
Genetic Variation in Schizothorax kozlovi Nikolsky in the Upper Reaches of the Chinese Yangtze River Based on Genotyping for Simplified Genome Sequencing


 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
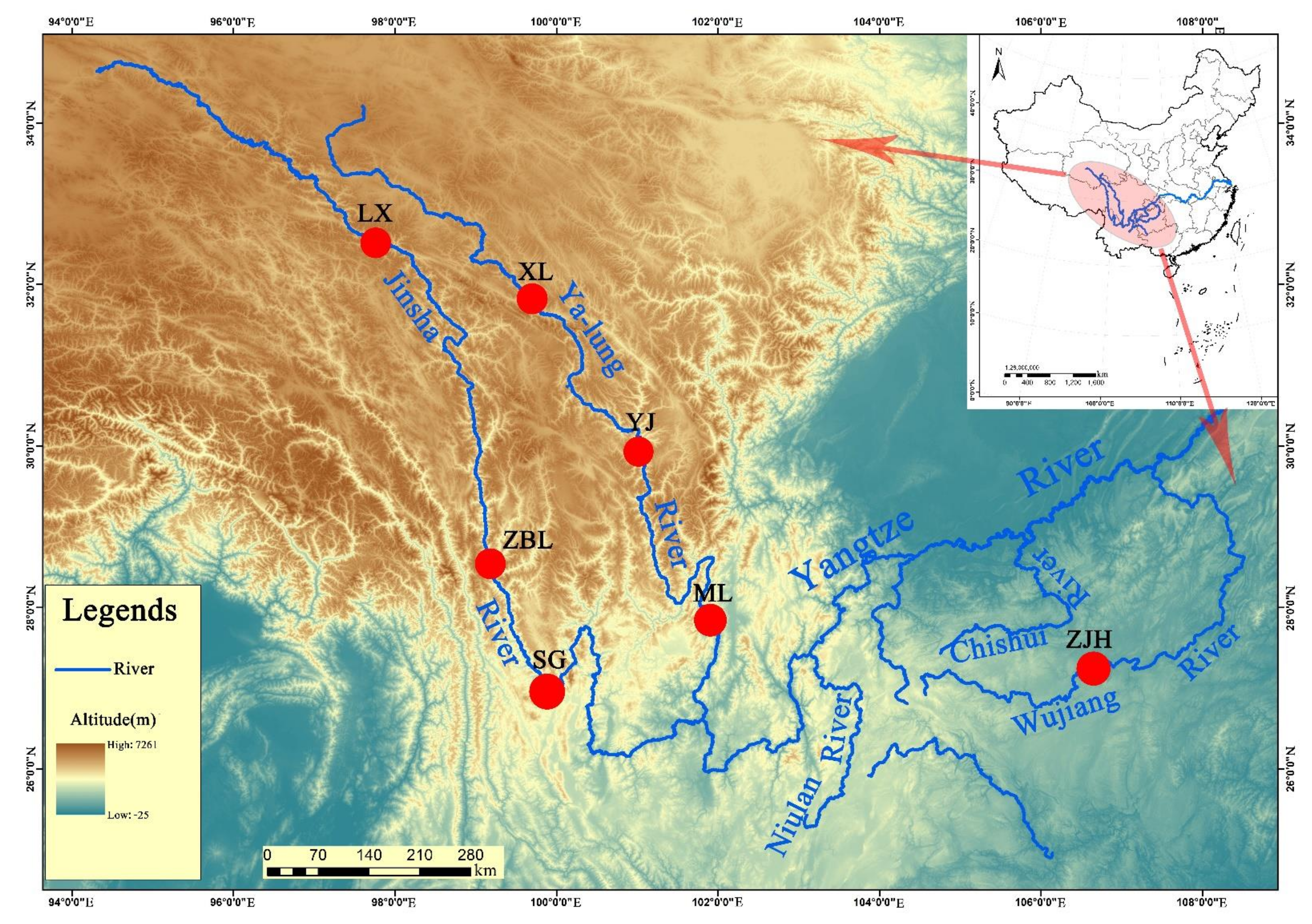
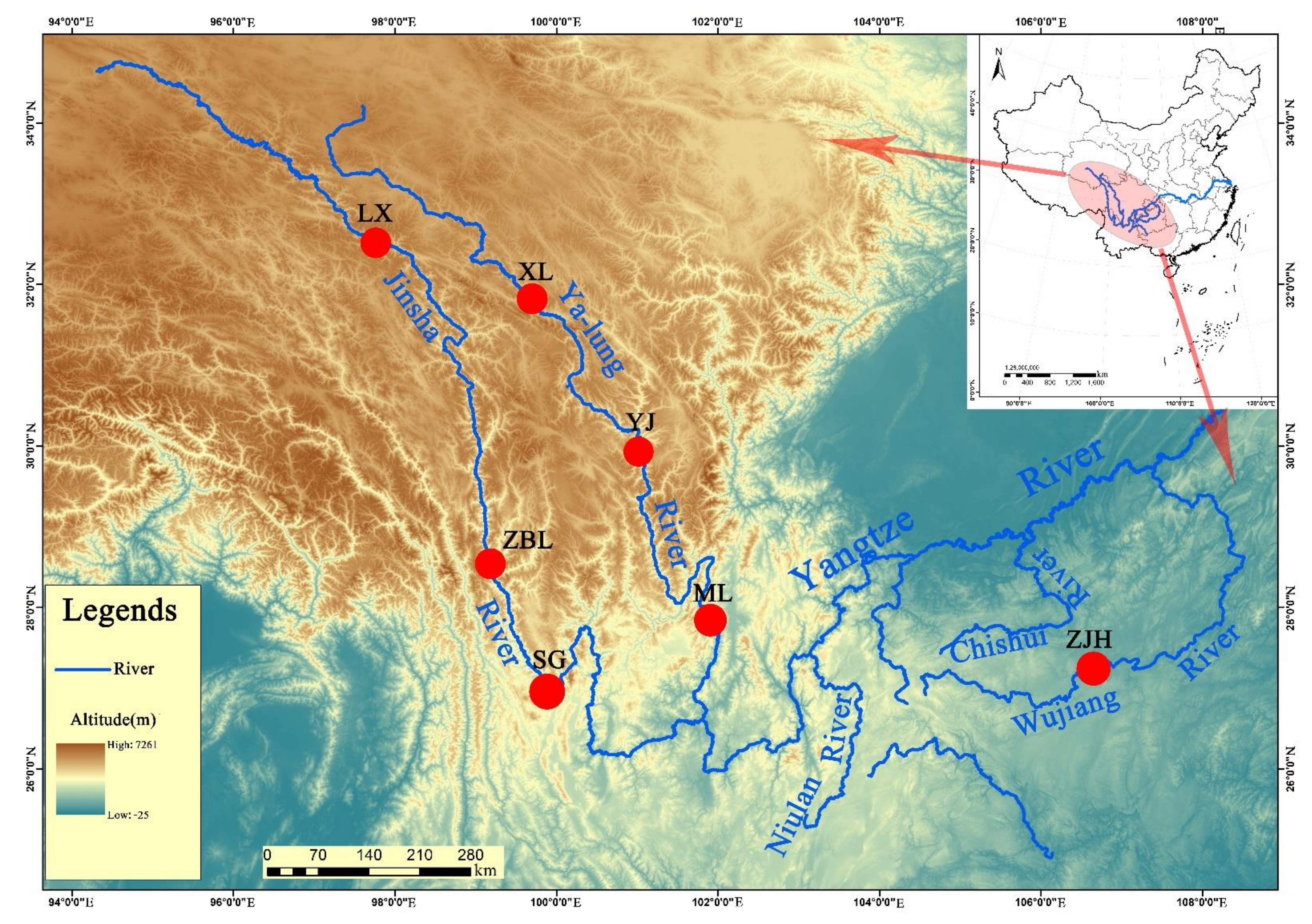
2.1. Sample Collection
2.2. Library Preparation
2.3. Sequencing and Genotyping
2.4. Genetic Diversity and Population Structure Analysis
2.5. Reconstructing the Phylogenetic Tree
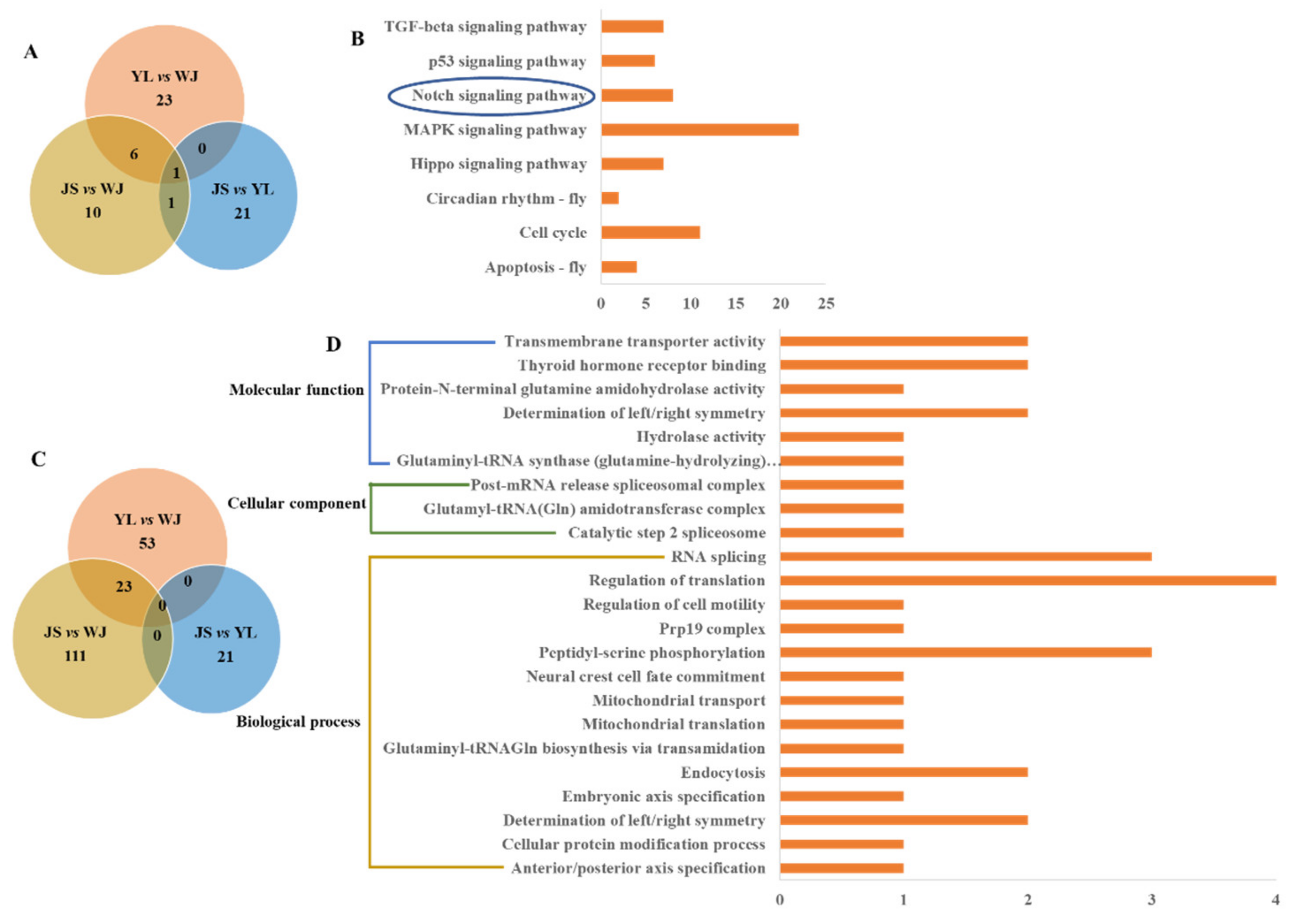
2.6. Functional Enrichment of the Selected SNPs
3. Results
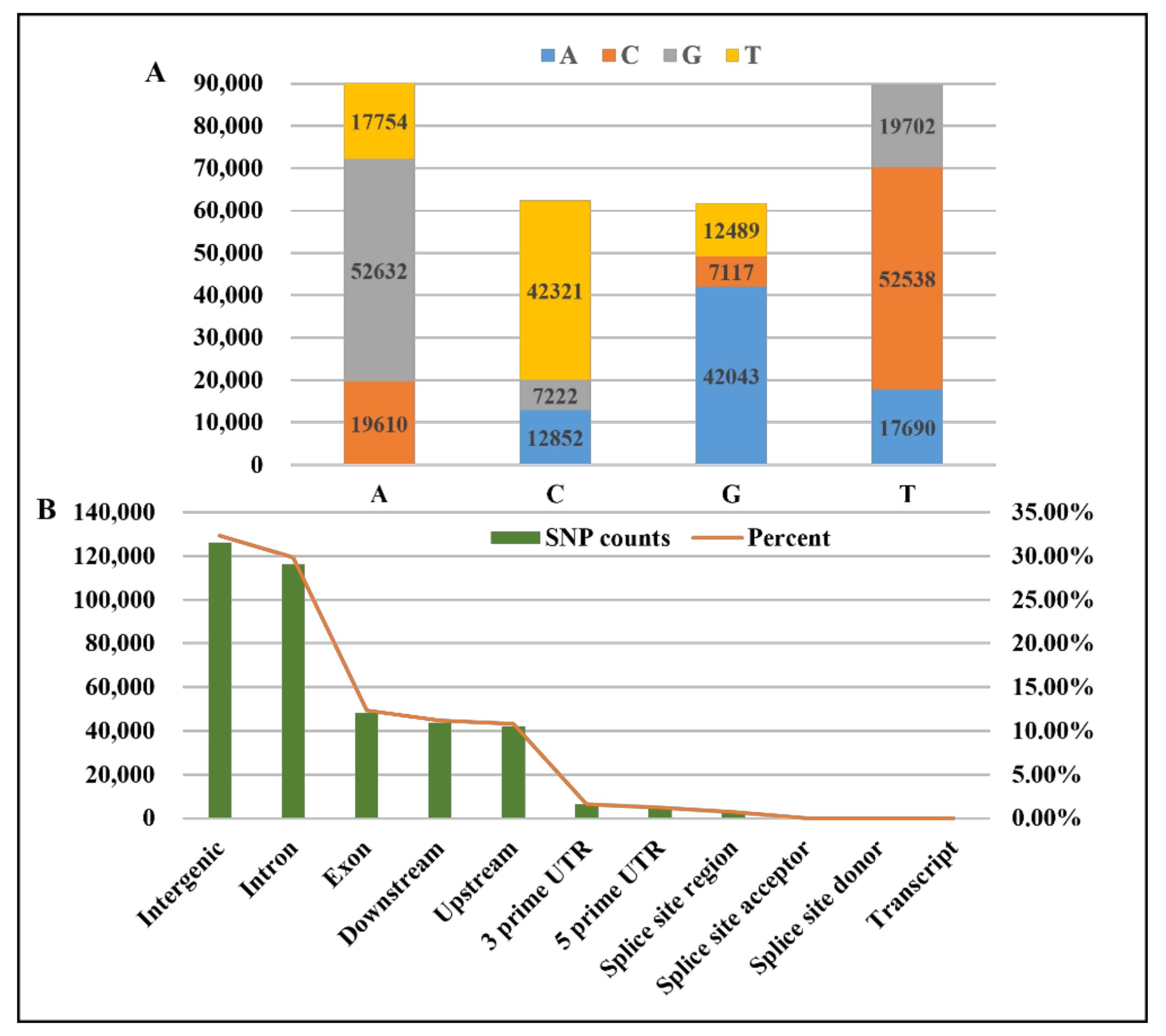
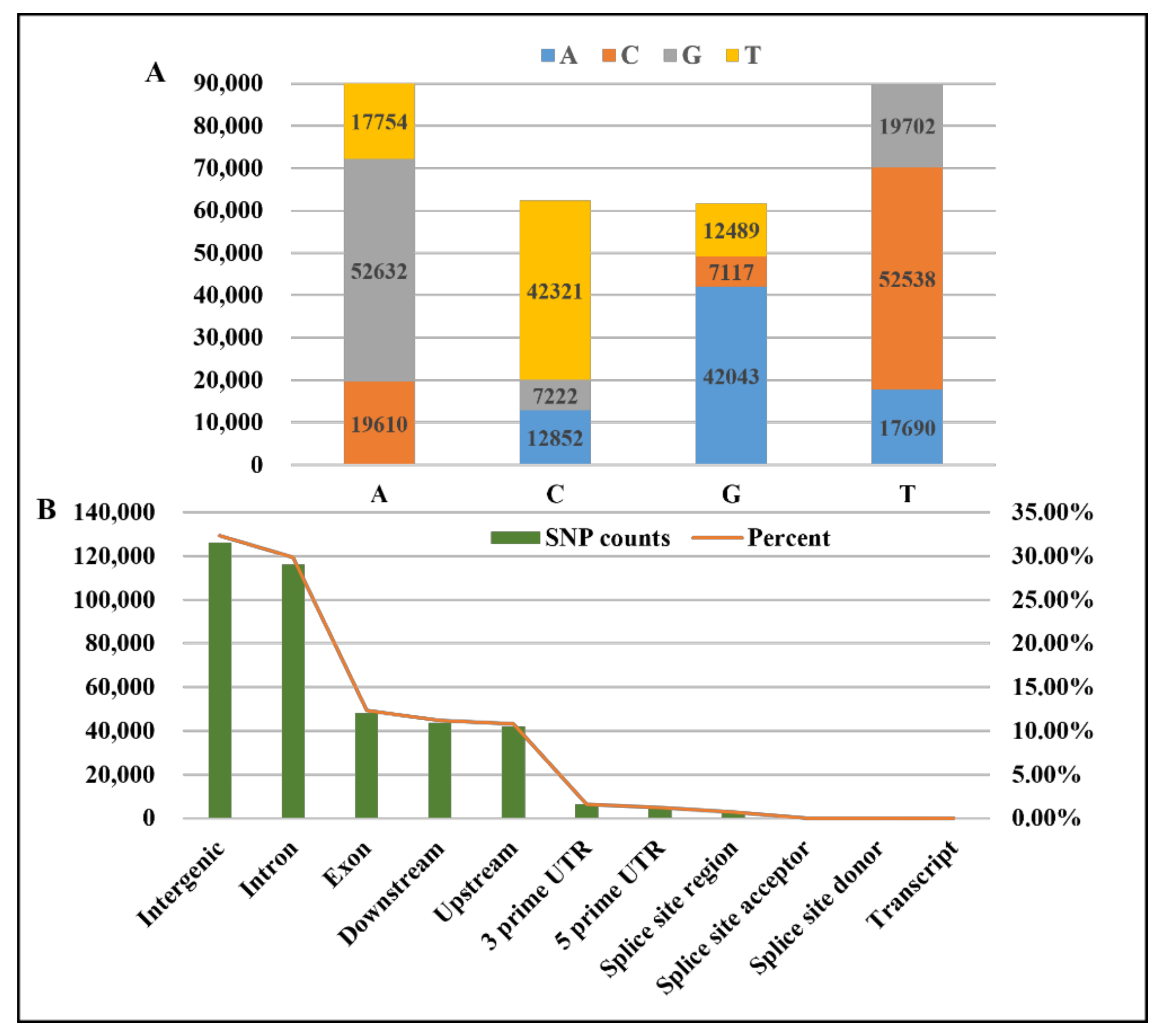
3.1. GBS Data and SNP Discovery
3.2. Clustering Analysis of Differential SNPs
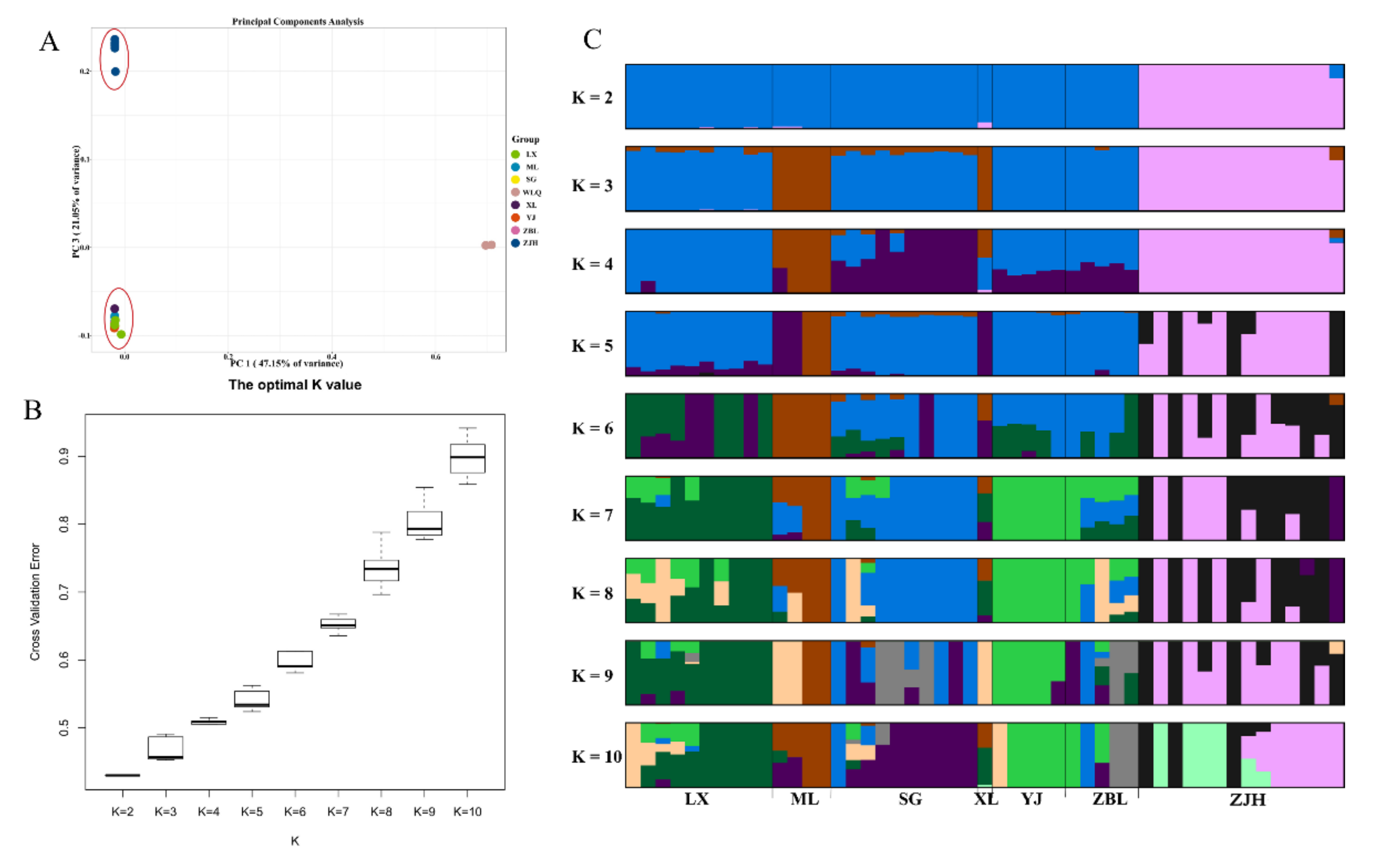
3.3. Analysis of Population Genetic Diversity
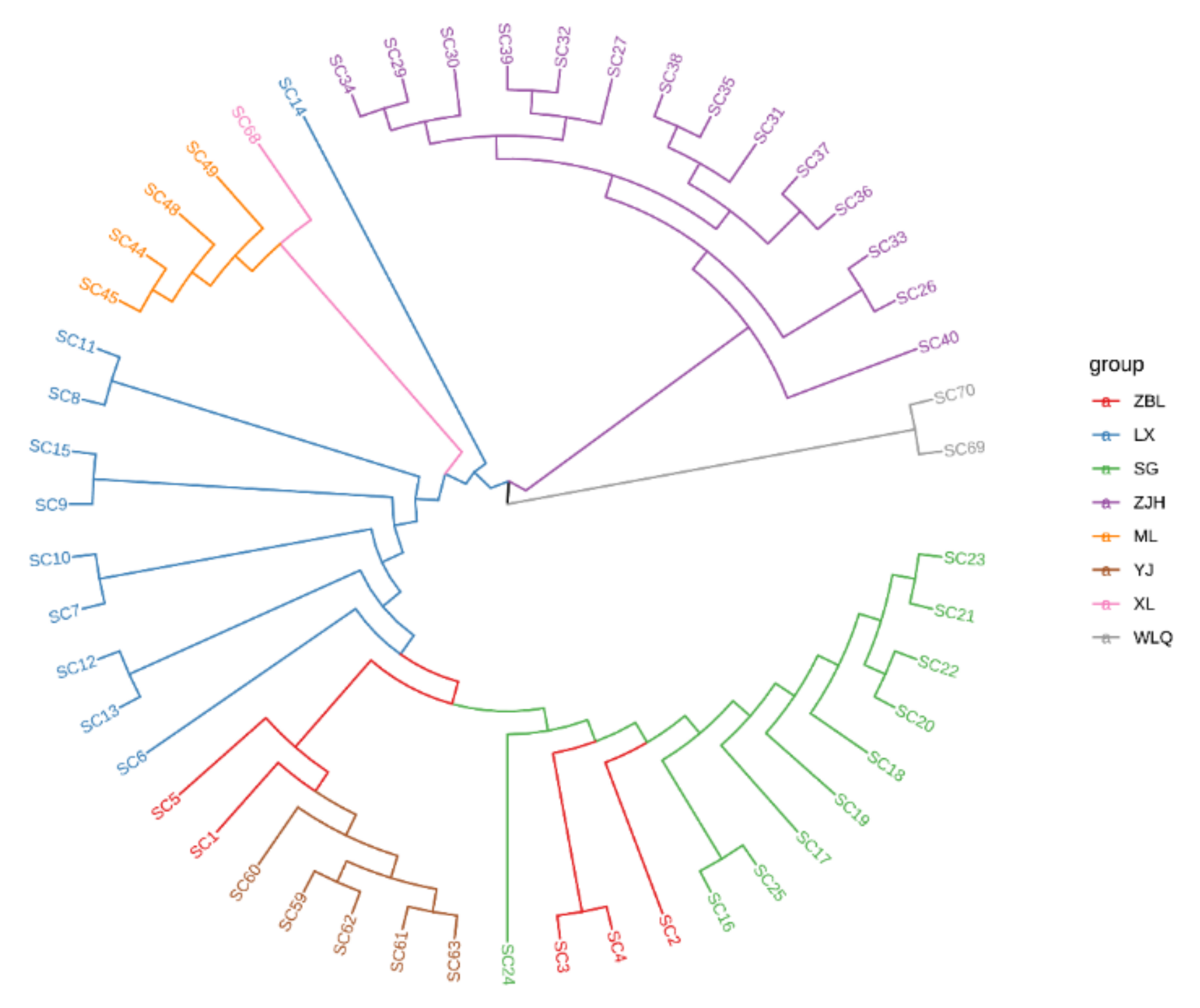
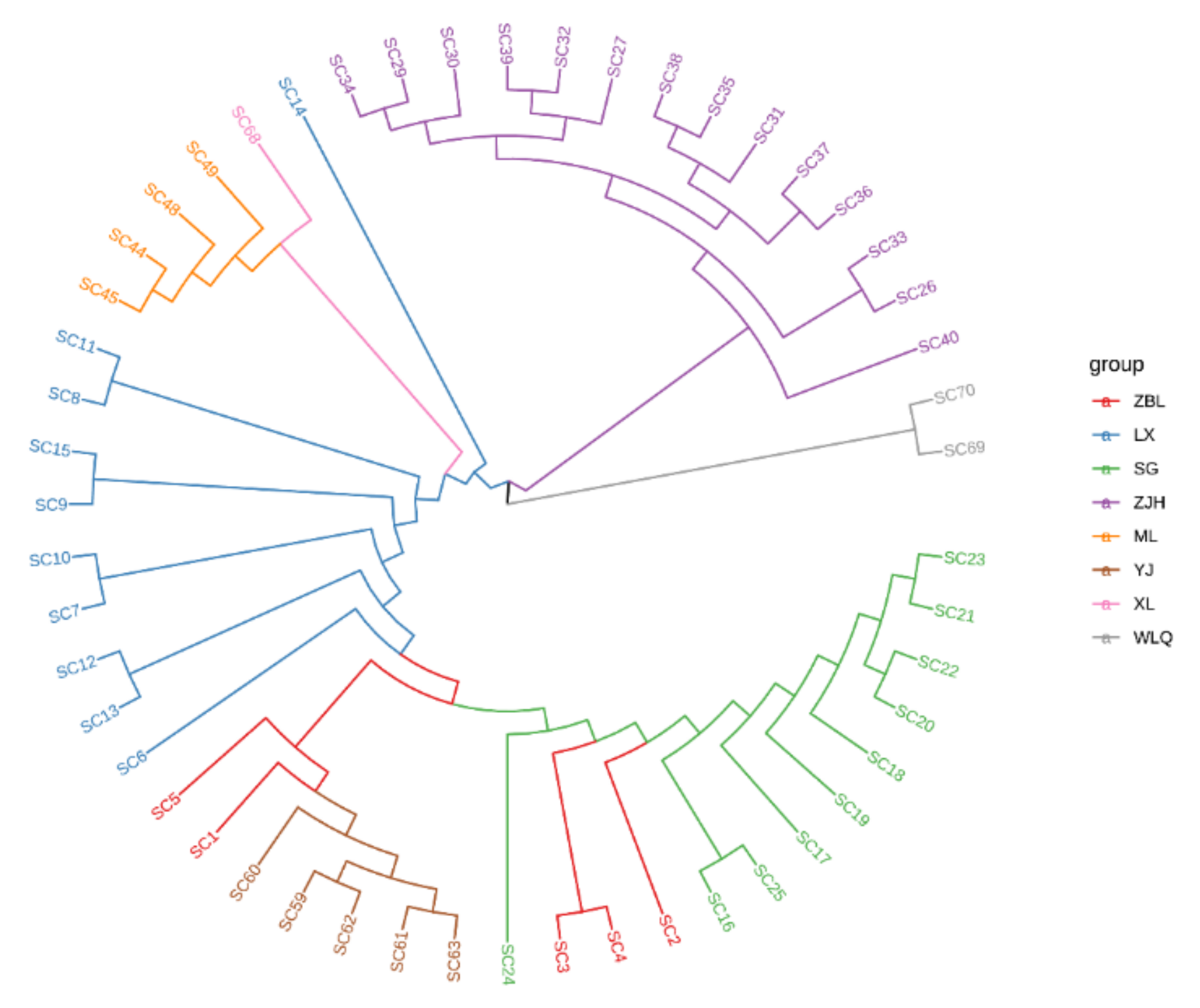
3.4. Phylogenetic Analysis
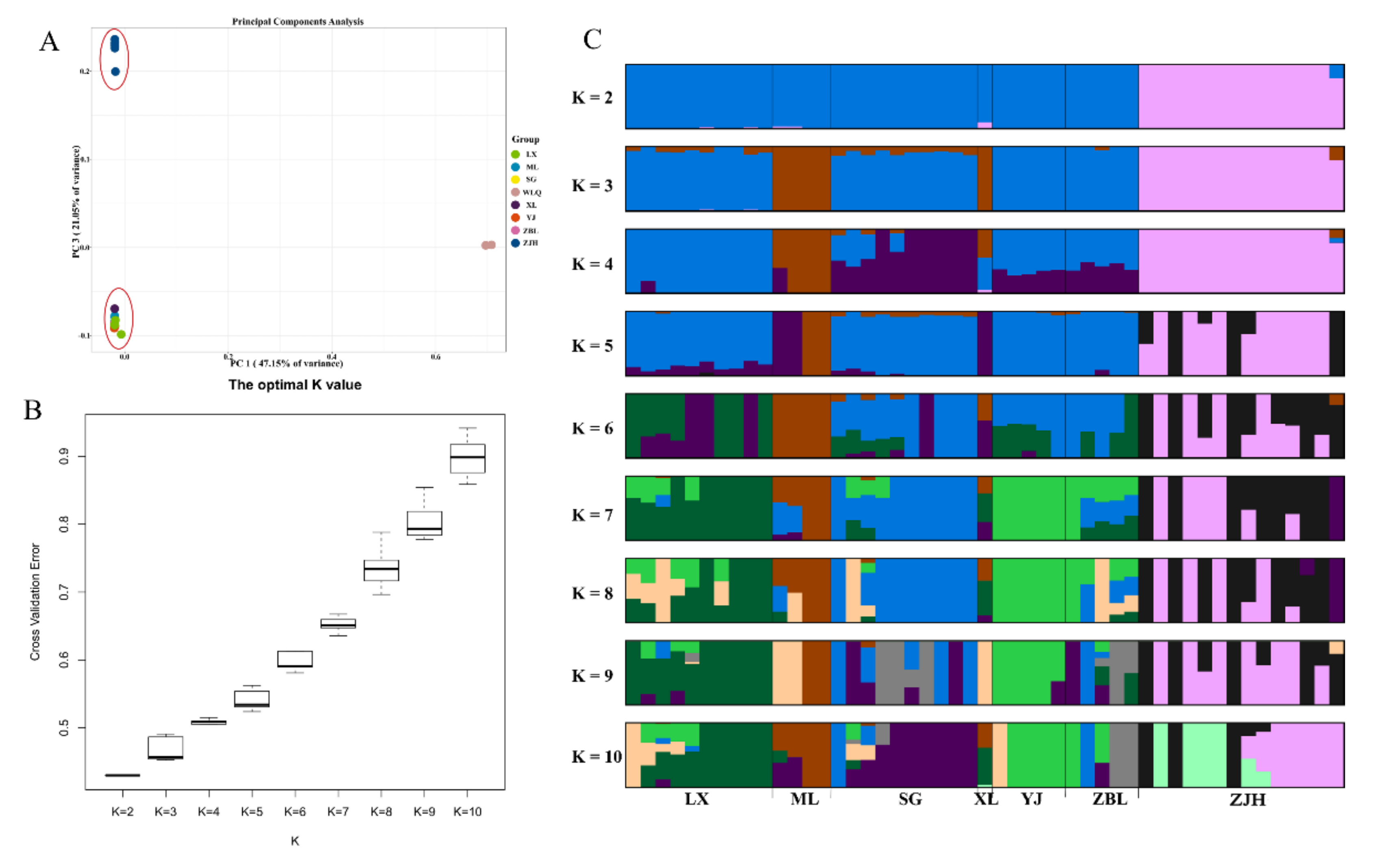
3.5. PCA
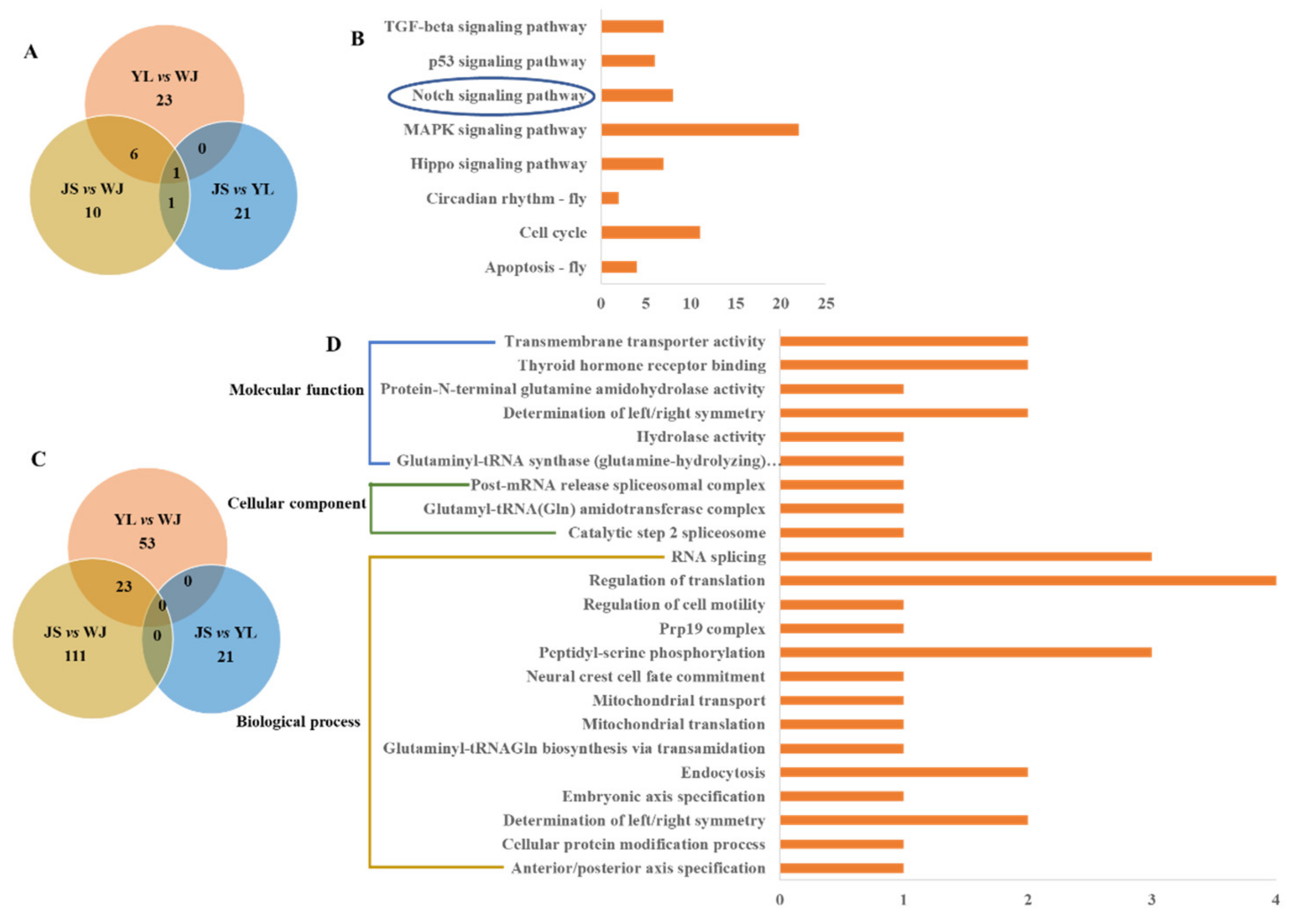
3.6. Functional Annotations of the Selected SNPs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, W.; Zeng, S.; Wang, X.; Zhan, H.X.; Zhou, L.; Xiong, P.; Xiang, Y. Genetic diversity in farmed Schizothorax kozlovi based on mitochondrial DNA D-loop and Cyt b gene sequence. Southwest China J. Agric. Sci. 2022, 35, 475–480. [Google Scholar]
- An, D.D.; Dai, Y.G.; Zou, X.J. AFLP Analysis of Genetic Structure and Diversity in Population of Schizothorax kolzovi from the Wujiang River. Prog. Fishery Sci. 2021, 42, 39–45. [Google Scholar]
- Chen, Y.X.; Luo, Q.S. The fecunidty of Schizothorax kozlovi from Wu River. Zool. Res. 1995, 4, 324–325. [Google Scholar]
- Zhang, X.J.; Dai, Y.G. Feeding habits and resources protection of Schizothorax kozlovi. J. Hydrol. 2011, 32, 110–114. [Google Scholar]
- Li, Z.L.; Hu, S.Y.; Chen, Y.X.; Zhao, H.T. Age Structure and Growth Characteristics of Schizothorax kozlovi in the Upper Wujiang River. J. Hydrol. 2015, 2, 75–80. [Google Scholar]
- Chen, D.; Fu, C.; Lei, Z.; Li, Y.; Zhao, T.; Fan, H.; Hu, T.; Wang, Q.; Zong, H. Complete mitochondrial genome and phylogenetic analysis of Schizothorax sinensis (Teleostei: Cypriniformes: Cyprinidae). Mitochondrial DNA Part B 2021, 6, 1118–1119. [Google Scholar] [CrossRef]
- Ma, Q.; He, K.; Wang, X.; Jiang, J.; Zhang, X.; Song, Z. Better Resolution for Cytochrome b than Cytochrome c Oxidase Subunit I to Identify Schizothorax Species (Teleostei: Cyprinidae) from the Tibetan Plateau and Its Adjacent Area. DNA Cell Biol. 2020, 39, 579–598. [Google Scholar] [CrossRef]
- Guo, X.-Z.; Zhang, G.-R.; Wei, K.-J.; Ji, W.; Yan, R.-J.; Wei, Q.-W.; Gardner, J.P.A. Phylogeography of the threatened tetraploid fish, Schizothorax waltoni, in the Yarlung Tsangpo River on the southern Qinghai-Tibet Plateau: Implications for conservation. Sci. Rep. 2019, 9, 2704. [Google Scholar] [CrossRef]
- Hassanin, A.; Ropiquet, A.; Couloux, A.; Cruaud, C. Evolution of the Mitochondrial Genome in Mammals Living at High Altitude: New Insights from a Study of the Tribe Caprini (Bovidae, Antilopinae). J. Mol. Evol. 2009, 68, 293–310. [Google Scholar] [CrossRef]
- Wang, N.; Yuan, Y.; Wang, H.; Yu, D.; Liu, Y.; Zhang, A.; Gowda, M.; Nair, S.K.; Hao, Z.; Lu, Y.; et al. Applications of genotyping-by-sequencing (GBS) in maize genetics and breeding. Sci. Rep. 2020, 10, 16308. [Google Scholar] [CrossRef]
- Han, Y.P. Genotyping-by-Sequencing-Based Genome-Wide Association Studies of Fusarium Wilt Resistance in Radishes (Raphanus sativus L.). Genes 2021, 12, 858. [Google Scholar]
- Jadhav, M.P.; Gangurde, S.S.; Hake, A.A.; Yadawad, A.; Mahadevaiah, S.S.; Pattanashetti, S.K.; Gowda, M.V.C.; Shirasawa, K.; Varshney, R.K.; Pandey, M.K.; et al. Genotyping-by-Sequencing Based Genetic Mapping Identified Major and Con-sistent Genomic Regions for Productivity and Quality Traits in Peanut. Front. Plant Sci. 2021, 12, 668020. [Google Scholar] [CrossRef] [PubMed]
- Szarmach, S.J.; Brelsford, A.; Witt, C.C.; Toews, D.P. Comparing divergence landscapes from reduced-representation and whole genome resequencing in the yellow-rumped warbler (Setophaga coronata) species complex. Mol. Ecol. 2021, 30, 5994–6005. [Google Scholar] [CrossRef] [PubMed]
- Yoon, D.; Sebastin, R.; Lee, G.A.; Lee, K.J.; Cho, G.T. Genome-Wide SNP Markers for Genotypic and Phenotypic Differentiation of Melon (Cucumis melo L.) Varieties Using Genotyping-by-Sequencing. Int. J. Mol. Sci. 2021, 22, 6722. [Google Scholar]
- Boulanger, E.; Benestan, L.; Guerin, P.; Dalongeville, A.; Mouillot, D.; Manel, S. Climate differently influences the genomic patterns of two sympatric marine fish species. J. Anim. Ecol. 2021, 91, 1180–1195. [Google Scholar] [CrossRef]
- Huo, B.; Liu, X.; Chen, S.; Liu, J.; Zhou, Q.; Shen, J.; Li, D.; Tang, R.; Chen, J.; Zhou, X. Population Structure, Genetic Diversity and Differentiation of Triplophysa tenuis in Xinjiang Tarim River. Front. Genet. 2022, 13, 860678. [Google Scholar] [CrossRef]
- Ding, R.H. The Fishes of Sichuan; Sichuan Science and Technology Press: Chengdu, China, 1994. [Google Scholar]
- Qin, P.; Davis, G.; Dipnarayan, S.; Stephan, S.; Debkanta, C.; Wang, X.; Mathews, D.M.; Malmberg, R.L.; Devos, K.M. UGbS-Flex, a novel bioinformatics pipeline for imputation-free SNP discovery in polyploids without a reference genome: Finger millet as a case study. BMC Plant Biol. 2018, 18, 117. [Google Scholar]
- Julian, C.; Paul, A.H.; Susan, B.; Angel, A.; William, A.C. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Liu, H.-P.; Xiao, S.-J.; Wu, N.; Wang, D.; Liu, Y.-C.; Zhou, C.-W.; Liu, Q.-Y.; Yang, R.-B.; Jiang, W.-K.; Liang, Q.-Q.; et al. The sequence and de novo assembly of Oxygymnocypris stewartii genome. Sci. Data 2019, 6, 190009. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Pickrell, J.K.; Pritchard, J.K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012, 8, e1002967. [Google Scholar] [CrossRef]
- Li, J. Methodology A fast neighbor joining method. Genet. Mol. Res. 2015, 14, 8733–8743. [Google Scholar] [CrossRef]
- Fabian, S.; Mateus, P.; Matthieu, M.; Miguel, P.; Alex, B. TreeFam v9: A new website, more species and orthology-on-the-fly. Nucleic Acids. Res. 2014, 42, D922–D925. [Google Scholar]
- Noma, H.; Nagashima, K.; Maruo, K.; Gosho, M.; Furukawa, T.A. Bartlett-type corrections and bootstrap adjustments of likelihood-based inference methods for network meta-analysis. Stat. Med. 2017, 37, 1178–1190. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Foll, M.; Gaggiotti, O.E. A Genome-Scan Method to Identify Selected Loci Appropriate for Both Dominant and Codominant Markers: A Bayesian Perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [Green Version]
- Jia, W.; Liu, H.; Zhang, B.; Chen, C.X.; Dai, Y.Y.; He, X.X.; Wang, Q.S. Analysis of genetic diversity of wild and cultivated populations of Cynoglossus semilaevis based on genotyping-by-sequencing technology. Trans. Oceanol. Limnol. 2020, 1, 107–114. [Google Scholar]
- Chen, Y.X. The Genetic Characterization and Population Genetic Diversity of Schizothorax Kozlovi Nikolsky. Ph.D. Thesis, Sichuan Agricultural University, Ya’an, China, 2013. [Google Scholar]
- Gao, L.; Bao, X.B.; Yu, S.Q.; Li, Z.; Li, Y.F.; He, C.B. Analysis of the genetic characteristics of Umbonium thomasi populations along the Yellow and Bohai Seas using GBS. J. Fish. Sci. China 2020, 27, 204–212. [Google Scholar]
- Bernatchez, L. On the maintenance of genetic variation and adaptation to environmental change: Considerations from popu-lation genomics in fishes. J. Fish Biol. 2016, 89, 2519–2556. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Huang, Y.; Jiang, J.; Guisan, A. Genetic diversity in frogs linked to past and future climate changes on the roof of the world. J. Anim. Ecol. 2019, 88, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Genetic Distance between Populations. Am. Nat. 1972, 106, 283–292. [Google Scholar] [CrossRef]
- Saugstad, L.F. From genetics to epigenetics. Nutr. Health 2006, 18, 285–300. [Google Scholar] [CrossRef]
- Jin, F.P.; Li, G.H.; Leng, Y.; Wu, J.X.; Zuo, P.X.; Lei, C.Y. Genetic diversity analysis of four Schizothoras lissolabiatus Sao popu-lations in the middle and Upper Rearches the Lancang River. Acta Hydrobiol. Sin. 2021, 45, 60–68. [Google Scholar]
- Kitada, S.; Nakamichi, R.; Kishino, H. Understanding population structure in an evolutionary context: Population-specific FST and pairwise FST. G3 Genes|Genomes|Genet. 2021, 11, jkab316. [Google Scholar] [CrossRef]
- Zhang, Z.S.; Hu, B.J.; Ye, X.Y.; Liu, G.J.; Zheng, S.M.; Hua, Y.E. Genetic diversity of the prenant’s Schizothoracin (Schizothorax prenanti) based on partial mtDNA CYTB sequences. Acta Hydrobiol. Sin. 2017, 41, 609–616. [Google Scholar]
- Ayelhan, H.S.; Guo, Y.; Wei, M.; Yang, T.; Karjan, A. Genetic diversity and population differentiation of Schizothorax biddulphi based on mtDNA control region sequences. J. Fish. Sci. China 2016, 23, 944–954. [Google Scholar]
- Dan, Y.U.; Zhang, Z.; Zhang, J.; Lin, P.C.; Xiong, S.R.; Tang, F.L.; Liu, H.Z. Genetic diversity and population demography of Schizothorax molesworthi from the MOTUO area of lower Reaches of the Yarlung Zangbo River and Lohit River. Acta Hydrobiol. Sin. 2019, 43, 923–930. [Google Scholar]
- Yi, S.K. The Phylogeny, Population Structure and Adaptive Evolution of Misgurnus Species in China. Ph.D. Thesis, Huazhong Agricultural University, Wuhan, China, 2017. [Google Scholar]
- Folio, D.M.; Gil, J.; Caudron, A.; Labonne, J. Genotype-by-environment interactions drive the maintenance of genetic variation in a Salmo trutta L. hybrid zone. Evol. Appl. 2021, 14, 2698–2711. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, D.R.; Reid, B.N.; Fitzpatrick, S.W. Genome-wide diversity and habitat underlie fine-scale phenotypic differentiation in the rainbow darter (Etheostoma caeruleum). Evol. Appl. 2020, 14, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Petty, J.T.; Thorne, D.; Huntsman, B.; Mazik, P.M. The temperature–productivity squeeze: Constraints on brook trout growth along an Appalachian river continuum. Hydrobiologia 2013, 727, 151–166. [Google Scholar] [CrossRef]
- Yang, T.Y.; Meng, W.; Guo, B.C. Population Genomic Analysis of Two Endemic Schizothoracins Reveals Their Genetic Dif-ferences and Underlying Selection Associated with Altitude and Temperature. Animals 2020, 10, 447. [Google Scholar] [CrossRef]
- Lu, L.U.; Wang, Q.; Wang, G.; Liu, Y.; Liu, C. Trend of climate change over the recent 60 years and its hydrological responses for Jinsha river basin. J. North China Univ. Water Resour. Electr. Power 2016, 37, 16–21. [Google Scholar]
- Huang, Y.Y.; Zhao, H.L.; Jiang, Y.Z.; Xin, L.U. Runoff and its influencing factors in the upper reaches of the Yalong River. Arid Land Geogr. 2018, 41, 127–133. [Google Scholar]
- Zhu, X.S. Preliminary Study on Water Area Changes and Climate Effects in Wujiang River Basin in Guizhou. Ph.D. Thesis, Guizhou Normal University, Guiyang, China, 2020. [Google Scholar]
- Luo, H.; Liu, H.; Zhang, J.; Hu, B.; Zhou, C.; Xiang, M.; Yang, Y.; Zhou, M.; Jing, T.; Li, Z.; et al. Full-length transcript sequencing accelerates the transcriptome research of Gymnocypris namensis, an iconic fish of the Tibetan Plateau. Sci. Rep. 2020, 10, 9668. [Google Scholar] [CrossRef]
- Feng, X.; Jia, Y.; Zhu, R.; Chen, K.; Chen, Y. Characterization and analysis of the transcriptome in Gymnocypris selincuoensis on the Qinghai-Tibetan Plateau using single-molecule long-read sequencing and RNA-seq. DNA Res. 2019, 26, 353–363. [Google Scholar] [CrossRef]
- Windisch, H.S.; Frickenhaus, S.; John, U.; Knust, R.; Prtner, H.O.; Lucassen, M. Stress response or beneficial temperature ac-climation: Transcriptomic signatures in antarctic fish (Pachycara brachycephalum). Mol. Ecol. 2014, 23, 3469–3482. [Google Scholar] [CrossRef]
- Pörtner, H.-O. Oxygen- and capacity-limitation of thermal tolerance: A matrix for integrating climate-related stressor effects in marine ecosystems. J. Exp. Biol. 2010, 213, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Múgica, M.; Sokolova, I.; Izagirre, U.; Marigómez, I. Season-dependent effects of elevated temperature on stress biomarkers, energy metabolism and gamete development in mussels. Mar. Environ. Res. 2015, 103, 1–10. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Sampling Site | Abbreviation | Tributary | Coordinates | Altitude | Sample Number |
|---|---|---|---|---|---|---|
| Schizothorax kozlovi | Luoxu, Sichuan | LX | Jinsha River | E 97°59′43.47″ N 32°27′41.20″ | 3288 | 10 |
| Zhubalong, Sichuan | ZBL | N 29°46′17.46″ E 99°0′38.54″ | 2476 | 5 | ||
| Shigu, Sichuan | SG | E 99°57′44.69″ N 26°52′12.82″ | 1818 | 10 | ||
| Xinlong, Sichuan | XL | Yalong River | E 100°18′48.15″ N 30°56′17.73″ | 3052 | 1 | |
| Yajiang, Sichuan | YJ | E 101° 0′58.49″ N 30° 2′0.01″ | 2572 | 5 | ||
| Muli, Sichuan | ML | E 101°15′59.47″ N 27°51′32.50″ | 1780 | 4 | ||
| Zongjihe, Guizhou | ZJH | Wujiang River | E 105°12′52.84″ N 27° 2′52.95″ | 1214 | 14 | |
| Schizopygopsis malacanthus | Yajiang, Sichuan | WLQ | Yalong River | E 101° 0′58.49″ N 30° 2′0.01″ | 2572 | 1 |
| Gymnodiptychus pachycheilus | Xinlong, Sichuan | WLQ | Yalong River | E 100°18′48.15″ N 30°56′17.73″ | 3052 | 1 |
| Populations | HE | HO | π | PIC | |
|---|---|---|---|---|---|
| Jinsha River | LX | 0.09693 | 0.07411 | 0.07827 | 0.06128 |
| ZBL | 0.09175 | 0.06706 | 0.07523 | 0.05439 | |
| SG | 0.09312 | 0.07058 | 0.07452 | 0.05801 | |
| Average value | 0.09393 | 0.07058 | 0.07601 | 0.05789 | |
| Yalong River | XL | 0.09802 | 0.04901 | 0.09202 | 0.03676 |
| YJ | 0.09437 | 0.06695 | 0.07486 | 0.05411 | |
| ML | 0.09576 | 0.06614 | 0.07676 | 0.05299 | |
| Average value | 0.09605 | 0.06070 | 0.08121 | 0.04795 | |
| Wujiang River | ZJH | 0.10054 | 0.07821 | 0.08132 | 0.06465 |
| Pop | ZBL | LX | SG | ZJH | ML | YJ | XL | WLQ | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Jiansha River | ZBL | − | 0.0038 | 0.0053 | 0.2014 | 0.0588 | 0.0053 | 0.0512 | 1.5535 | |
| LX | 0.0038 | − | 0.0110 | 0.1974 | 0.0543 | 0.0074 | 0.0315 | 1.6972 | ||
| SG | 0.0053 | 0.0109 | − | 0.2073 | 0.0549 | 0.0108 | 0.0533 | 1.7441 | ||
| Wujiang River | ZJH | 0.1824 | 0.1791 | 0.1872 | − | 0.2095 | 0.2094 | 0.1720 | 1.7522 | |
| Yalong River | ML | 0.0571 | 0.0529 | 0.0534 | 0.1890 | − | 0.0606 | 1.4727 | ||
| YJ | 0.0053 | 0.0073 | 0.0107 | 0.1889 | 0.0636 | − | 0.0718 | 1.5669 | ||
| XL | 0.0499 | 0.0310 | 0.0519 | 0.1580 | 0.0588 | 0.0693 | − | 0.7783 | ||
| Outgroup | WLQ | 0.7885 | 0.8168 | 0.8252 | 0.8266 | 0.7707 | 0.7913 | 0.5408 | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, J.; He, Z.; Yang, D.; Ma, Z.; Chen, H.; Zhang, Q.; Deng, F.; Ye, L.; Pu, Y.; Zhang, M.; et al. Genetic Variation in Schizothorax kozlovi Nikolsky in the Upper Reaches of the Chinese Yangtze River Based on Genotyping for Simplified Genome Sequencing. Animals 2022, 12, 2181. https://doi.org/10.3390/ani12172181
He J, He Z, Yang D, Ma Z, Chen H, Zhang Q, Deng F, Ye L, Pu Y, Zhang M, et al. Genetic Variation in Schizothorax kozlovi Nikolsky in the Upper Reaches of the Chinese Yangtze River Based on Genotyping for Simplified Genome Sequencing. Animals. 2022; 12(17):2181. https://doi.org/10.3390/ani12172181
Chicago/Turabian StyleHe, Jiayang, Zhi He, Deying Yang, Zhijun Ma, Hongjun Chen, Qian Zhang, Faqiang Deng, Lijuan Ye, Yong Pu, Mingwang Zhang, and et al. 2022. "Genetic Variation in Schizothorax kozlovi Nikolsky in the Upper Reaches of the Chinese Yangtze River Based on Genotyping for Simplified Genome Sequencing" Animals 12, no. 17: 2181. https://doi.org/10.3390/ani12172181
APA StyleHe, J., He, Z., Yang, D., Ma, Z., Chen, H., Zhang, Q., Deng, F., Ye, L., Pu, Y., Zhang, M., Yang, S., Yang, S., & Yan, T. (2022). Genetic Variation in Schizothorax kozlovi Nikolsky in the Upper Reaches of the Chinese Yangtze River Based on Genotyping for Simplified Genome Sequencing. Animals, 12(17), 2181. https://doi.org/10.3390/ani12172181