, R.; Verbrugghe, E.; Vandenbroucke, V.; Li, S.; Haesebrouck, F.; Immerseel, F.V.; Croubels, S. The Impact of Fusarium Mycotoxins on Human and Animal Host Susceptibility to Infectious Diseases. Toxins 2014, 6, 430–452. [Google Scholar] [CrossRef]
- Shukla, A.K. Emerging Infectious Diseases Caused by Fungi in Animals and Their Prevention. In Recent Developments in Fungal Diseases of Laboratory Animals; Gupta, A., Singh, N.P., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 1–5. [Google Scholar]
- Lans, C. Possible similarities between the folk medicine historically used by First Nations and American Indians in North America and the ethnoveterinary knowledge currently used in British Columbia, Canada. J. Ethnopharmacol. 2016, 192, 53–66. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, T.; Liu, Q.; Zheng, C.; Meng, X.; Bai, X.; Li, D.; Wang, T.; Guo, J.; Xu, Z.; Jie, H. An Attempted Correlation Between the Fecal Microbial Community of Chinese Forest Musk Deer (Moschus berezovskii) and Differences in Musk Production and Quality. Animals 2025, 15, 1622. https://doi.org/10.3390/ani15111622
Zheng T, Liu Q, Zheng C, Meng X, Bai X, Li D, Wang T, Guo J, Xu Z, Jie H. An Attempted Correlation Between the Fecal Microbial Community of Chinese Forest Musk Deer (Moschus berezovskii) and Differences in Musk Production and Quality. Animals. 2025; 15(11):1622. https://doi.org/10.3390/ani15111622
Chicago/Turabian StyleZheng, Tingting, Qian Liu, Chengli Zheng, Xiuxiang Meng, Xue Bai, Diyan Li, Tao Wang, Jun Guo, Zhongxian Xu, and Hang Jie. 2025. "An Attempted Correlation Between the Fecal Microbial Community of Chinese Forest Musk Deer (Moschus berezovskii) and Differences in Musk Production and Quality" Animals 15, no. 11: 1622. https://doi.org/10.3390/ani15111622
APA StyleZheng, T., Liu, Q., Zheng, C., Meng, X., Bai, X., Li, D., Wang, T., Guo, J., Xu, Z., & Jie, H. (2025). An Attempted Correlation Between the Fecal Microbial Community of Chinese Forest Musk Deer (Moschus berezovskii) and Differences in Musk Production and Quality. Animals, 15(11), 1622. https://doi.org/10.3390/ani15111622