
Microbial Community Composition Associated with Potato Plants Displaying Early Dying Syndrome

 ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Sampling Location
2.2. Soil Type
2.3. Sample Processing and DNA Extraction
2.4. Quantitative Real-Time Polymerase Chain Reaction
2.5. Stem and Soil Sample Amplicon-Targeted Next-Generation Sequencing
2.6. Microbiome Workflow and Data Analysis
3. Results
3.1. Soil Microbiota Around Healthy- and Diseased-Looking Potato Plants
3.1.1. V. dahliae Incidence and Abundance in Soil Collected from the Proximity of Healthy- and Diseased-Looking Potato Plants
3.1.2. Bacterial Diversity and Abundance in Soil Samples Collected from the Proximity of Healthy- and Diseased-Looking Potato Plants
3.1.3. Fungal Diversity and Abundance in Soil Samples Collected from the Proximity of Healthy and Diseased-Looking Potato Plants
3.1.4. Eukaryote Diversity and Abundance in Soil Samples Collected from the Proximity of Healthy- and Diseased-Looking Potato Plants
3.2. Microbiota Associated with the Stems of Healthy- and Diseased-Looking Potato Plants
3.2.1. V. dahliae Incidence and Abundance in the Stems of Healthy and Diseased Potato Plants
3.2.2. Bacterial Diversity and Abundance in the Stems of Healthy- and Diseased-Looking Potato Plants
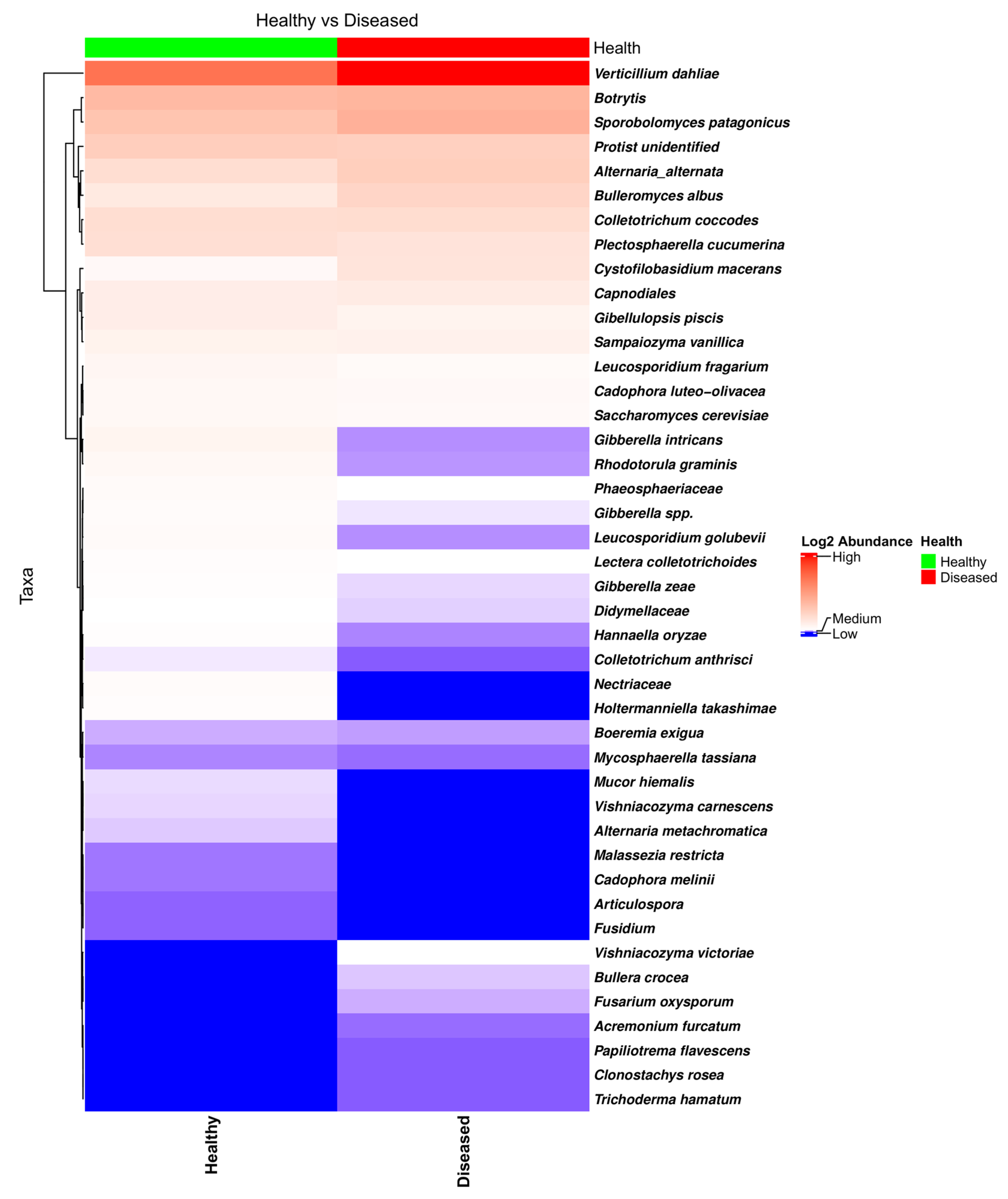
3.2.3. Fungal Diversity and Abundance in the Stems of Healthy- and Diseased-Looking Potato Plants
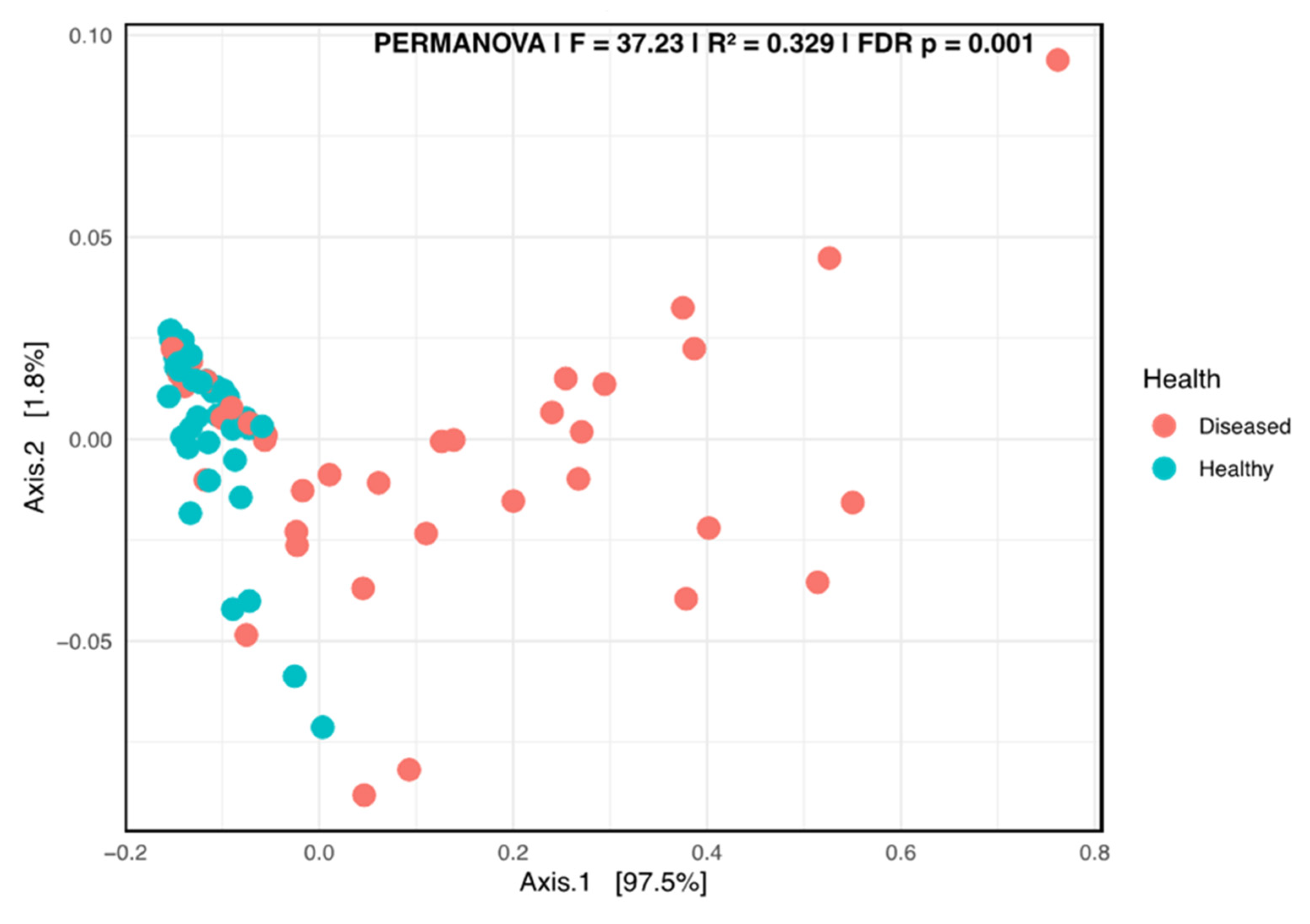
3.2.4. Fungal Network Co-Occurrence and Differential Structure Related to Plant Health Status
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Powelson, M.L.; Rowe, R.C. Biology and management of early dying of potatoes. Annu. Rev. Phytopathol. 1993, 31, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.C.; Powelson, M.L. Potato early dying: Management challenges in a changing production environment. Plant Dis. 2002, 86, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Inderbitzin, P.; Subbarao, K.V. Verticillium systematics and evolution: How confusion impedes Verticillium wilt management and how to resolve it. Phytopathology 2014, 104, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Borza, T.; Beaton, B.; Govindarajan, A.; Gao, X.; Liu, Y.; Ganga, Z.; Wang-Pruski, G. Incidence and abundance of Verticillium dahliae in soil from various agricultural fields in Prince Edward Island, Canada. Eur. J. Plant Pathol. 2018, 151, 825–830. [Google Scholar] [CrossRef]
- Borza, T.; Govindarajan, A.; Stephen, J.; Best, K.; Pruski, K.; Wang-Pruski, G. Verticillium dahliae and Verticillium nonalfalfae occurrence and abundance in several agricultural fields from Nova Scotia, Canada, assessed by real-time quantitative PCR. Eur. J. Plant Pathol. 2019, 154, 1171–1177. [Google Scholar] [CrossRef]
- Chen, D.; Barrett, R.; Mimee, B.; Arseneault, T.; Comeau, L.-P.; Nahar, K.; Jimenez, S.I.; Zebarth, B.J. Prevalence of Verticillium spp. and Pratylenchus spp. in commercial potato fields in Atlantic Canada. Am. J. Potato Res. 2024, 101, 191–305. [Google Scholar] [CrossRef]
- Schlatter, D.; Kinkel, L.; Thomashow, L.; Weller, D.; Paulitz, T. Disease suppressive soils: New insights from the soil microbiome. Phytopathology 2017, 107, 1284–1297. [Google Scholar] [CrossRef]
- Andargie, Y.E.; Lee, G.; Jeong, M.; Tagele, S.B.; Shin, J.-H. Deciphering key factors in pathogen-suppressive microbiome assembly in the rhizosphere. Front. Plant Sci. 2023, 14, 1301698. [Google Scholar] [CrossRef]
- Millican, M.; Shan, S.; Lankau, R.; Kinkel, L. Changes in potato rhizosphere microbiome alpha-and beta-diversity are correlated with shifts in the frequency of pathogen inhibitory populations. Phytopathology 2022, 112, 171. [Google Scholar]
- Klasek, S.A.; Crants, J.E.; Abbas, T.; Ashley, K.; Bolton, M.L.; Celovsky, M.; Gudmestead, N.C.; Hao, J.; Caballero, J.R.I.; Jahn, C.E.; et al. Potato soil core microbiomes are regionally variable across the continental United States. Phytobiomes J. 2024, 8, 168–178. [Google Scholar] [CrossRef]
- Reiter, B.; Pfeifer, U.; Schwab, H.; Sessitsch, A. Response of endophytic bacterial communities in potato plants to infection with Erwinia carotovora subsp. atroseptica. Appl. Env. Microbiol. 2002, 68, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Pavlo, A.; Leonid, O.; Iryna, Z.; Natalia, K.; Maria, P.A. Endophytic bacteria enhancing growth and disease resistance of potato (Solanum tuberosum L.). Biol. Control 2011, 56, 43–49. [Google Scholar] [CrossRef]
- Bahmani, K.; Hasanzadeh, N.; Harighi, B.; Marefat, A. Isolation and identification of endophytic bacteria from potato tissues and their effects as biological control agents against bacterial wilt. Physiol. Mol. Plant Pathol. 2021, 116, 101692. [Google Scholar] [CrossRef]
- Götz, M.; Nirenberg, H.; Krause, S.; Wolters, H.; Draeger, S.; Buchner, A.; Lottmann, J.; Berg, G.; Smalla, K. Fungal endophytes in potato roots studied by traditional isolation and cultivation-independent DNA-based methods. FEMS Microbiol. Ecol. 2006, 58, 404–413. [Google Scholar] [CrossRef]
- Sorokan, A.V.; Benkovskaya, G.V.; Maksimov, I.V. The influence of potato endophytes on Leptinotarsa decemlineata endosymbionts promotes mortality of the pest. J. Invertebr. Pathol. 2016, 136, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Bilodeau, G.J.; Koike, S.T.; Uribe, P.; Martin, F.N. Development of an assay for rapid detection and quantification of Verticillium dahliae in soil. Phytopathology 2012, 102, 331–343. [Google Scholar] [CrossRef]
- Alkher, H.; El Hadrami, A.; Adam, L.R.; Daayf, F. Cross-pathogenicity of Verticillium dahliae between potato and sunflower. Eur. J. Plant Pathol. 2009, 124, 505–519. [Google Scholar] [CrossRef]
- El-Bebany, A.F.; Alkher, H.; Adam, L.R.; Daayf, F. Vegetative compatibility of Verticillium dahliae isolates from potato and sunflower using nitrate non-utilizing (nit) mutants and PCR-based approaches. Can. J. Plant Pathol. 2013, 35, 1–9. [Google Scholar] [CrossRef]
- Borza, T.; Peters, R.D.; Gao, X.; Wang-Pruski, G. Effects of phosphite on the in vitro growth of Verticillium nonalfalfae and Verticillium dahliae and on their in vivo ability to infect potato plants. Eur. J. Plant Pathol. 2019, 155, 1333–1344. [Google Scholar] [CrossRef]
- Klosterman, S.J.; Subbarao, K.V.; Kang, S.; Veronese, P.; Gold, S.E.; Thomma, B.P.; Chen, Z.; Henrissat, B.; Lee, Y.H.; Park, J.; et al. Comparative genomics yields insights into niche adaptation of plant vascular wilt pathogens. PLoS Pathog. 2011, 7, e1002137. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Douglas, G.M.; Langille, M.G. Microbiome Helper: A custom and streamlined workflow for microbiome research. mSystems 2017, 2, e00127-16. [Google Scholar] [CrossRef] [PubMed]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems 2017, 2, e00191-16. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, L.; Lun, A.T.L.; Baldoni, P.L.; Smyth, G.K. edgeR v4: Powerful differential analysis of sequencing data with expanded functionality and improved support for small counts and larger datasets. Nucleic Acids Res. 2025, 53, gkaf018. [Google Scholar] [CrossRef]
- Kurtz, Z.D.; Müller, C.L.; Miraldi, E.R.; Littman, D.R.; Blaser, M.J.; Bonneau, R.A. Sparse and compositionally robust inference of microbial ecological networks. PLOS Comput. Biol. 2015, 11, e1004226. [Google Scholar] [CrossRef]
- Csárdi, G.; Nepusz, T.; Müller, K.; Horvát, S.; Traag, V.; Zanini, F.; Noom, D. igraph for R: R interface of the igraph library for graph theory and network analysis (v2.1.4). Zenodo 2025. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Liu, C.; Cui, Y.; Li, X.; Yao, M. microeco: An R package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 2020, 97, fiaa255. [Google Scholar] [CrossRef]
- Inderbitzin, P.; Bostock, R.M.; Davis, R.M.; Usami, T.; Platt, H.W.; Subbarao, K.V. Phylogenetics and taxonomy of the fungal vascular wilt pathogen Verticillium, with the descriptions of five new species. PLoS ONE 2011, 6, e28341. [Google Scholar] [CrossRef]
- Zare, R.; Starink, M.; Summerbell, R. Gibellulopsis, a suitable genus for Verticillium nigrescens, and Musicillium, a new genus for V. theobromae. Nova Hedwig. 2007, 85, 463–489. [Google Scholar] [CrossRef]
- Agrios, G.N. Plant Pathology, 5th ed.; Elsevier Academic Press: Cambridge, MA, USA, 2005. [Google Scholar]
- Celetti, M.J.; Platt, H.W. A new cause for an old disease: Verticillium dahliae found on Prince Edward Island. Am. Potato J. 1987, 64, 209–212. [Google Scholar] [CrossRef]
- Robb, J.; Moukhamedov, R.; Hu, X.; Platt, H.; Nazar, R. Putative subgroups of Verticillium albo-atrum distinguishable by PCR-based assays. Physiol. Mol. Plant Pathol. 1993, 43, 423–436. [Google Scholar] [CrossRef]
- Kimpinski, J.; Platt, H.W.; Perley, S.; Walsh, J.R. Pratylenchus spp. and Verticillium spp. in New Brunswick potato fields. Am. J. Potato Res. 1998, 75, 87–91. [Google Scholar] [CrossRef]
- Mahuku, G.S.; Platt, H.W. Molecular evidence that Verticillium albo-atrum Grp 2 isolates are distinct from V. albo-atrum Grp 1 and V. tricorpus. Mol. Plant Pathol. 2002, 3, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.C.; Davis, J.R.; Powelson, M.L.; Rouse, D.I. Potato early dying: Causal agents and management strategies. Plant Dis. 1987, 71, 482–489. [Google Scholar] [CrossRef]
- Hao, J.; Wang, D.; Wang, Y.; Zhou, H. Attenuated isolate Gibellulopsis nigrescens Vn-1 enhances resistance against Verticillium dahliae in potato. Agronomy 2022, 12, 3082. [Google Scholar] [CrossRef]
- Carlucci, A.; Raimondo, M.L.; Santos, J.; Phillips, A.J. Plectosphaerella species associated with root and collar rots of horticultural crops in southern Italy. Persoonia 2012, 28, 34–48. [Google Scholar] [CrossRef]
- Mullen, J.M.; Sikora, E.J. First report of Plectosporium blight on pumpkin caused by Plectosporium tabacinum in Alabama. Plant Dis. 2003, 87, 749. [Google Scholar] [CrossRef]
- Sato, T.; Inaba, T.; Mori, M.; Watanabe, K.; Tomioka, K.; Hamaya, E. Plectosporium blight of pumpkin and ranunculus caused by Plectosporium tabacinum. J. Gen. Plant Pathol. 2005, 71, 127–132. [Google Scholar] [CrossRef]
- Zhang, W.; Sulz, M.; Bailey, K.L. Growth and spore production of Plectosporium tabacinum. Can. J. Bot. 2001, 79, 1297–1306. [Google Scholar] [CrossRef]
- Atkins, S.D.; Clark, I.M.; Sosnowska, D.; Hirsch, P.R.; Kerry, B.R. Detection and quantification of Plectosphaerella cucumerina, a potential biological control agent of potato cyst nematodes, by using conventional PCR, real-time PCR, selective media, and baiting. Appl. Environ. Microbiol. 2003, 69, 4788–4793. [Google Scholar] [CrossRef]
- Golubev, W.; Ikeda, R.; Shinoda, T.; Nakase, T. Antifungal activity of Bullera alba (Hanna) Derx. Mycoscience 1997, 38, 25–29. [Google Scholar] [CrossRef]
- Kristjuhan, A.; Kristjuhan, K.; Tamm, T. Richness of yeast community associated with apple fruits in Estonia. Heliyon 2024, 10, e27885. [Google Scholar] [CrossRef] [PubMed]
- Chreptowicz, K.; Mierzejewska, J.; Tkáčová, J.; Młynek, M.; Čertik, M. Carotenoid-producing yeasts: Identification and characteristics of environmental isolates with a valuable extracellular enzymatic activity. Microorganisms 2019, 7, 653. [Google Scholar] [CrossRef]
- Wuczkowski, M.; Passoth, V.; Turchetti, B.; Andersson, A.-C.; Olstorpe, M.; Laitila, A.; Theelen, B.; van Broock, M.; Buzzini, P.; Prillinger, H.; et al. Description of Holtermanniella gen. nov., including Holtermanniella takashimae sp. nov. and four new combinations, and proposal of the order Holtermanniales to accommodate tremellomycetous yeasts of the Holtermannia clade. Int. J. Syst. Evol. Microbiol. 2011, 61, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.S.; Cook, R.J. Suppression of Fusarium roseum “Avenaceum” by soil microorganisms. Phytopathology 1979, 69, 384–388. [Google Scholar] [CrossRef]
- Ziedan, E.-S.H.E.; Farrag, E.S.H.; Sahab, A.F. First record and preliminary evaluation of Mucor hiemalis as biocontrol agent on inflorescence brown rot incidence of date palm. Arch. Phytopathol. Plant Prot. 2013, 46, 617–626. [Google Scholar] [CrossRef]
- Ezrari, S.; Lahlali, R.; Radouane, N.; Tahiri, A.; Lazraq, A. First report of Fusarium equiseti causing pre- and postharvest fruit rot on zucchini in Morocco. J. Plant Pathol. 2020, 102, 251. [Google Scholar] [CrossRef]
- El Hazzat, N.; Adnani, M.; Msairi, S.; El Alaoui, M.A.; Mouden, N.; Chliyeh, M.; Boughribil, S.; Selmaoui, K.; Amina, O.T.; Douira, A. Fusarium equiseti as one of the main Fusarium species causing wilt and root rot of chickpeas in Morocco. Acta Mycol. 2023, 58, 1–10. [Google Scholar] [CrossRef]
- Firrincieli, A.; Otillar, R.; Salamov, A.; Schmutz, J.; Khan, Z.; Redman, R.S.; Fleck, N.D.; Lindquist, E.; Grigoriev, I.V.; Doty, S.L. Genome sequence of the plant growth promoting endophytic yeast Rhodotorula graminis WP1. Front. Microbiol. 2015, 6, 978. [Google Scholar] [CrossRef] [PubMed]
- Xin, G.; Glawe, D.; Doty, S.L. Characterization of three endophytic, indole-3-acetic acid-producing yeasts occurring in Populus trees. Mycol. Res. 2009, 113, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Li, W.K.W.; Tremblay, J.-É.; Carmack, E.C.; Lovejoy, C. Arctic ocean microbial community structure before and after the 2007 record sea ice minimum. PLoS ONE 2011, 6, e27492. [Google Scholar] [CrossRef] [PubMed]
- Op De Beeck, M.; Lievens, B.; Busschaert, P.; Declerck, S.; Vangronsveld, J.; Colpaert, J.V. Comparison and validation of some ITS primer pairs useful for fungal metabarcoding studies. PLoS ONE 2014, 9, e97629. [Google Scholar] [CrossRef]
- Paliy, O.; Kenche, H.; Abernathy, F.; Michail, S. High-throughput quantitative analysis of the human intestinal microbiota with a phylogenetic microarray. Appl. Environ. Microbiol. 2009, 75, 3572–3579. [Google Scholar] [CrossRef]
- Toju, H.; Tanabe, A.S.; Yamamoto, S.; Sato, H. High-coverage ITS primers for the DNA-based identification of ascomycetes and basidiomycetes in environmental samples. PLoS ONE 2012, 7, e40863. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Field # | Rotation Crop in the Previous Year | Sample Type | Average Number of Vd Cells/g Soil | SE | Incidence |
|---|---|---|---|---|---|
| 1 | Oat | healthy | 1104.5 | 370.1 | 70 |
| diseased | 743.9 | 212.1 | 70 | ||
| 2 | Oat | healthy | 2511.3 | 1203.6 | 100 |
| diseased | 2450.5 | 1288.8 | 90 | ||
| 3 | Barley | healthy | 1266.5 | 495.2 | 100 |
| diseased | 1602.9 | 388.5 | 100 | ||
| 4 | Barley | healthy | 152.9 | 36.7 | 60 |
| diseased | 1030.8 | 489.0 | 90 |
| Field # | Rotation Crop in the Previous Year | Sample Type | Average Number of Vd Cells x 103/g Fresh Tissue | SE | Incidence |
|---|---|---|---|---|---|
| 1 | oat | healthy | 23.8 | 7.3 | 100 |
| diseased | 35.1 | 13.7 | 80 | ||
| 2 | oat | healthy | 51.7 | 14.4 | 100 |
| diseased | 986.9 | 531.9 | 100 | ||
| 3 | barley | healthy |  | 26.8 | 100 |
| diseased | 430.5 | 100 | |||
| 4 | barley | healthy |  | 32.0 | 100 |
| diseased | 364.7 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borza, T.; Lumactud, R.A.; Shim, S.Y.; Al-Mughrabi, K.; Prithiviraj, B. Microbial Community Composition Associated with Potato Plants Displaying Early Dying Syndrome. Microorganisms 2025, 13, 1482. https://doi.org/10.3390/microorganisms13071482
Borza T, Lumactud RA, Shim SY, Al-Mughrabi K, Prithiviraj B. Microbial Community Composition Associated with Potato Plants Displaying Early Dying Syndrome. Microorganisms. 2025; 13(7):1482. https://doi.org/10.3390/microorganisms13071482
Chicago/Turabian StyleBorza, Tudor, Rhea Amor Lumactud, So Yeon Shim, Khalil Al-Mughrabi, and Balakrishnan Prithiviraj. 2025. "Microbial Community Composition Associated with Potato Plants Displaying Early Dying Syndrome" Microorganisms 13, no. 7: 1482. https://doi.org/10.3390/microorganisms13071482
APA StyleBorza, T., Lumactud, R. A., Shim, S. Y., Al-Mughrabi, K., & Prithiviraj, B. (2025). Microbial Community Composition Associated with Potato Plants Displaying Early Dying Syndrome. Microorganisms, 13(7), 1482. https://doi.org/10.3390/microorganisms13071482