Comparative Transcriptomics Reveals Distinct Gene Expressions of a Model Ciliated Protozoan Feeding on Bacteria-Free Medium, Digestible, and Digestion-Resistant Bacteria


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Organisms and Cultures
2.2. Isolation and Identification of DRB Candidate Strains and Feeding Experiments
2.3. Fluorescence In Situ Hybridization (FISH) Assays
2.4. RNA Extraction, cDNA Library Construction and Transcriptome Sequencing
2.5. Processing and Analysis of Transcriptomic Data
3. Results
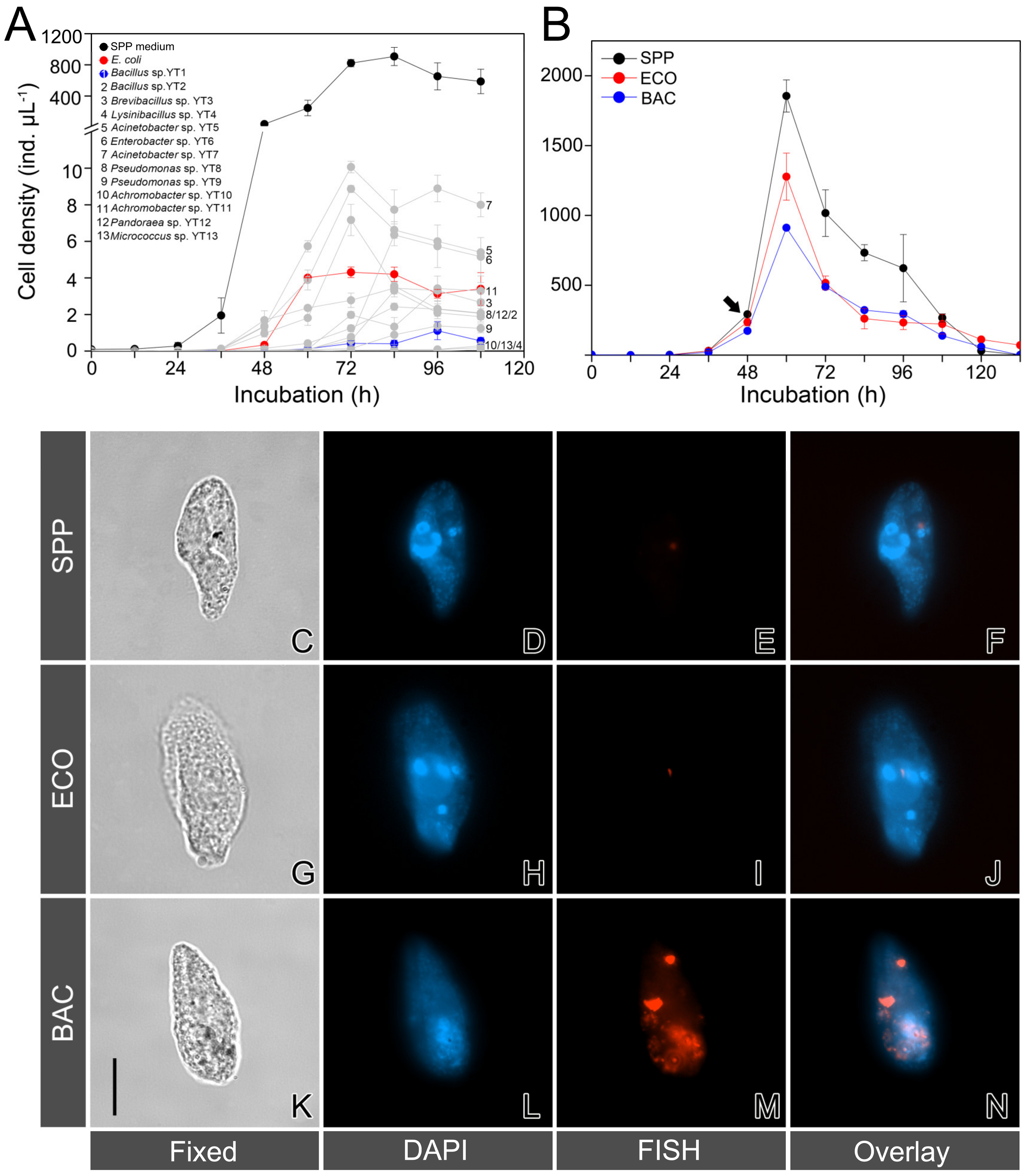
3.1. Growth Performance of T. thermophila Feeding on Digestion-Resistant Bacteria
3.2. Validation of Digestion-Resistant Bacteria Using FISH
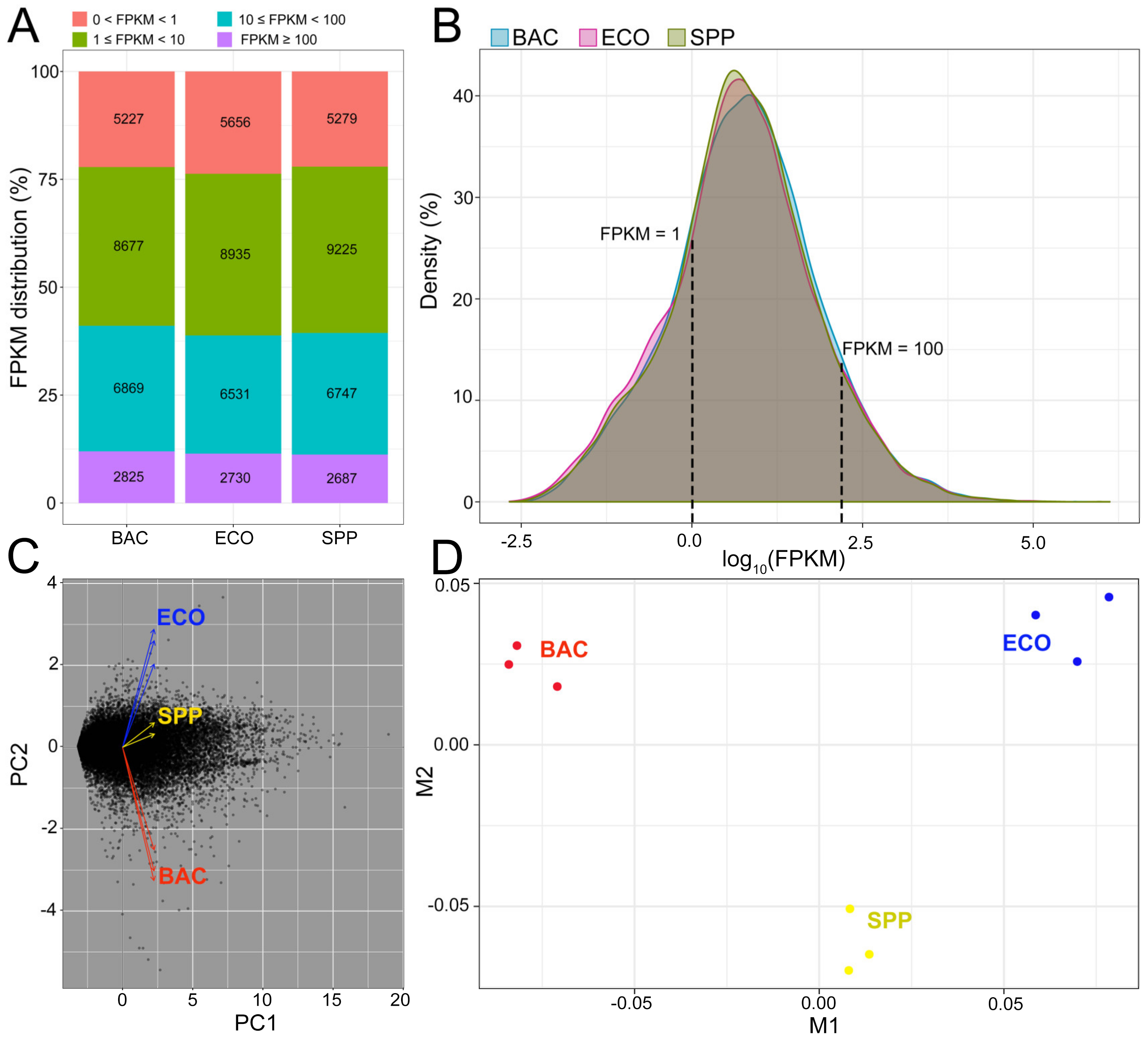
3.3. Transcriptome Overview
3.4. Overview of Differentially Expressed Genes (DEGs)
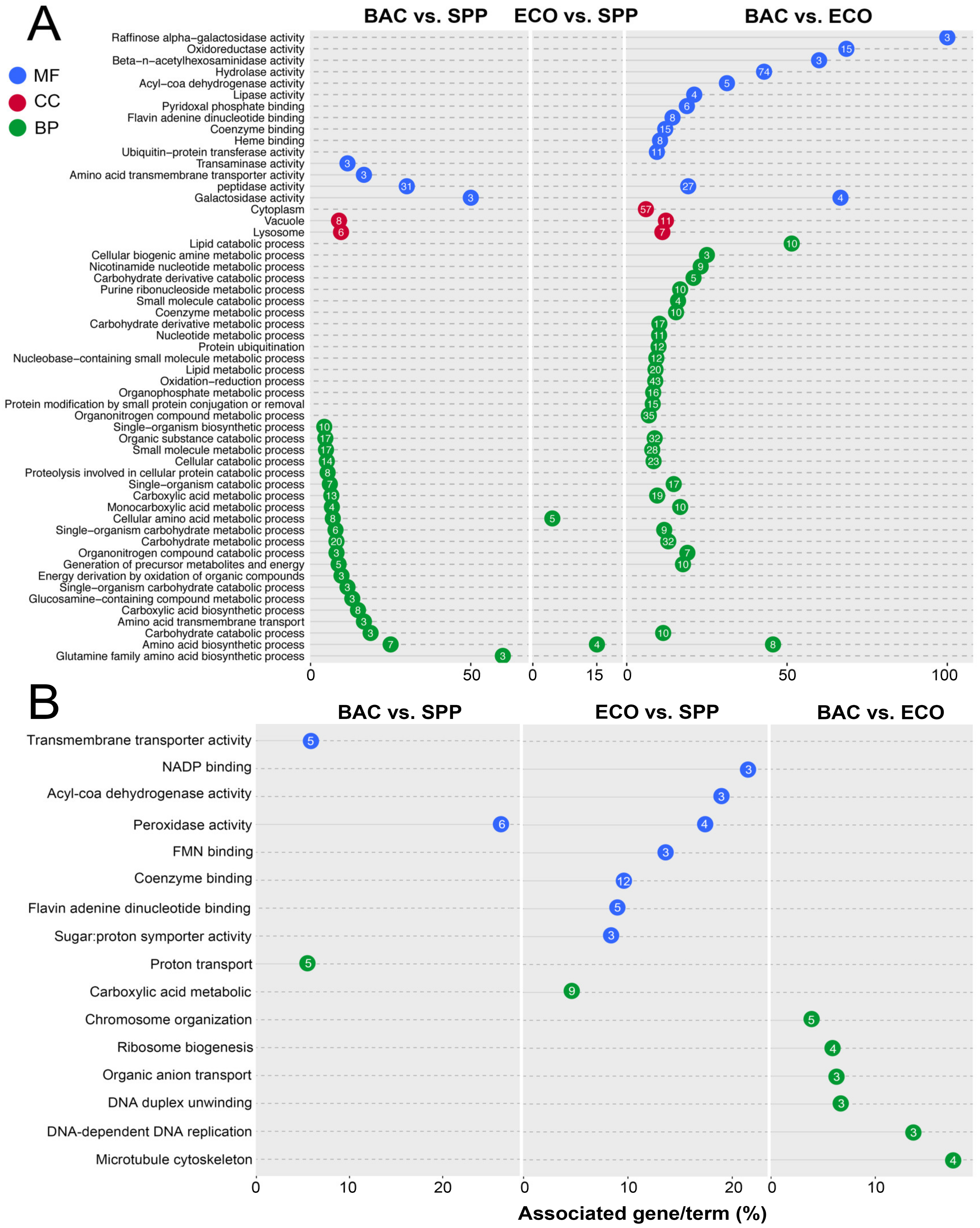
3.5. Functional Annotation and Enrichment of DEGs
3.6. Transcriptional Expression Patterns Inferred from KEGG Annotations
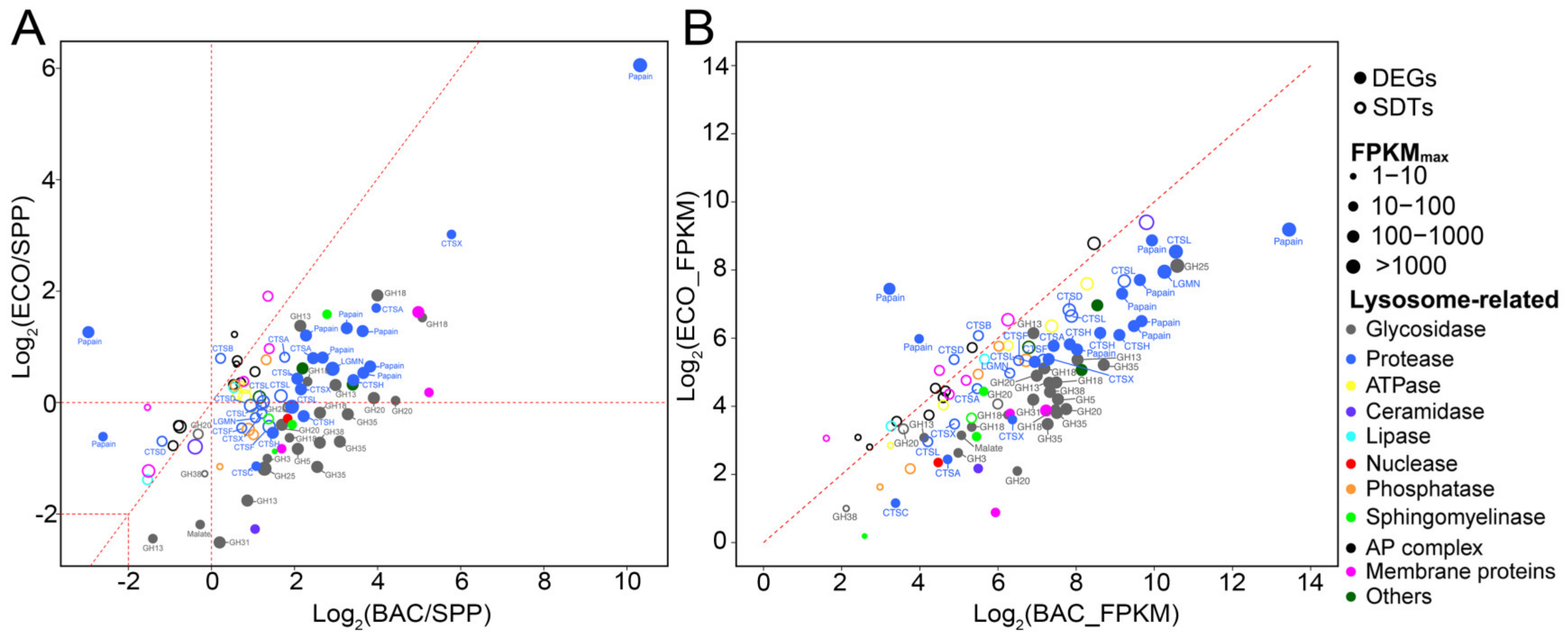
3.6.1. Differential Expression of Lysosomal Genes
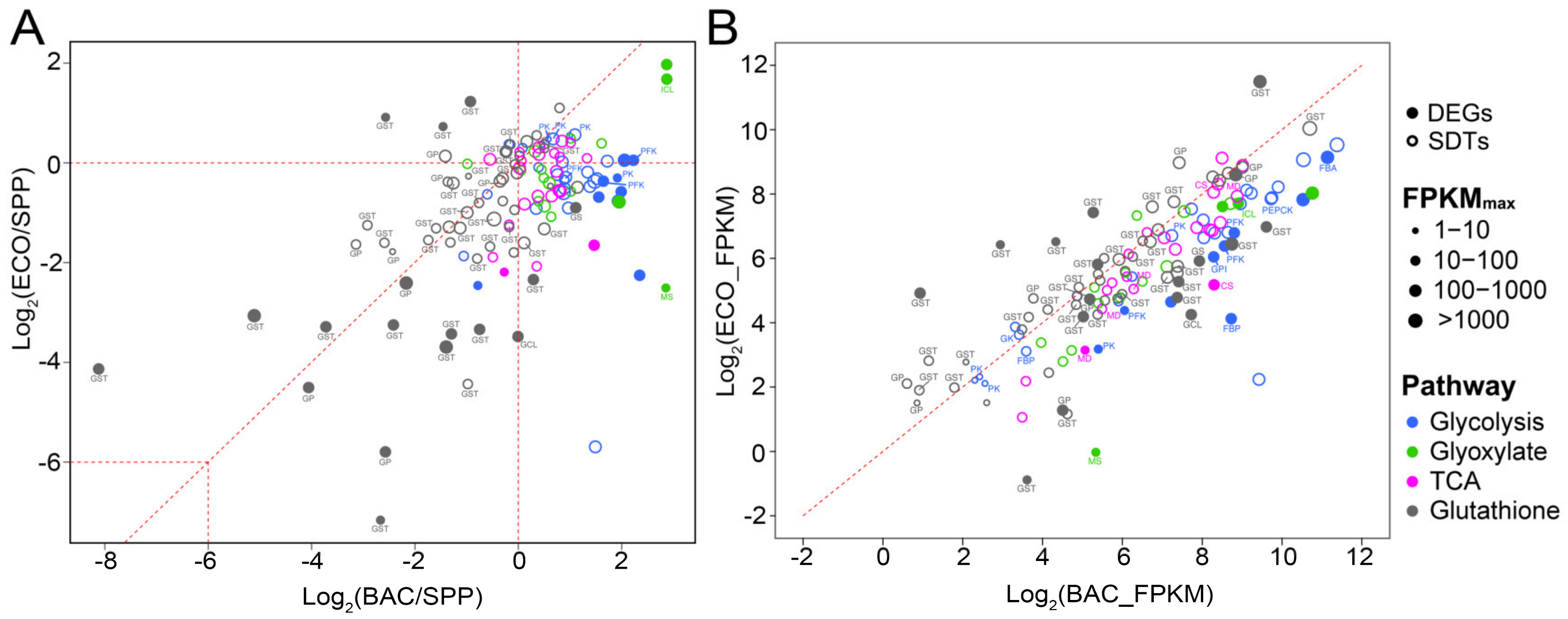
3.6.2. Differential Expression of Genes Involved in Glycolytic, TCA and Glyoxylate Cycle
3.6.3. Glutathione Metabolism
4. Discussion
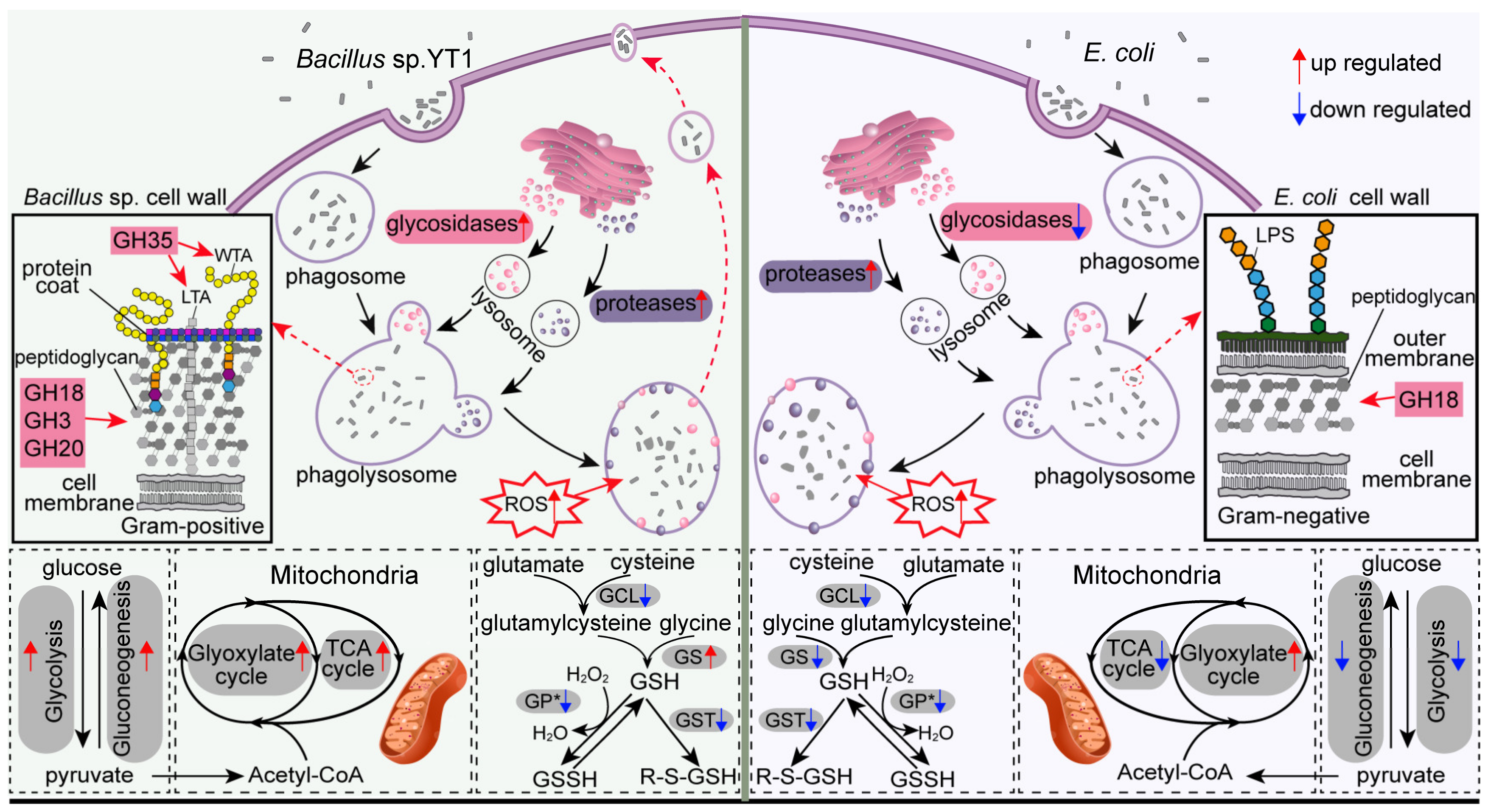
4.1. Increased Protistan Lysosomal Protease, Chitinase, and Glyoxylate Cycle-Related Activities for Digesting Bacterial Preys
4.2. Higher Expression Levels of Protease, Glycosidase, and Carbon Metabolism-Related Genes in Response to DRB vs. Digestible Bacteria
4.3. Lower Expression of Glutathione (GSH) Metabolism in Tetrahymena feeding on Bacteria
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Azam, F.; Fenchel, T.; Field, J.G.; Gray, J.S.; Meyer-Reil, L.A.; Thingstad, F. The ecological role of water-column microbes in the sea. Mar. Ecol. Prog. Ser. 1983, 10, 257–263. [Google Scholar] [CrossRef]
- Massana, R.; Unrein, F.; Rodríguez-Martínez, R.; Forn, I.; Lefort, T.; Pinhassi, J.; Not, F. Grazing rates and functional diversity of uncultured heterotrophic flagellates. ISME J. 2009, 3, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Matz, C.; Kjelleberg, S. Off the hook-how bacteria survive protozoan grazing. Trends Microbiol. 2005, 13, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Pernthaler, J. Predation on prokaryotes in the water column and its ecological implications. Nat. Rev. Microbiol. 2005, 3, 537–546. [Google Scholar] [CrossRef]
- Jousset, A. Ecological and evolutive implications of bacterial defences against predators. Environ. Microbiol. 2012, 14, 1830–1843. [Google Scholar] [CrossRef] [PubMed]
- First, M.R.; Park, N.Y.; Berrang, M.E.; Meinersmann, R.J.; Bernhard, J.M.; Gast, R.J.; Hollibaugh, J.T. Ciliate ingestion and digestion: Flow cytometric measurements and regrowth of a digestion-resistant Campylobacter jejuni. Front. Microbiol. 2012, 59, 12–19. [Google Scholar] [CrossRef]
- Martinez-Garcia, M.; Brazel, D.; Poulton, N.J.; Swan, B.K.; Gomez, M.L.; Masland, D.; Sieracki, M.E.; Stepanauskas, R. Unveiling in situ interactions between marine protists and bacteria through single cell sequencing. ISME J. 2012, 6, 703–707. [Google Scholar] [CrossRef]
- Gong, J.; Qing, Y.; Zou, S.; Fu, R.; Su, L.; Zhang, X.; Zhang, Q. Protist-bacteria associations: Gammaproteobacteria and alphaproteobacteria are prevalent as digestion-resistant bacteria in ciliated protozoa. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef]
- Šimek, K.; Kasalický, V.; Jezbera, J.; Horňák, K.; Nedoma, J.; Hahn, M.W.; Bass, D.; Jost, S.; Boenigk, J. Differential freshwater flagellate community response to bacterial food quality with a focus on Limnohabitans bacteria. ISME J. 2013, 7, 1519–1530. [Google Scholar] [CrossRef]
- Jousset, A.; Rochat, L.; Scheu, S.; Bonkowski, M.; Keel, C. Predator-prey chemical warfare determines the expression of biocontrol genes by rhizosphere-associated Pseudomonas fluorescens. Appl. Environ. Microbiol. 2010, 76, 5263. [Google Scholar] [CrossRef]
- Song, C.; Mazzola, M.; Cheng, X.; Oetjen, J.; Alexandrov, T.; Dorrestein, P.; Watrous, J.; van der Voort, M.; Raaijmakers, J.M. Molecular and chemical dialogues in bacteria-protozoa interactions. Sci. Rep. 2015, 5, 12837. [Google Scholar] [CrossRef]
- Matz, C.; Deines, P.; Boenigk, J.; Arndt, H.; Eberl, L.; Kjelleberg, S.; Jürgens, K. Impact of violacein-producing bacteria on survival and feeding of bacterivorous nanoflagellates. Appl. Environ. Microbiol. 2004, 70, 1593. [Google Scholar] [CrossRef]
- Caron, D.A.; Alexander, H.; Allen, A.E.; Archibald, J.M.; Armbrust, E.V.; Bachy, C.; Bell, C.J.; Bharti, A.; Dyhrman, S.T.; Guida, S.M.; et al. Probing the evolution, ecology and physiology of marine protists using transcriptomics. Nat. Rev. Microbiol. 2017, 15, 6–20. [Google Scholar] [CrossRef]
- Moustafa, A.; Evans, A.N.; Kulis, D.M.; Hackett, J.D.; Erdner, D.L.; Anderson, D.M.; Bhattacharya, D. Transcriptome profiling of a toxic dinoflagellate reveals a gene-rich protist and a potential impact on gene expression due to bacterial presence. PLoS ONE 2010, 5, e9688. [Google Scholar] [CrossRef] [PubMed]
- Lie, A.A.Y.; Liu, Z.; Terrado, R.; Tatters, A.O.; Heidelberg, K.B.; Caron, D.A. Effect of light and prey availability on gene expression of the mixotrophic chrysophyte, Ochromonas sp. BMC Genom. 2017, 18, 163. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jones, A.C.; Campbell, V.; Hambright, K.D.; Heidelberg, K.B.; Caron, D.A. Gene expression in the mixotrophic prymnesiophyte, Prymnesium parvum, responds to prey availability. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef]
- Lie, A.A.Y.; Liu, Z.; Terrado, R.; Tatters, A.O.; Heidelberg, K.B.; Caron, D.A. A tale of two mixotrophic chrysophytes: Insights into the metabolisms of two Ochromonas species (Chrysophyceae) through a comparison of gene expression. PLoS ONE 2018, 13, e0192439. [Google Scholar] [CrossRef] [PubMed]
- Sillo, A.; Bloomfield, G.; Balest, A.; Balbo, A.; Pergolizzi, B.; Peracino, B.; Skelton, J.; Ivens, A.; Bozzaro, S. Genome-wide transcriptional changes induced by phagocytosis or growth on bacteria in Dictyostelium. BMC Genom. 2008, 9, 291. [Google Scholar] [CrossRef]
- Eisen, J.A.; Coyne, R.S.; Wu, M.; Wu, D.; Thiagarajan, M.; Wortman, J.R.; Badger, J.H.; Ren, Q.; Amedeo, P.; Jones, K.M.; et al. Macronuclear genome sequence of the ciliate Tetrahymena thermophila, a model eukaryote. PLoS Biol. 2006, 4, e286. [Google Scholar] [CrossRef]
- Xiong, J.; Lu, X.; Zhou, Z.; Chang, Y.; Yuan, D.; Tian, M.; Zhou, Z.; Wang, L.; Fu, C.; Orias, E.; et al. Transcriptome analysis of the model protozoan, Tetrahymena thermophila, using deep RNA sequencing. PLoS ONE 2012, 7, e30630. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Marsh, T.; Garrity, G.M.; et al. The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis. Nucl. Acids Res. 2009, 37, D141–D145. [Google Scholar] [CrossRef] [PubMed]
- Fried, J.; Ludwig, W.; Psenner, R.; Schleifer, K.H. Improvement of ciliate identification and quantification: A new protocol for fluorescence in situ hybridization (FISH) in combination with silver stain techniques. Syst. Appl. Microbiol. 2002, 25, 555–571. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Qing, Y.; Guo, X.; Warren, A. “Candidatus Sonnebornia yantaiensis”, a member of candidate division OD1, as intracellular bacteria of the ciliated protist Paramecium bursaria (Ciliophora, Oligohymenophorea). Syst. Appl. Microbiol. 2014, 37, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucl. Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed]
- Kirschke, H.; Langner, J.; Wiederanders, B.; Ansorge, S.; Bohley, P.; Hanson, H. Cathepsin H: An endoaminopeptidase from rat liver lysosomes. Acta Biol. Med. Ger. 1977, 36, 185–199. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta Proteins Proteom. 2012, 1824, 68–88. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Sudek, S.; McMullan, D.; Miller, M.D.; Geierstanger, B.; Jones, D.H.; Krishna, S.S.; Spraggon, G.; Bursalay, B.; Abdubek, P.; et al. Structural basis of murein peptide specificity of a γ-D-glutamyl-L-diamino acid endopeptidase. Structure 2009, 17, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Klemenčič, I.; Carmona, A.K.; Cezari, M.H.S.; Juliano, M.A.; Juliano, L.; Gunčar, G.; Turk, D.; Križaj, I.; Turk, V.; Turk, B. Biochemical characterization of human cathepsin X revealed that the enzyme is an exopeptidase, acting as carboxymonopeptidase or carboxydipeptidase. Eur. J. Biochem. 2000, 267, 5404–5412. [Google Scholar] [CrossRef]
- Azzo, A.; Hoogeveen, A.; Reuser, A.J.; Robinson, D.; Galjaard, H. Molecular defect in combined beta-galactosidase and neuraminidase deficiency in man. Proc. Natl. Acad. Sci. USA 1982, 79, 4535. [Google Scholar] [CrossRef]
- Barrett, A.J.; Rawlings, N.D. Families and clans of cysteine peptidases. Perspect. Drug. Discov. 1996, 6, 1–11. [Google Scholar] [CrossRef]
- Ahn, Y.O.; Zheng, M.; Bevan, D.R.; Esen, A.; Shiu, S.; Benson, J.; Peng, H.; Miller, J.T.; Cheng, C.; Poulton, J.E.; et al. Functional genomic analysis of Arabidopsis thaliana glycoside hydrolase family 35. Phytochemistry 2007, 68, 1510–1520. [Google Scholar] [CrossRef]
- Stam, M.R.; Danchin, E.G.J.; Rancurel, C.; Coutinho, P.M.; Henrissat, B. Dividing the large glycoside hydrolase family 13 into subfamilies: Towards improved functional annotations of α-amylase-related proteins. Protein Eng. Des. Sel. 2006, 19, 555–562. [Google Scholar] [CrossRef]
- Aspeborg, H.; Coutinho, P.M.; Wang, Y.; Brumer, H.; Henrissat, B. Evolution, substrate specificity and subfamily classification of glycoside hydrolase family 5 (GH5). BMC Evol. Biol. 2012, 12, 186. [Google Scholar] [CrossRef]
- Cobucci-Ponzano, B.; Conte, F.; Strazzulli, A.; Capasso, C.; Fiume, I.; Pocsfalvi, G.; Rossi, M.; Moracci, M. The molecular characterization of a novel GH38 α-mannosidase from the crenarchaeon Sulfolobus solfataricus revealed its ability in de-mannosylating glycoproteins. Biochimie 2010, 92, 1895–1907. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, W.; Blanot, D.; De Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Gunsalus, I.C. Isocitritase: A new tricarboxylic acid cleavage system. J. Am. Chem. Soc. 1954, 76, 5002–5003. [Google Scholar] [CrossRef]
- Tjaden, B.; Plagens, A.; Dörr, C.; Siebers, B.; Hensel, R. Phosphoenolpyruvate synthetase and pyruvate, phosphate dikinase of Thermoproteus tenax: Key pieces in the puzzle of archaeal carbohydrate metabolism. Mol. Microbiol. 2006, 60, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Mokrasch, L.C.; McGilvery, R.W. Purification and properties of fructose-1, 6-diphosphatase. J. Biol. Chem. 1956, 221, 909–917. [Google Scholar] [CrossRef]
- Yang, J.; Kalhan, S.C.; Hanson, R.W. What is the metabolic role of phosphoenolpyruvate carboxykinase? J. Biol. Chem. 2009, 284, 27025–27029. [Google Scholar] [CrossRef]
- Mendoza-Cózatl, D.; Loza-Tavera, H.; Hernández-Navarro, A.; Moreno-Sánchez, R. Sulfur assimilation and glutathione metabolism under cadmium stress in yeast, protists and plants. FEMS Microbiol. Rev. 2005, 29, 653–671. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Aspects Med. 2009, 30, 42–59. [Google Scholar] [CrossRef]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef]
- Boenigk, J.; Matz, C.; Jürgens, K.; Arndt, H. The influence of preculture conditions and food quality on the ingestion and digestion process of three species of heterotrophic nanoflagellates. Microb. Ecol. 2001, 42, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Apple, J.K.; Strom, S.L.; Palenik, B.; Brahamsha, B. Variability in protist grazing and growth on different marine Synechococcus isolates. Appl. Environ. Microbiol. 2011, 77, 3074. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, T.R.; Rappitt, A.F. Endocytosis and the adaptive acid hydrolases in Tetrahymena pyriformis GL. Arch. Microbiol. 1974, 98, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Que, X.; Brinen, L.S.; Perkins, P.; Herdman, S.; Hirata, K.; Torian, B.E.; Rubin, H.; McKerrow, J.H.; Reed, S.L. Cysteine proteinases from distinct cellular compartments are recruited to phagocytic vesicles by Entamoeba histolytica. Mol. Biochem. Parasitol. 2002, 119, 23–32. [Google Scholar] [CrossRef]
- Ishihama, Y.; Schmidt, T.; Rappsilber, J.; Mann, M.; Hartl, F.U.; Kerner, M.J.; Frishman, D. Protein abundance profiling of the Escherichia coli cytosol. BMC Genom. 2008, 9, 102. [Google Scholar] [CrossRef]
- Mullineaux, C.W. How bacteria keep proteins moving. eLife 2017, 6, e33590. [Google Scholar] [CrossRef]
- Hosoda, N.; Kurokawa, Y.; Sako, Y.; Nagasaki, K.; Yoshida, T.; Hiroishi, S. The functional effect of Gly209 and Ile213 substitutions on lysozyme activity of family 19 chitinase encoded by cyanophage Ma-LMM01. Fish. Sci. 2011, 77, 665–670. [Google Scholar] [CrossRef]
- Fukamizo, F. Chitinolytic enzymes: Catalysis, substrate binding, and their application. Curr. Protein Pept. Sci. 2000, 1, 105–124. [Google Scholar] [CrossRef]
- Ensign, S.A. Revisiting the glyoxylate cycle: Alternate pathways for microbial acetate assimilation. Mol. Microbiol. 2006, 61, 274–276. [Google Scholar] [CrossRef]
- Eastmond, P.J.; Graham, I.A. Re-examining the role of the glyoxylate cycle in oilseeds. Trends Plant Sci. 2001, 6, 72–78. [Google Scholar] [CrossRef]
- Prasad Maharjan, R.; Yu, P.L.; Seeto, S.; Ferenci, T. The role of isocitrate lyase and the glyoxylate cycle in Escherichia coli growing under glucose limitation. Res. Microbiol. 2005, 156, 178–183. [Google Scholar] [CrossRef]
- Liu, F.; Thatcher, J.D.; Epstein, H.F. Induction of glyoxylate cycle expression in Caenorhabditis elegans: A fasting response throughout larval development. Biochemistry 1997, 36, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Cruz, A.H.; Brock, M.; Zambuzzi-Carvalho, P.F.; Santos-Silva, L.K.; Troian, R.F.; Góes, A.M.; de Almeida Soares, C.M.; Pereira, M. Phosphorylation is the major mechanism regulating isocitrate lyase activity in Paracoccidioides brasiliensis yeast cells. FEBS J. 2011, 278, 2318–2332. [Google Scholar] [CrossRef] [PubMed]
- Hayhurst, E.J.; Kailas, L.; Hobbs, J.K.; Foster, S.J. Cell wall peptidoglycan architecture in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2008, 105, 14603. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.M.; Phadke, N.D.; Kachman, M.T.; Giorno, R.; Vazquez, S.; Vazquez, J.A.; Maddock, J.R.; Driks, A. Proteomic analysis of the spore coats of Bacillus subtilis and Bacillus anthracis. J. Bacteriol. 2003, 185, 1443. [Google Scholar] [CrossRef]
- Zhang, J.; Fitz-James, P.C.; Aronson, A.I. Cloning and characterization of a cluster of genes encoding polypeptides present in the insoluble fraction of the spore coat of Bacillus subtilis. J. Bacteriol. 1993, 175, 3757. [Google Scholar] [CrossRef]
- Driks, A. Bacillus subtilis spore coat. Microbiol. Mol. Biol. Rev. 1999, 63, 1–20. [Google Scholar] [CrossRef]
- Filion, G.; Laflamme, C.; Turgeon, N.; Ho, J.; Duchaine, C. Permeabilization and hybridization protocols for rapid detection of Bacillus spores using fluorescence in situ hybridization. J. Microbiol. Meth. 2009, 77, 29–36. [Google Scholar] [CrossRef]
- Caron, D.A.; Goldman, J.C.; Dennett, M.R. Experimental demonstration of the roles of bacteria and bacterivorous protozoa in plankton nutrient cycles. Hydrobiologia 1988, 159, 27–40. [Google Scholar] [CrossRef]
- Parry, B.R.; Surovtsev, I.V.; Cabeen, M.T.; O’Hern, C.S.; Dufresne, E.R.; Jacobs-Wagner, C. The bacterial cytoplasm has glass-like properties and is fluidized by metabolic activity. Cell 2014, 156, 183–194. [Google Scholar] [CrossRef]
- Brown, S.; Santa Maria, J.P.; Walker, S. Wall teichoic acids of gram-positive bacteria. Annu. Rev. Microbiol. 2013, 67, 313–336. [Google Scholar] [CrossRef]
- Ezzell, J.W.; Abshire, T.G.; Little, S.F.; Lidgerding, B.C.; Brown, C. Identification of Bacillus anthracis by using monoclonal antibody to cell wall galactose-N-acetylglucosamine polysaccharide. J. Clin. Microbiol. 1990, 28, 223. [Google Scholar] [CrossRef] [PubMed]
- Shibaev, V.N.; Duckworth, M.; Archibald, A.R.; Baddiley, J. The structure of a polymer containing galactosamine from walls of Bacillus subtilis 168. Biochem. J. 1973, 135, 383–384. [Google Scholar] [CrossRef]
- Brandl, M.T.; Rosenthal, B.M.; Haxo, A.F.; Berk, S.G. Enhanced survival of Salmonella enterica in vesicles released by a soilborne Tetrahymena species. Appl. Environ. Microbiol. 2005, 71, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Bouyer, S.; Imbert, C.; Rodier, M.-H.; Héchard, Y. Long-term survival of Legionella pneumophila associated with Acanthamoeba castellanii vesicles. Environ. Microbiol. 2007, 9, 1341–1344. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gu, J.; Zhou, Q. Review of aerobic glycolysis and its key enzymes-new targets for lung cancer therapy. Thorac. Cancer 2015, 6, 17–24. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef]
- Cotgreave, I.A. Recent trends in glutathione biochemistry glutathione-protein interactions: A molecular link between oxidative stress and cell proliferation. Biochem. Biophys. Res. Commun. 1998, 242, 1–9. [Google Scholar] [CrossRef]
- Allen, R.G.; Balin, A.K. Oxidative influence on development and differentiation: An overview of a free radical theory of development. Free Radic. Biol. Med. 1989, 6, 631–661. [Google Scholar] [CrossRef]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496. [Google Scholar] [CrossRef]
- Kim, J.M.; Kim, H.; Kwon, S.B.; Lee, S.Y.; Chung, S.C.; Jeong, D.W.; Min, B.M. Intracellular glutathione status regulates mouse bone marrow monocyte-derived macrophage differentiation and phagocytic activity. Biochem. Biophys. Res. Commun. 2004, 325, 101–108. [Google Scholar] [CrossRef]
- Nathan, C.; Shiloh, M.U. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl. Acad. Sci. USA 2000, 97, 8841. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Chen, C.; Zeng, Z.; Yang, H.; Oh, J.; Chen, L.; Lu, S.C. Mechanism and significance of increased glutathione level in human hepatocellular carcinoma and liver regeneration. FASEB J. 2000, 15, 19–21. [Google Scholar] [CrossRef]
- Markovic, J.; Borrás, C.; Ortega, Á.; Sastre, J.; Viña, J.; Pallardó, F.V. Glutathione is recruited into the nucleus in early phases of cell proliferation. J. Biol. Chem. 2007, 282, 20416–20424. [Google Scholar] [CrossRef] [PubMed]
- Pallardó, F.V.; Markovic, J.; García, J.L.; Viña, J. Role of nuclear glutathione as a key regulator of cell proliferation. Mol. Aspects Med. 2009, 30, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A. Regulation of ribonucleotide reductase. In Current Topics in Cellular Regulation; Horecker, B.L., Stadtman, E.R., Eds.; Academic Press: Cambridge, MA, USA, 1981; Volume 19, pp. 47–76. [Google Scholar]
- Avval, F.Z.; Holmgren, A. Molecular mechanisms of thioredoxin and glutaredoxin as hydrogen donors for mammalians phase ribonucleotide reductase. J. Biol. Chem. 2009, 284, 8233–8240. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zou, S.; Zhang, Q.; Gong, J. Comparative Transcriptomics Reveals Distinct Gene Expressions of a Model Ciliated Protozoan Feeding on Bacteria-Free Medium, Digestible, and Digestion-Resistant Bacteria. Microorganisms 2020, 8, 559. https://doi.org/10.3390/microorganisms8040559
Zou S, Zhang Q, Gong J. Comparative Transcriptomics Reveals Distinct Gene Expressions of a Model Ciliated Protozoan Feeding on Bacteria-Free Medium, Digestible, and Digestion-Resistant Bacteria. Microorganisms. 2020; 8(4):559. https://doi.org/10.3390/microorganisms8040559
Chicago/Turabian StyleZou, Songbao, Qianqian Zhang, and Jun Gong. 2020. "Comparative Transcriptomics Reveals Distinct Gene Expressions of a Model Ciliated Protozoan Feeding on Bacteria-Free Medium, Digestible, and Digestion-Resistant Bacteria" Microorganisms 8, no. 4: 559. https://doi.org/10.3390/microorganisms8040559
APA StyleZou, S., Zhang, Q., & Gong, J. (2020). Comparative Transcriptomics Reveals Distinct Gene Expressions of a Model Ciliated Protozoan Feeding on Bacteria-Free Medium, Digestible, and Digestion-Resistant Bacteria. Microorganisms, 8(4), 559. https://doi.org/10.3390/microorganisms8040559