Tick-Borne Pathogens Shape the Native Microbiome Within Tick Vectors

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Tick Collection and Identification
2.2. Genomic DNA Extraction
2.3. Detection of Pathogen and Endosymbiont
2.4. 16S rRNA Library Preparation and Sequencing
2.5. Sequence Analysis
2.6. Alpha Diversity
2.7. Beta Diversity
2.8. Statistical Analysis
3. Results
3.1. Pathogen and Symbiont Prevalence
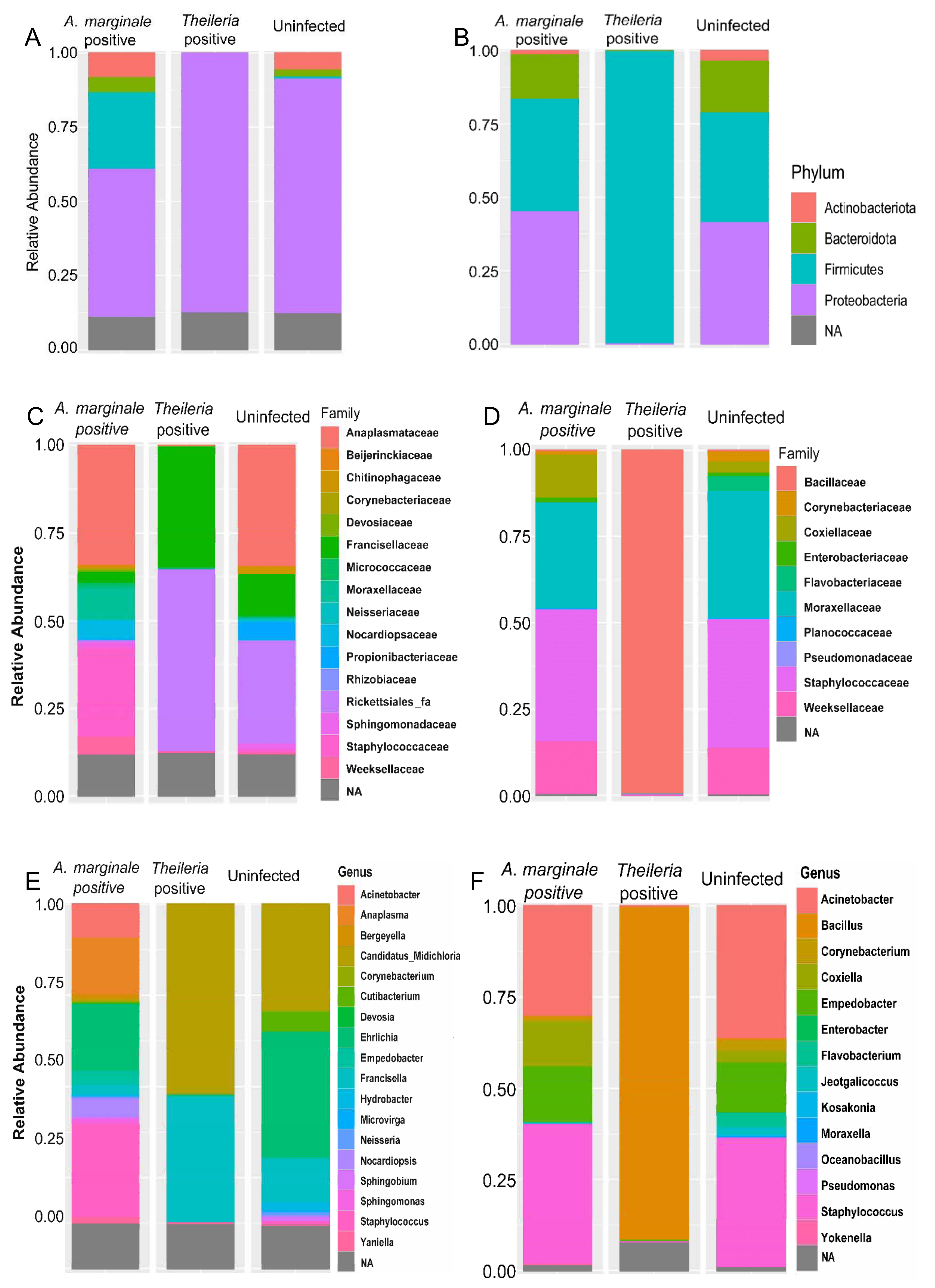
3.2. Bacteria 16S rRNA Abundance Profile
3.3. Bacteria Relative Abundance
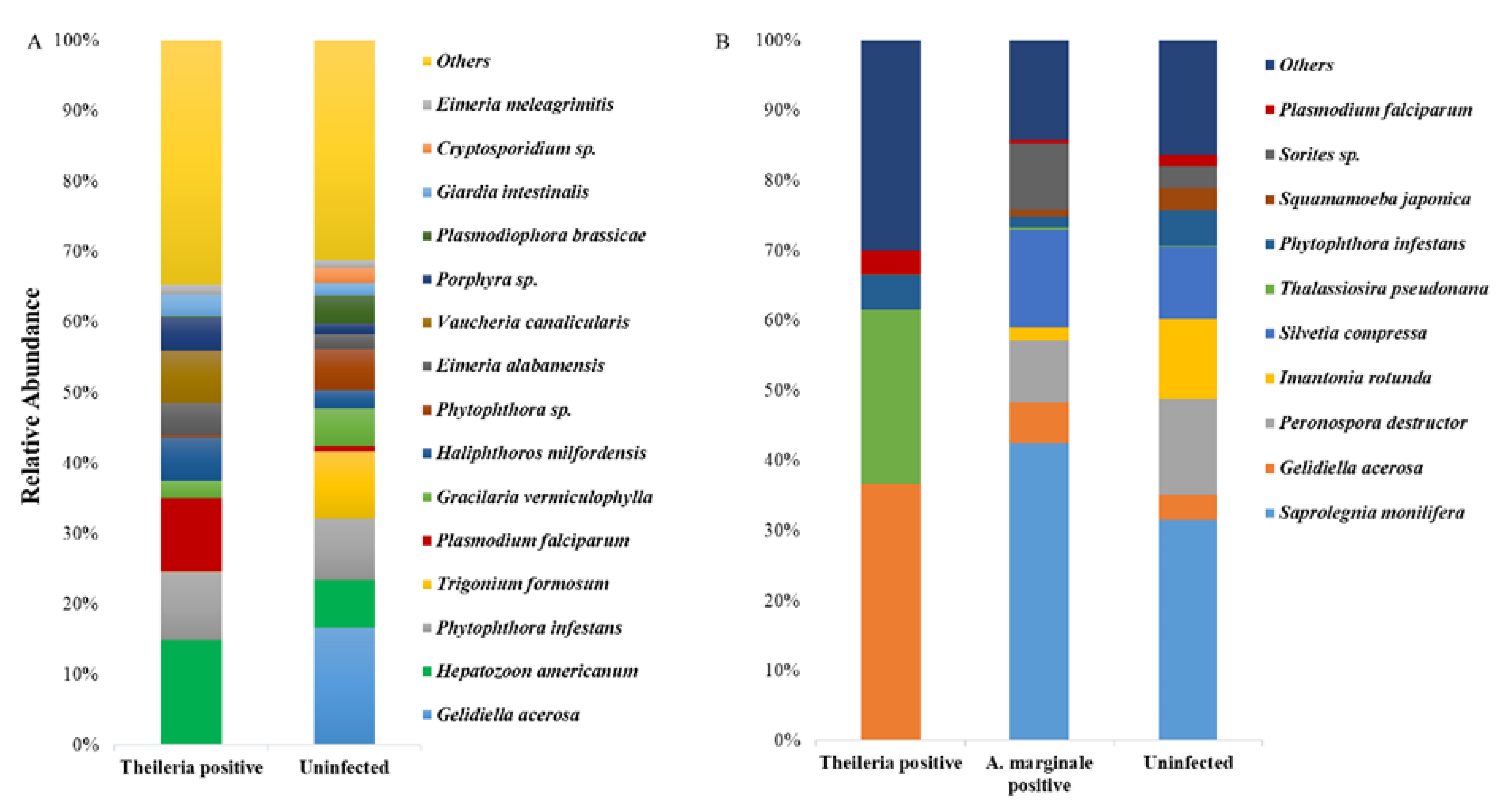
3.4. Eukaryote 18S rRNA Abundance
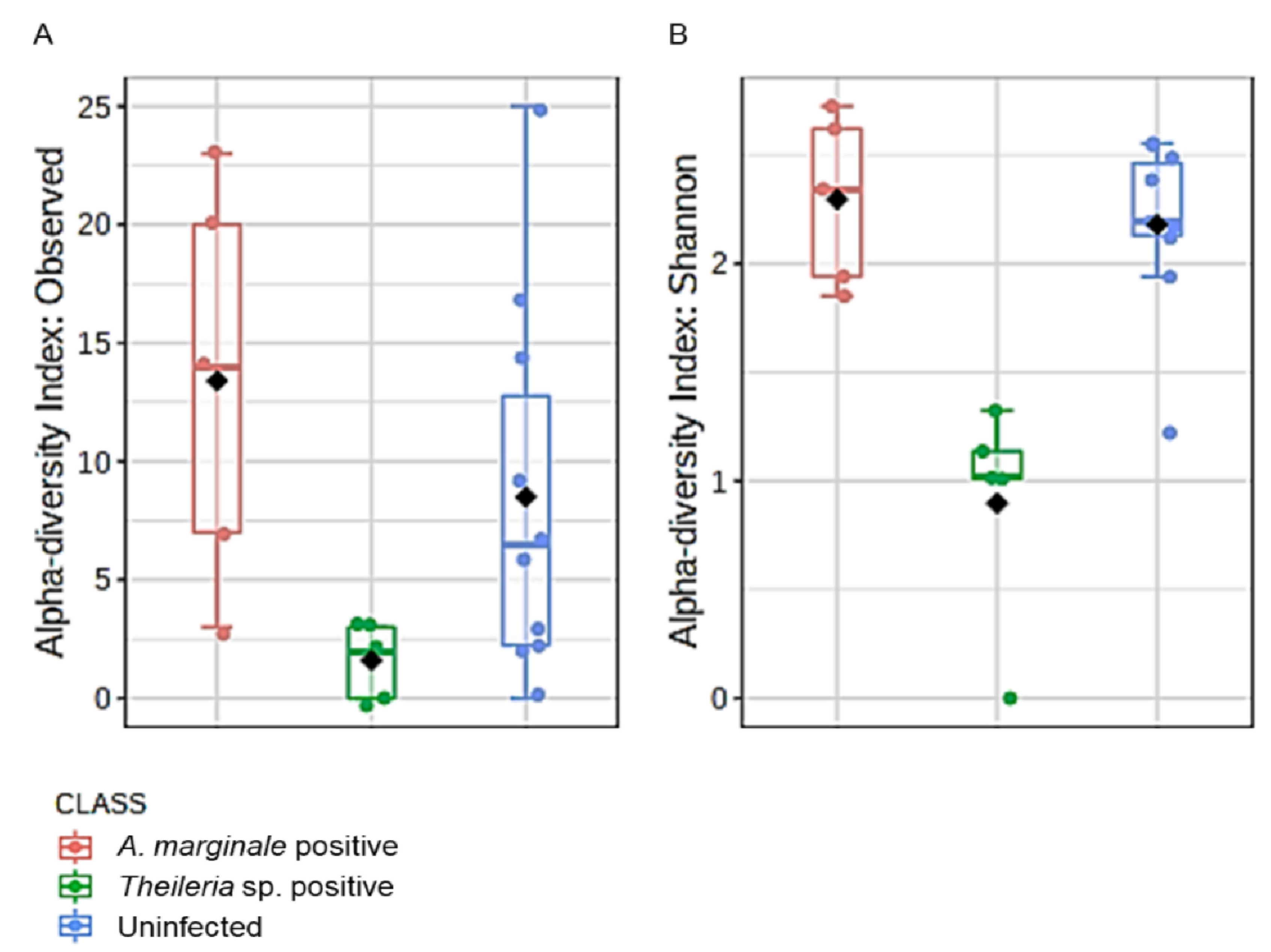
3.5. Microbial Richness and Evenness
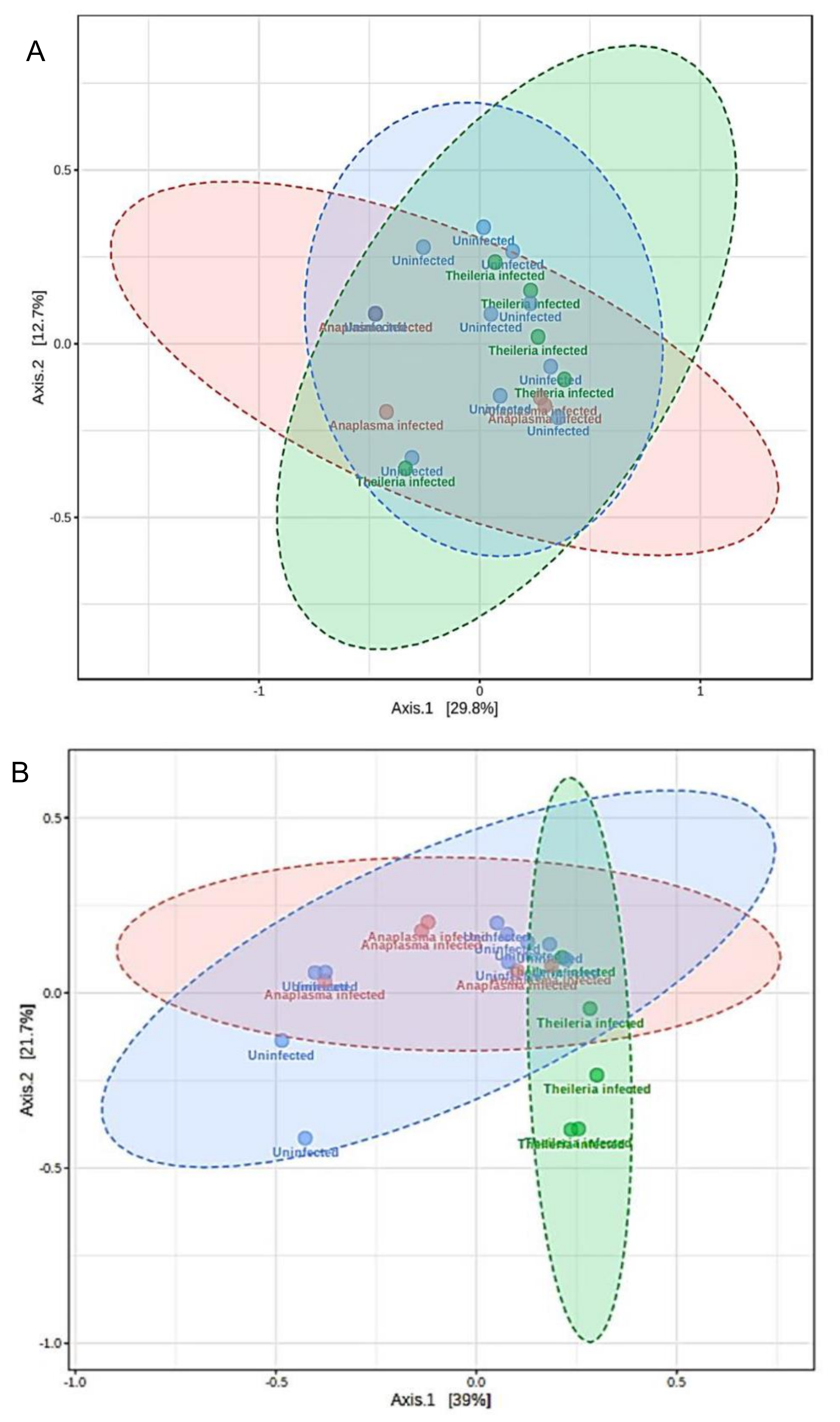
3.6. Microbial Similarity/Dissimilarity Patterns
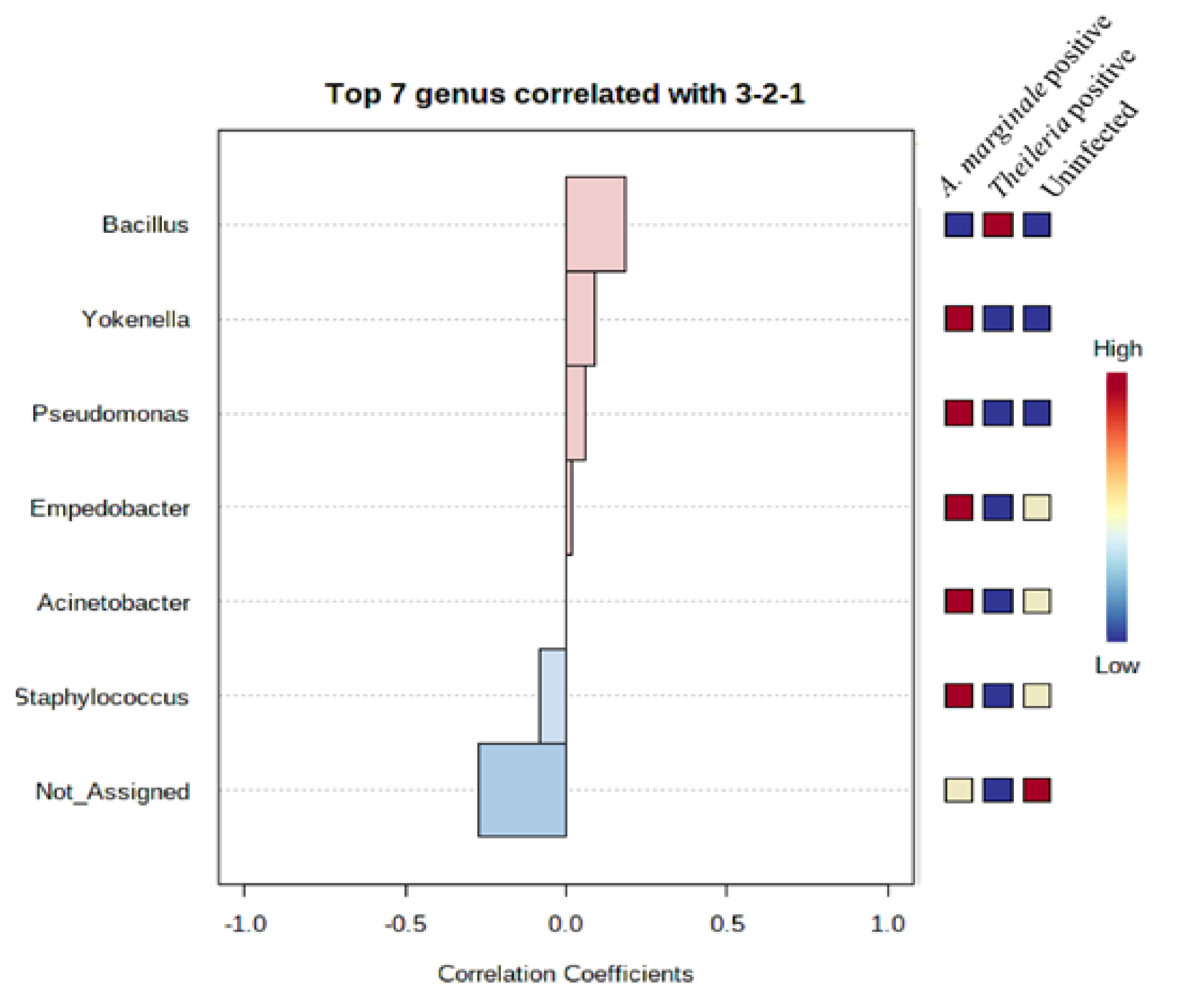
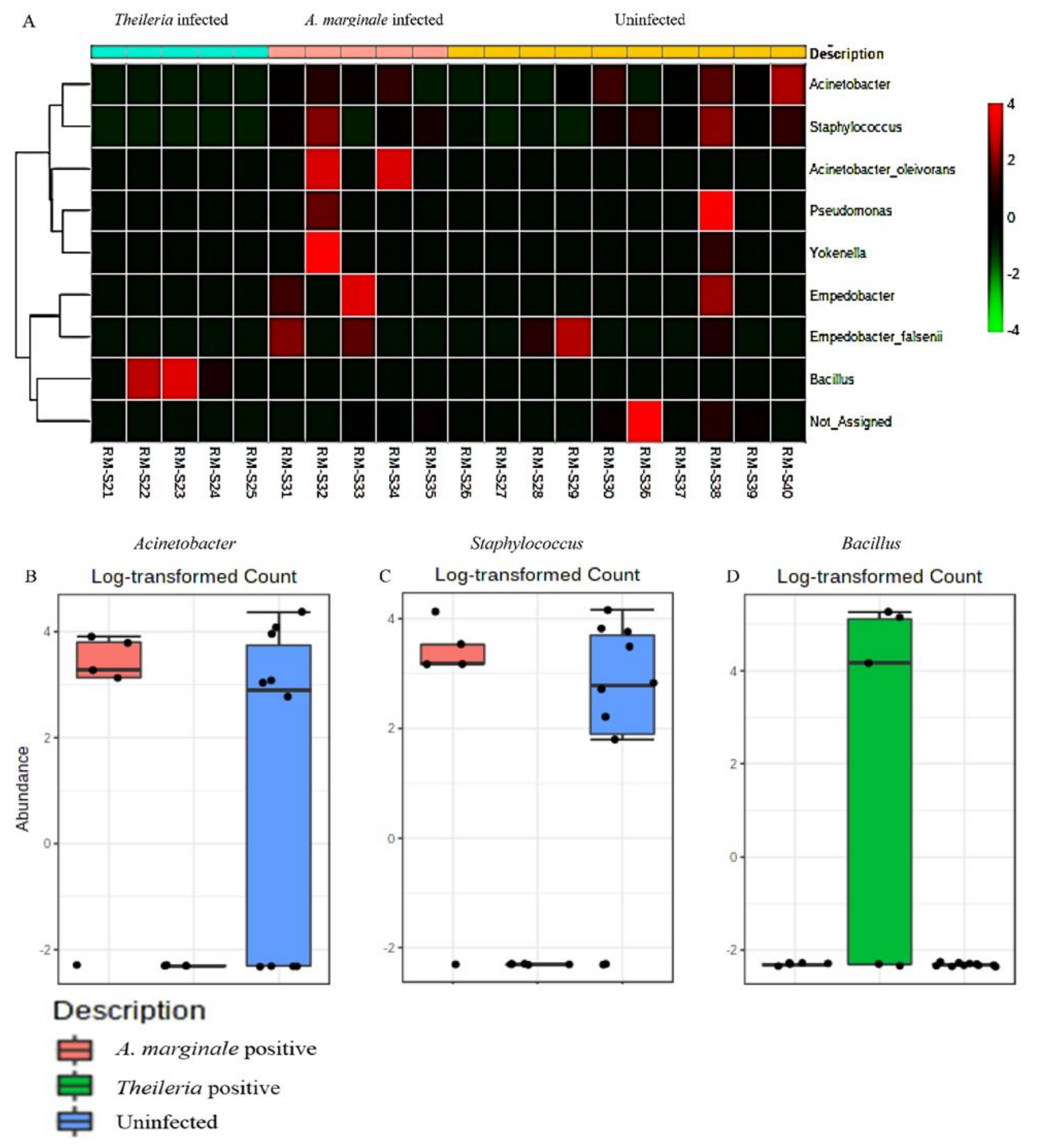
3.7. Community Profiling and Correlation Analysis of R. microplus Ticks
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Availability of data and material
References
- Ministry of Finance, Government of Pakistan. Available online: http://www.finance.gov.pk/survey_1819.html (accessed on 11 July 2020).
- Hoogstraal, H.; Varma, M.G.R. Haemaphysalis cornupunctata sp. n. and H. kashmirensis sp. n. from Kashmir, with Notes on H. sundrai Sharif and H. sewelli Sharif of India and Pakistan (Ixodoidea, Ixodidae). J. Parasitol. 1962, 48, 185. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.G.; Wisseman, C.L.; Traub, R. Tick-borne rickettsiae of the spotted fever group in West Pakistan: I. Isolation of strains from ticks in different habitats. Am. J. Epidemiol. 1970, 92, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.; Budachetri, K.; Mukherjee, N.; Williams, J.; Kausar, A.; Hassan, M.J.; Adamson, S.; Dowd, S.E.; Apanskevich, D.; Arijo, A.; et al. A study of ticks and tick-borne livestock pathogens in Pakistan. PLoS Negl. Trop. Dis. 2017, 11, e0005681. [Google Scholar] [CrossRef]
- Zeb, J.; Szekeres, S.; Takács, N.; Kontschán, J.; Shams, S.; Ayaz, S.; Hornok, S. Genetic diversity, piroplasms and trypanosomes in Rhipicephalus microplus and Hyalomma anatolicum collected from cattle in northern Pakistan. Exp. Appl. Acarol. 2019, 79, 233–243. [Google Scholar] [CrossRef]
- Jabbar, A.; Abbas, T.; Sandhu, Z.U.D.; Saddiqi, H.A.; Qamar, M.F.; Gasser, R.B. Tick-borne diseases of bovines in Pakistan: Major scope for future research and improved control. Parasites Vectors 2015, 8, 283. [Google Scholar] [CrossRef]
- Atif, M.; Saqib, A.; Ikram, R.; Sarwar, M.R.; Scahill, S. The reasons why Pakistan might be at high risk of Crimean Congo hemorrhagic fever epidemic; a scoping review of the literature. Virol. J. 2017, 14, 63. [Google Scholar] [CrossRef]
- Sjödin, A.; Svensson, K.; Ohrman, C.; Ahlinder, J.; Lindgren, P.; Duodu, S.; Johansson, A.; Colquhoun, D.J.; Larsson, P.; Forsman, M. Genome characterization of the genus Francisella reveals insight into similar evolutionary paths in pathogens of mammals and fish. BMC Genomics 2012, 13, 268. [Google Scholar] [CrossRef]
- Gottlieb, Y.; Lalzar, I.; Klasson, L. Distinctive genome reduction rates revealed by genomic analyses of two Coxiella-like endosymbionts in ticks. Genome Biol. Evol. 2015, 7, 1779–1796. [Google Scholar] [CrossRef]
- Smith, T.A.; Driscoll, T.; Gillespie, J.J.; Raghavan, R. A Coxiella-like endosymbiont is a potential vitamin source for the lone star tick. Genome Biol. Evol. 2015, 7, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, S.; Rajeevan, N.; Liu, L.; Zhao, Y.O.; Heisig, J.; Pan, J.; Eppler-Epstein, R.; Deponte, K.; Fish, D.; Fikrig, E. Gut microbiota of the tick vector Ixodes scapularis modulate colonization of the Lyme disease spirochete. Cell Host Microbe 2014, 15, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Abraham, N.M.; Liu, L.; Jutras, B.L.; Yadav, A.K.; Narasimhan, S.; Gopalakrishnan, V.; Ansari, J.M.; Jefferson, K.K.; Cava, F.; Jacobs-Wagner, C.; et al. Pathogen-mediated manipulation of arthropod microbiota to promote infection. Proc. Natl. Acad. Sci. USA 2017, 114, E781–E790. [Google Scholar] [CrossRef] [PubMed]
- Dowd, S.E.; Callaway, T.R.; Wolcott, R.D.; Sun, Y.; McKeehan, T.; Hagevoort, R.G.; Edrington, T.S. Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol. 2008, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, N.; Beati, L.; Sellers, M.; Burton, L.; Adamson, S.; Robbins, R.G.; Moore, F.; Karim, S. Importation of exotic ticks and tick-borne spotted fever group rickettsiae into the United States by migrating songbirds. Ticks Tick-borne Dis. 2014, 5, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, L.; Li, J. Molecular phylogeny and morphological distinctions of two popular bivalves, Ctenoides scabei and Ctenoides mitis. J. Mar. Boil. 2017, 2017, 1–9. [Google Scholar] [CrossRef]
- Kundave, V.; Ram, H.; Banerjee, P.S.; Garg, R.; Mahendran, K.; Ravikumar, G.; Tiwari, A.K. Development of multiplex PCR assay for concurrent detection of tick-borne haemoparasitic infections in bovines. Acta Parasitol. 2018, 63, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Masui, S.; Sasaki, T.; Ishikawa, H. groE-Homologous Operon of Wolbachia, an Intracellular Symbiont of Arthropods: A New Approach for Their Phylogeny. Zool. Sci. 1997, 14, 701–706. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using Microbiome Analyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. Microbiome Analyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]
- Budachetri, K.; Kumar, D.; Crispell, G.; Beck, C.; Dasch, G.; Karim, S. The tick endosymbiont Candidatus Midichloria mitochondrii and selenoproteins are essential for the growth of Rickettsia parkeri in the Gulf Coast tick vector. Microbiome 2018, 6, 141. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, S.; Schuijt, T.J.; Abraham, N.M.; Rajeevan, N.; Coumou, J.; Graham, M.; Robson, A.; Wu, M.-J.; Daffre, S.; Hovius, J.W.; et al. Modulation of the tick gut milieu by a secreted tick protein favors Borrelia burgdorferi colonization. Nat. Commun. 2017, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Fryxell, R.T.T.; DeBruyn, J.M. The Microbiome of Ehrlichia-infected and uninfected lone star ticks (Amblyomma americanum). PLoS ONE 2016, 11, e0146651. [Google Scholar]
- Durrani, A.Z.; Kamal, N. Identification of ticks and detection of blood protozoa in Friesian cattle by polymerase chain reaction test and estimation of blood parameters in district Kasur, Pakistan. Trop. Anim. Heal. Prod. 2008, 40, 441–447. [Google Scholar] [CrossRef]
- Rehman, A.; Nijhof, A.M.; Sauter-Louis, C.; Schauer, B.; Staubach, C.; Conraths, F.J. Distribution of ticks infesting ruminants and risk factors associated with high tick prevalence in livestock farms in the semi-arid and arid agro-ecological zones of Pakistan. Parasites Vectors 2017, 10, 1–15. [Google Scholar] [CrossRef]
- Budachetri, K.; Browning, R.E.; Adamson, S.W.; Dowd, S.E.; Chao, C.-C.; Ching, W.-M.; Karim, S. An insight into the microbiome of the Amblyomma maculatum (Acari: Ixodidae). J. Med. Entomol. 2014, 51, 119–129. [Google Scholar] [CrossRef]
- Travanty, N.V.; Ponnusamy, L.; Kakumanu, M.L.; Nicholson, W.L.; Apperson, C.S. Diversity and structure of the bacterial microbiome of the American dog tick, Dermacentor variabilis, is dominated by the endosymbiont Francisella. Symbiosis 2019, 79, 1–12. [Google Scholar] [CrossRef]
- Swei, A.; Kwan, J.Y. Tick microbiome and pathogen acquisition altered by host blood meal. ISME J. 2017, 11, 813–816. [Google Scholar] [CrossRef]
- Epis, S.; Mandrioli, M.; Genchi, M.; Montagna, M.; Sacchi, L.; Pistone, D.; Sassera, D. Localization of the bacterial symbiont Candidatus Midichloria mitochondrii within the hard tick Ixodes ricinus by whole-mount FISH staining. Ticks Tick-borne Dis. 2013, 4, 39–45. [Google Scholar] [CrossRef]
- Dergousoff, S.J.; Chilton, N.B. Association of different genetic types of Francisella-like organisms with the rocky mountain wood tick (Dermacentor andersoni) and the American Dog Tick (Dermacentor variabilis) in Localities Near Their Northern Distributional Limits. Appl. Environ. Microbiol. 2011, 78, 965–971. [Google Scholar] [CrossRef]
- Chakraborty, S.; Roy, S.; Mistry, H.U.; Murthy, S.; George, N.; Bhandari, V.; Sharma, P. Potential sabotage of host cell physiology by apicomplexan parasites for their survival benefits. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Bahia, A.C.; Oliveira, J.H.M.; Kubota, M.S.; Araújo, H.R.C.; Lima, J.B.P.; Ríos-Velásquez, C.M.; Lacerda, M.V.G.; De Oliveira, P.L.; Traub-Csekö, Y.M.; Pimenta, P.F.P. The Role of Reactive Oxygen Species in Anopheles aquasalis Response to Plasmodium vivax Infection. PLoS ONE 2013, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mann, A.E.; Mitchell, E.A.; Zhang, Y.; Curtis-Robles, R.; Thapa, S.; Hamer, S.A.; Allen, M.S. Comparison of the bacterial gut microbiome of North American Triatoma spp. With and without Trypanosoma cruzi. Front. Microbiol. 2020, 11, 364. [Google Scholar] [CrossRef] [PubMed]
- Hilgenboecker, K.; Hammerstein, P.; Schlattmann, P.; Telschow, A.; Werren, J.H. How many species are infected with Wolbachia? A statistical analysis of current data. FEMS Microbiol. Lett. 2008, 281, 215–220. [Google Scholar] [CrossRef]
- Sicard, M.; Bonneau, M.; Weill, M. Wolbachia prevalence, diversity, and ability to induce cytoplasmic incompatibility in mosquitoes. Curr. Opin. Insect Sci. 2019, 34, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Stouthamer, R.; Breeuwer, J.A.J.; Hurst, G.D.D. Wolbachia Pipientis: Microbial manipulator of arthropod reproduction. Annu. Rev. Microbiol. 1999, 53, 71–102. [Google Scholar] [CrossRef]
- Plantard, O.; Bouju-Albert, A.; Malard, M.-A.; Hermouet, A.; Capron, G.; Verheyden, H. Detection of Wolbachia in the tick Ixodes ricinus is due to the presence of the Hymenoptera endoparasitoid Ixodiphagus hookeri. PLoS ONE 2012, 7, e30692. [Google Scholar] [CrossRef]
- Tijsse-Klasen, E.; Braks, M.; Scholte, E.J.; Sprong, H. Parasites of vectors—Ixodiphagus hookeri and its Wolbachia symbionts in ticks in The Netherlands. Parasites Vectors 2011, 4, 228. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Genes | Primer Sequence (5’–3’) | Amplicon Size (bp) | References |
|---|---|---|---|
| Amar 16S-F | GGC GGT GAT CTG TAG CTG GTC TGA | 270 bp | [16] |
| Amar 16S-R | GCC CAA TAA TTC CGA ACA ACG CTT | ||
| Theileria sp 18S-F | GGT AAT TCC AGC TCCAAT AG | 300 bp | [4] |
| Theileria sp 18S-R | ACC AAC AAA ATA GAA CCA AAG TC | ||
| 16S rRNA 27F | AGR GTT TGA TCM TGG CTC AG | V1–V3 | [13] |
| 16S rRNA 519R | GTN TTA CNG CGG CKG CTG | ||
| COI-F | GGT CAA CAA ATC ATA AAG ATA TTG G | 708 bp | [15] |
| COI-R | TAA ACT TCA GGG TGA CCA AAA AAT CA | ||
| Wolbachia sp GroEL-F | TGT ATT AGA TGA TAA CGT GC | 800 bp | [17] |
| Wolbachia sp GroEL-R | CCA TTT GCA GAA ATT ATT GCA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adegoke, A.; Kumar, D.; Bobo, C.; Rashid, M.I.; Durrani, A.Z.; Sajid, M.S.; Karim, S. Tick-Borne Pathogens Shape the Native Microbiome Within Tick Vectors. Microorganisms 2020, 8, 1299. https://doi.org/10.3390/microorganisms8091299
Adegoke A, Kumar D, Bobo C, Rashid MI, Durrani AZ, Sajid MS, Karim S. Tick-Borne Pathogens Shape the Native Microbiome Within Tick Vectors. Microorganisms. 2020; 8(9):1299. https://doi.org/10.3390/microorganisms8091299
Chicago/Turabian StyleAdegoke, Abdulsalam, Deepak Kumar, Cailyn Bobo, Muhammad Imran Rashid, Aneela Zameer Durrani, Muhammad Sohail Sajid, and Shahid Karim. 2020. "Tick-Borne Pathogens Shape the Native Microbiome Within Tick Vectors" Microorganisms 8, no. 9: 1299. https://doi.org/10.3390/microorganisms8091299
APA StyleAdegoke, A., Kumar, D., Bobo, C., Rashid, M. I., Durrani, A. Z., Sajid, M. S., & Karim, S. (2020). Tick-Borne Pathogens Shape the Native Microbiome Within Tick Vectors. Microorganisms, 8(9), 1299. https://doi.org/10.3390/microorganisms8091299