
Comparative Analysis of Bacteriome in Hair Follicle Layers of Patients with Female Pattern Androgenic Alopecia

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Patient Demographics and Hair Follicle Layer Sample Collection
2.2. Participant Exclusion Criteria and Sample Processing
2.3. Bacterial Genomic DNA Extraction and Quality Control
2.4. Illumina 16S Ribosomal RNA Gene V3–V4 Amplicon Sequencing Library Preparation and Sequencing
2.5. 16S rRNA Gene V3–V4 ASV Data Processing Workflow
2.6. Evaluation of Alpha-Diversity and Beta-Diversity in FPHL, MPHL, and Control Groups
2.7. Bacterial Relative Abundance in FPHL, MPHL, and Control Groups
2.8. Taxonomic and Functional Profiling of Microbiome Using LEfSe and PICRUSt2 Analysis
3. Results
3.1. 16S rRNA Gene V3–V4 Sequencing Data Processing of Clinical Samples
3.2. Alpha-Diversity and Beta-Diversity of Hair Follicle Layer Microbiome
3.3. Relative Abundance in Hair Follicle Layer Microbiome
3.4. Significant Microbiota Biomarkers Identified Using LEfSe Analysis
3.5. In Silico Functional Analysis Using PICRUSt2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oiwoh, S.O.; Enitan, A.O.; Adegbosin, O.T.; Akinboro, A.O.; Onayemi, E.O. Androgenetic Alopecia: A Review. Niger. Postgrad. Med. J. 2024, 31, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Sibbald, C. Alopecia Areata: An Updated Review for 2023. J. Cutan. Med. Surg. 2023, 27, 241–259. [Google Scholar] [CrossRef]
- Cummins, D.M.; Chaudhry, I.H.; Harries, M. Scarring Alopecias: Pathology and an Update on Digital Developments. Biomedicines 2021, 9, 1755. [Google Scholar] [CrossRef]
- Asghar, F.; Shamim, N.; Farooque, U.; Sheikh, H.; Aqeel, R. Telogen Effluvium: A Review of the Literature. Cureus 2020, 12, e8320. [Google Scholar] [CrossRef]
- Otberg, N.; Finner, A.M.; Shapiro, J. Androgenetic alopecia. Endocrinol. Metab. Clin. N. Am. 2007, 36, 379–398. [Google Scholar] [CrossRef]
- Ho, C.-Y.; Chen, J.Y.; Hsu, W.-L.; Yu, S.; Chen, W.-C.; Chiu, S.-H.; Yang, H.-R.; Lin, S.-Y.; Wu, C.-Y. Female Pattern Hair Loss: An Overview with Focus on the Genetics. Genes 2023, 14, 1326. [Google Scholar] [CrossRef] [PubMed]
- Ntshingila, S.; Oputu, O.; Arowolo, A.T.; Khumalo, N.P. Androgenetic alopecia: An update. JAAD Int. 2023, 13, 150–158. [Google Scholar] [CrossRef]
- Yip, L.; Rufaut, N.; Sinclair, R. Role of genetics and sex steroid hormones in male androgenetic alopecia and female pattern hair loss: An update of what we now know. Australas. J. Dermatol. 2011, 52, 81–88. [Google Scholar] [CrossRef]
- Suchonwanit, P.; Iamsumang, W.; Leerunyakul, K. Topical finasteride for the treatment of male androgenetic alopecia and female pattern hair loss: A review of the current literature. J. Dermatol. Treat. 2022, 33, 643–648. [Google Scholar] [CrossRef]
- Carmina, E.; Azziz, R.; Bergfeld, W.; Escobar-Morreale, H.F.; Futterweit, W.; Huddleston, H.; Lobo, R.; Olsen, E. Female Pattern Hair Loss and Androgen Excess: A Report From the Multidisciplinary Androgen Excess and PCOS Committee. J. Clin. Endocrinol. Metab. 2019, 104, 2875–2891. [Google Scholar] [CrossRef]
- Byrd, A.L.; Belkaid, Y.; Segre, J.A. The human skin microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155. [Google Scholar] [CrossRef]
- Lunjani, N.; Ahearn-Ford, S.; Dube, F.S.; Hlela, C.; O’Mahony, L. Mechanisms of microbe-immune system dialogue within the skin. Genes Immun. 2021, 22, 276–288. [Google Scholar] [CrossRef]
- Prajapati, S.K.; Lekkala, L.; Yadav, D.; Jain, S.; Yadav, H. Microbiome and Postbiotics in Skin Health. Biomedicines 2025, 13, 791. [Google Scholar] [CrossRef]
- Truglio, M.; Sivori, F.; Cavallo, I.; Abril, E.; Licursi, V.; Fabrizio, G.; Cardinali, G.; Pignatti, M.; Toma, L.; Valensise, F.; et al. Modulating the skin mycobiome-bacteriome and treating seborrheic dermatitis with a probiotic-enriched oily suspension. Sci. Rep. 2024, 14, 2722. [Google Scholar] [CrossRef]
- Jung, D.R.; Yoo, H.Y.; Kim, M.J.; Singh, V.; Park, S.H.; Jeong, M.; Park, B.J.; Shin, J.H. Comparative analysis of scalp and gut microbiome in androgenetic alopecia: A Korean cross-sectional study. Front. Microbiol. 2022, 13, 1076242. [Google Scholar] [CrossRef]
- Suzuki, K.; Inoue, M.; Cho, O.; Mizutani, R.; Shimizu, Y.; Nagahama, T.; Sugita, T. Scalp Microbiome and Sebum Composition in Japanese Male Individuals with and without Androgenetic Alopecia. Microorganisms 2021, 9, 2132. [Google Scholar] [CrossRef]
- Ho, B.S.; Ho, E.X.P.; Chu, C.W.; Ramasamy, S.; Bigliardi-Qi, M.; de Sessions, P.F.; Bigliardi, P.L. Microbiome in the hair follicle of androgenetic alopecia patients. PLoS ONE 2019, 14, e0216330. [Google Scholar] [CrossRef]
- Constantinou, A.; Kanti, V.; Polak-Witka, K.; Blume-Peytavi, U.; Spyrou, G.M.; Vogt, A. The Potential Relevance of the Microbiome to Hair Physiology and Regeneration: The Emerging Role of Metagenomics. Biomedicines 2021, 9, 236. [Google Scholar] [CrossRef]
- Paus, R.; Cotsarelis, G. The biology of hair follicles. N. Engl. J. Med. 1999, 341, 491–497. [Google Scholar] [CrossRef]
- Lin, X.; Zhu, L.; He, J. Morphogenesis, Growth Cycle and Molecular Regulation of Hair Follicles. Front. Cell Dev. Biol. 2022, 10, 899095. [Google Scholar] [CrossRef]
- An, S.Y.; Kim, H.-S.; Kim, S.Y.; Van, S.Y.; Kim, H.J.; Lee, J.-H.; Han, S.W.; Kwon, I.K.; Lee, C.-K.; Do, S.H.; et al. Keratin-mediated hair growth and its underlying biological mechanism. Commun. Biol. 2022, 5, 1270. [Google Scholar] [CrossRef]
- Rogers, G.E. Hair follicle differentiation and regulation. Int. J. Dev. Biol. 2004, 48, 163–170. [Google Scholar] [CrossRef]
- Rogers, G.E. Known and Unknown Features of Hair Cuticle Structure: A Brief Review. Cosmetics 2019, 6, 32. [Google Scholar] [CrossRef]
- Abreu, C.M.; Reis, R.L.; Marques, A.P. Dermal papilla cells and melanocytes response to physiological oxygen levels depends on their interactions. Cell Prolif. 2021, 54, e13013. [Google Scholar] [CrossRef]
- Zerbinati, N.; Sommatis, S.; Maccario, C.; Capillo, M.C.; Di Francesco, S.; Rauso, R.; Protasoni, M.; D’Este, E.; Gasperina, D.D.; Mocchi, R. In Vitro Hair Growth Promoting Effect of a Noncrosslinked Hyaluronic Acid in Human Dermal Papilla Cells. BioMed Res. Int. 2021, 2021, 5598110. [Google Scholar] [CrossRef]
- Cundell, A.M. Microbial Ecology of the Human Skin. Microb. Ecol. 2018, 76, 113–120. [Google Scholar] [CrossRef]
- Lousada, M.B.; Lachnit, T.; Edelkamp, J.; Rouillé, T.; Ajdic, D.; Uchida, Y.; Di Nardo, A.; Bosch, T.C.G.; Paus, R. Exploring the human hair follicle microbiome. Br. J. Dermatol. 2021, 184, 802–815. [Google Scholar] [CrossRef]
- Skowron, K.; Bauza-Kaszewska, J.; Kraszewska, Z.; Wiktorczyk-Kapischke, N.; Grudlewska-Buda, K.; Kwiecińska-Piróg, J.; Wałecka-Zacharska, E.; Radtke, L.; Gospodarek-Komkowska, E. Human Skin Microbiome: Impact of Intrinsic and Extrinsic Factors on Skin Microbiota. Microorganisms 2021, 9, 543. [Google Scholar] [CrossRef]
- Chen, C.-L.; Huang, W.-Y.; Wang, E.H.C.; Tai, K.-Y.; Lin, S.-J. Functional complexity of hair follicle stem cell niche and therapeutic targeting of niche dysfunction for hair regeneration. J. Biomed. Sci. 2020, 27, 43. [Google Scholar] [CrossRef]
- Kiselev, A.; Park, S. Immune niches for hair follicle development and homeostasis. Front. Physiol. 2024, 15, 1397067. [Google Scholar] [CrossRef]
- Lousada, M.B.; Edelkamp, J.; Lachnit, T.; Fehrholz, M.; Jimenez, F.; Paus, R. Laser capture microdissection as a method for investigating the human hair follicle microbiome reveals region-specific differences in the bacteriome profile. BMC Res. Notes 2023, 16, 29. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Ro, B.I.; Hong, S.P.; Bak, H.; Sim, W.Y.; Kim, D.W.; Park, J.K.; Ihm, C.W.; Eun, H.C.; Kwon, O.S.; et al. A new classification of pattern hair loss that is universal for men and women: Basic and specific (BASP) classification. J. Am. Acad. Dermatol. 2007, 57, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Norwood, O.T. Male pattern baldness: Classification and incidence. South. Med. J. 1975, 68, 1359–1365. [Google Scholar] [CrossRef]
- Hamilton, J.B. Patterned loss of hair in man; types and incidence. Ann. N. Y. Acad. Sci. 1951, 53, 708–728. [Google Scholar] [CrossRef]
- Ludwig, E. Classification of the types of androgenetic alopecia (common baldness) occurring in the female sex. Br. J. Dermatol. 1977, 97, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.R. The Declaration of Helsinki and public health. Bull. World Health Organ. 2008, 86, 650–652. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Cameron, E.S.; Schmidt, P.J.; Tremblay, B.J.M.; Emelko, M.B.; Müller, K.M. Enhancing diversity analysis by repeatedly rarefying next generation sequencing data describing microbial communities. Sci. Rep. 2021, 11, 22302. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.H. Measurement of Diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric Estimation of the Number of Classes in a Population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Heip, C. A New Index Measuring Evenness. J. Mar. Biol. Assoc. United Kingd. 1974, 54, 555–557. [Google Scholar] [CrossRef]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Anderson, M.J. Permutational Multivariate Analysis of Variance (PERMANOVA). In Wiley StatsRef: Statistics Reference Online; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 1–15. [Google Scholar]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: https://www.R-project.org/ (accessed on 10 March 2023).
- Wickham, H. ggplot2. WIREs Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Grimshaw, S.G.; Smith, A.M.; Arnold, D.S.; Xu, E.; Hoptroff, M.; Murphy, B. The diversity and abundance of fungi and bacteria on the healthy and dandruff affected human scalp. PLoS ONE 2019, 14, e0225796. [Google Scholar] [CrossRef]
- Woo, Y.R.; Cho, M.; Han, Y.; Lee, S.H.; Cho, S.H.; Lee, J.D.; Kim, H.S. Characterization of Distinct Microbiota Associated with Scalp Dermatitis in Patients with Atopic Dermatitis. J. Clin. Med. 2022, 11, 1735. [Google Scholar] [CrossRef]
- Li, X.; Yang, F.; Yan, H.; Shi, Y.; Chang, X.; Zhang, M.; Zhang, Y.; Zhang, M. Microbiota profiling on itchy scalp with undetermined origin. Arch. Microbiol. 2022, 204, 446. [Google Scholar] [CrossRef]
- Pinto, D.; Sorbellini, E.; Marzani, B.; Rucco, M.; Giuliani, G.; Rinaldi, F. Scalp bacterial shift in Alopecia areata. PLoS ONE 2019, 14, e0215206. [Google Scholar] [CrossRef] [PubMed]
- Won, E.J.; Jang, H.H.; Park, H.; Kim, S.J. A Potential Predictive Role of the Scalp Microbiome Profiling in Patients with Alopecia Areata: Staphylococcus caprae, Corynebacterium, and Cutibacterium Species. Microorganisms 2022, 10, 864. [Google Scholar] [CrossRef]
- Wang, L.; Clavaud, C.; Bar-Hen, A.; Cui, M.; Gao, J.; Liu, Y.; Liu, C.; Shibagaki, N.; Guéniche, A.; Jourdain, R.; et al. Characterization of the major bacterial-fungal populations colonizing dandruff scalps in Shanghai, China, shows microbial disequilibrium. Exp. Dermatol. 2015, 24, 398–400. [Google Scholar] [CrossRef]
- Soares, R.C.; Camargo-Penna, P.H.; de Moraes, V.C.; De Vecchi, R.; Clavaud, C.; Breton, L.; Braz, A.S.; Paulino, L.C. Dysbiotic Bacterial and Fungal Communities Not Restricted to Clinically Affected Skin Sites in Dandruff. Front. Cell. Infect. Microbiol. 2016, 6, 157. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuji, T.; Cheng, J.Y.; Gallo, R.L. Mechanisms for control of skin immune function by the microbiome. Curr. Opin. Immunol. 2021, 72, 324–330. [Google Scholar] [CrossRef]
- Fournière, M.; Latire, T.; Souak, D.; Feuilloley, M.G.J.; Bedoux, G. Staphylococcus epidermidis and Cutibacterium acnes: Two Major Sentinels of Skin Microbiota and the Influence of Cosmetics. Microorganisms 2020, 8, 1752. [Google Scholar] [CrossRef]
- Rozas, M.; Hart de Ruijter, A.; Fabrega, M.J.; Zorgani, A.; Guell, M.; Paetzold, B.; Brillet, F. From Dysbiosis to Healthy Skin: Major Contributions of Cutibacterium acnes to Skin Homeostasis. Microorganisms 2021, 9, 628. [Google Scholar] [CrossRef] [PubMed]
- Fitz-Gibbon, S.; Tomida, S.; Chiu, B.H.; Nguyen, L.; Du, C.; Liu, M.; Elashoff, D.; Erfe, M.C.; Loncaric, A.; Kim, J.; et al. Propionibacterium acnes strain populations in the human skin microbiome associated with acne. J. Investig. Dermatol. 2013, 133, 2152–2160. [Google Scholar] [CrossRef] [PubMed]
- Park, T.; Kim, H.J.; Myeong, N.R.; Lee, H.G.; Kwack, I.; Lee, J.; Kim, B.J.; Sul, W.J.; An, S. Collapse of human scalp microbiome network in dandruff and seborrhoeic dermatitis. Exp. Dermatol. 2017, 26, 835–838. [Google Scholar] [CrossRef] [PubMed]
- Matard, B.; Donay, J.L.; Resche-Rigon, M.; Tristan, A.; Farhi, D.; Rousseau, C.; Mercier-Delarue, S.; Cavelier-Balloy, B.; Assouly, P.; Petit, A.; et al. Folliculitis decalvans is characterized by a persistent, abnormal subepidermal microbiota. Exp. Dermatol. 2020, 29, 295–298. [Google Scholar] [CrossRef]
- Lin, Q.; Panchamukhi, A.; Li, P.; Shan, W.; Zhou, H.; Hou, L.; Chen, W. Malassezia and Staphylococcus Dominate Scalp Microbiome for Seborrheic Dermatitis. Bioproc. Biosyst. Eng. 2021, 44, 965–975. [Google Scholar] [CrossRef]
- Krishna, S.; Miller, L.S. Host-pathogen interactions between the skin and Staphylococcus aureus. Curr. Opin. Microbiol. 2012, 15, 28–35. [Google Scholar] [CrossRef]
- Parlet, C.P.; Brown, M.M.; Horswill, A.R. Commensal Staphylococci Influence Staphylococcus aureus Skin Colonization and Disease. Trends Microbiol. 2019, 27, 497–507. [Google Scholar] [CrossRef]
- Heath, V.; Cloutman-Green, E.; Watkin, S.; Karlikowska, M.; Ready, D.; Hatcher, J.; Pearce-Smith, N.; Brown, C.; Demirjian, A. Staphylococcus capitis: Review of Its Role in Infections and Outbreaks. Antibiotics 2023, 12, 669. [Google Scholar] [CrossRef]
- Otto, M. Staphylococcus epidermidis—The ‘accidental’ pathogen. Nat. Rev. Microbiol. 2009, 7, 555–567. [Google Scholar] [CrossRef]
- Yu, J.; Ma, X.; Wang, X.; Cui, X.; Ding, K.; Wang, S.; Han, C. Application and mechanism of probiotics in skin care: A review. J. Cosmet. Dermatol. 2022, 21, 886–894. [Google Scholar] [CrossRef]
- Soccol, C.R.; Vandenberghe, L.P.S.; Spier, M.R.; Medeiros, A.B.P.; Yamaguishi, C.T.; Lindner, J.D.D.; Pandey, A.; Thomaz-Soccol, V.J.F.T. The potential of probiotics: A review. Food Technol. Biotechnol. 2010, 48, 413–434. [Google Scholar]
- Ryan, L.A.M.; Zannini, E.; Dal Bello, F.; Pawlowska, A.; Koehler, P.; Arendt, E.K. Lactobacillus amylovorus DSM 19280 as a novel food-grade antifungal agent for bakery products. Int. J. Food Microbiol. 2011, 146, 276–283. [Google Scholar] [CrossRef]
- Jain, R.; Voss, A.L.; Tagg, J.R.; Hale, J.D.F. Evaluation of the Preliminary Safety, Tolerability and Colonisation Efficacy of Topical Probiotic Formulations Containing Micrococcus luteus Q24 in Healthy Human Adults. Cosmetics 2022, 9, 121. [Google Scholar] [CrossRef]
- Mei, F.; Liu, J.; Wu, J.; Duan, Z.; Chen, M.; Meng, K.; Chen, S.; Shen, X.; Xia, G.; Zhao, M. Collagen Peptides Isolated from Salmo salar and Tilapia nilotica Skin Accelerate Wound Healing by Altering Cutaneous Microbiome Colonization via Upregulated NOD2 and BD14. J. Agric. Food Chem. 2020, 68, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Zamlynska, K.; Komaniecka, I.; Zebracki, K.; Mazur, A.; Sroka-Bartnicka, A.; Choma, A. Studies on lipid A isolated from Phyllobacterium trifolii PETP02T lipopolysaccharide. Antonie Leeuwenhoek 2017, 110, 1413–1433. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Jope, R.S. Lipopolysaccharide-induced interleukin-6 production is controlled by glycogen synthase kinase-3 and STAT3 in the brain. J. Neuroinflammation 2009, 6, 9. [Google Scholar] [CrossRef]
- Chang, C.Y.; Tucci, M.; Baker, R.C. Lipopolysaccharide-stimulated nitric oxide production and inhibition of cell proliferation is antagonized by ethanol in a clonal macrophage cell line. Alcohol 2000, 20, 37–43. [Google Scholar] [CrossRef]
- Butler, M.; Chaudhary, R.; van Heel, D.A.; Playford, R.J.; Ghosh, S. NOD2 activity modulates the phenotype of LPS-stimulated dendritic cells to promote the development of T-helper type 2-like lymphocytes—Possible implications for NOD2-associated Crohn’s disease. J. Crohn’s Colitis 2007, 1, 106–115. [Google Scholar] [CrossRef]
- Young, J.W.; Conte, E.T.; Leavitt, M.L.; Nafz, M.A.; Schroeter, A.L. Cutaneous immunopathology of androgenetic alopecia. J. Am. Osteopath. Assoc. 1991, 91, 765–771. [Google Scholar] [CrossRef]
- Chekanov, K.; Danko, D.; Tlyachev, T.; Kiselev, K.; Hagens, R.; Georgievskaya, A. State-of-the-Art in Skin Fluorescent Photography for Cosmetic and Skincare Research: From Molecular Spectra to AI Image Analysis. Life 2024, 14, 1271. [Google Scholar] [CrossRef]
- Schanzenbacher, J.; Hendrika Kähler, K.; Mesler, E.; Kleingarn, M.; Marcel Karsten, C.; Leonard Seiler, D. The role of C5a receptors in autoimmunity. Immunobiology 2023, 228, 152413. [Google Scholar] [CrossRef]
- Rodríguez-Tamez, G.; Herz-Ruelas, M.E.; Gómez-Flores, M.; Ocampo-Candiani, J.; Chavez-Alvarez, S. Hair Disorders in Autoimmune Diseases. Ski. Appendage Disord. 2023, 9, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Sanford, J.A.; Zhang, L.J.; Williams, M.R.; Gangoiti, J.A.; Huang, C.M.; Gallo, R.L. Inhibition of HDAC8 and HDAC9 by microbial short-chain fatty acids breaks immune tolerance of the epidermis to TLR ligands. Sci. Immunol. 2016, 1, eaah4609. [Google Scholar] [CrossRef] [PubMed]
- Bomar, L.; Brugger, S.D.; Yost, B.H.; Davies, S.S.; Lemon, K.P. Corynebacterium accolens Releases Antipneumococcal Free Fatty Acids from Human Nostril and Skin Surface Triacylglycerols. mBio 2016, 7, e01725-15. [Google Scholar] [CrossRef]
- Vom Steeg, L.G.; Klein, S.L. Sex Steroids Mediate Bidirectional Interactions Between Hosts and Microbes. Horm. Behav. 2017, 88, 45–51. [Google Scholar] [CrossRef]
- Hatanaka, T.; Lulic, Z.; Mefo, T.; Booth, C.; Harrison, E.; Ong, G. Change in hair growth-related gene expression profile in human isolated hair follicles induced by 5-alpha reductase inhibitors–dutasteride and finasteride–in the presence of testosterone. Singap. Med. J. 2022, 63, 552–558. [Google Scholar] [CrossRef]
- Owecka, B.; Tomaszewska, A.; Dobrzeniecki, K.; Owecki, M. The Hormonal Background of Hair Loss in Non-Scarring Alopecias. Biomedicines 2024, 12, 513. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Xi, H.; Huang, W.; Liu, M.-f.; Yuan, L.; Chen, Q.; Xiao, Y.; Zhu, Q.; Zhao, R.; Sheng, Y.-y. The role of male hormones in bacterial infections: Enhancing Staphylococcus aureus virulence through testosterone-induced Agr activation. Arch. Microbiol. 2024, 206, 401. [Google Scholar] [CrossRef]
- Gümüş, D.; Kalaycı Yüksek, F.; Sefer, Ö.; Yörük, E.; Uz, G.; Anğ Küçüker, M. The roles of hormones in the modulation of growth and virulence genes’ expressions in UPEC strains. Microb. Pathog. 2019, 132, 319–324. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
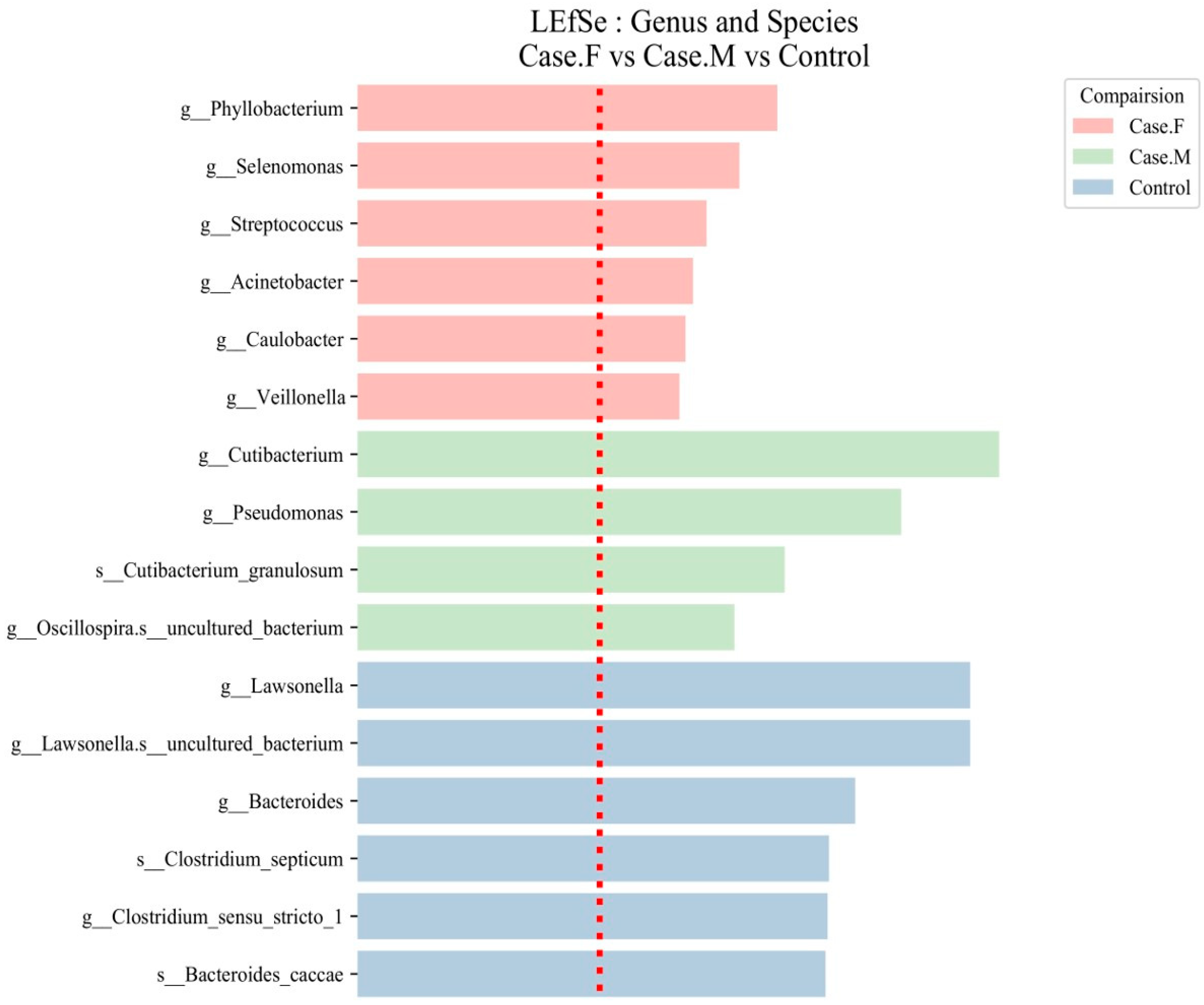
| Group | Genus and Species | LDA Score | Case.F | Case.M | Control |
|---|---|---|---|---|---|
| FPHL | Phyllobacterium | 3.466 | 0.488 | 0.000 | 0.000 |
| Selenomonas | 3.153 | 0.038 | 0.000 | 0.010 | |
| Streptococcus | 2.882 | 0.173 | 0.047 | 0.168 | |
| Acinetobacter | 2.771 | 0.038 | 0.018 | 0.022 | |
| Caulobacter | 2.708 | 0.057 | 0.019 | 0.033 | |
| Veillonella | 2.658 | 0.086 | 0.001 | 0.038 | |
| MPHL | Cutibacterium | 5.299 | 62.579 | 78.954 | 42.135 |
| Pseudomonas | 4.490 | 0.143 | 5.810 | 4.072 | |
| Cutibacterium granulosum | 3.528 | 0.152 | 0.659 | 0.030 | |
| Oscillospira uncultured bacterium | 3.113 | 0.000 | 0.000 | 0.007 | |
| Control | Lawsonella | 5.060 | 16.398 | 3.542 | 27.096 |
| Lawsonella uncultured bacterium | 5.060 | 16.394 | 3.542 | 27.091 | |
| Bacteroides | 4.110 | 0.172 | 0.093 | 2.446 | |
| Clostridiumsepticum | 3.894 | 0.005 | 0.000 | 1.519 | |
| Clostridium sensu stricto 1 | 3.882 | 0.145 | 0.212 | 1.606 | |
| Bacteroidescaccae | 3.865 | 0.020 | 0.007 | 1.372 |
| Disease | Androgenetic Alopecia | Alopecia Areata | Atopic Dermatitis | Scalp Pruritus | Dandruff | Seborrheic Dermatitis | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Skin area | Hair follicle layers | Scalp | |||||||||
| Sex | Female | Male | Female | Male | Male | Male | Female, Male | Male | |||
| Country | Korea | Japan | Korea | Italia | Korea | China | |||||
| Author | Our study | Jung et al., 2022 [15] | Suzuki et al., 2021 [16] | Won et al., 2022 [58] | Pinto et al., 2019 [57] | Woo et al., 2022 [55] | Li et al., 2022 [56] | Grimshaw et al., 2019 [54] | Lin et al., 2021 [67] | ||
| Cutibacterium | ↑ | ↑ | ↓ | ↓ | ↑ | ns | ↑ | ↓ | ↓ | ↓ | ns |
| Lawsonella | ↓ | ↓ | ↓ | - | - | - | ↓ | ns | - | - | |
| Staphylococcus | ↑ | ↓ | ↓ | ↓ | ↑ | ↓ | ↓ | ↑ | ↓ | ↑ | ↑ |
| Pseudomonas | ↓ | ↑ | ↓ | ↑ | ↓ | - | - | - | ↓ | - | ↓ |
| Corynebacterium | ↓ | ↑ | ↓ | ↑ | ↓ | ↑ | - | ↑ | ↑ | - | - |
| Micrococcus | ↓ | ↓ | ↓ | ↑ | ↓ | - | - | - | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, Y.; Kyung, S.; Mun, S.; Yu, B.S.; Yun, K.; Baek, C.; Lee, D.-G.; Kang, S.; Kim, S.R.; Kim, J.-H.; et al. Comparative Analysis of Bacteriome in Hair Follicle Layers of Patients with Female Pattern Androgenic Alopecia. Microorganisms 2025, 13, 1365. https://doi.org/10.3390/microorganisms13061365
Park Y, Kyung S, Mun S, Yu BS, Yun K, Baek C, Lee D-G, Kang S, Kim SR, Kim J-H, et al. Comparative Analysis of Bacteriome in Hair Follicle Layers of Patients with Female Pattern Androgenic Alopecia. Microorganisms. 2025; 13(6):1365. https://doi.org/10.3390/microorganisms13061365
Chicago/Turabian StylePark, Yujun, Seoyeon Kyung, Seyoung Mun, Byung Sun Yu, Kyengeui Yun, Chaeyun Baek, Dong-Geol Lee, Seunghyun Kang, Soon Re Kim, Ju-Hee Kim, and et al. 2025. "Comparative Analysis of Bacteriome in Hair Follicle Layers of Patients with Female Pattern Androgenic Alopecia" Microorganisms 13, no. 6: 1365. https://doi.org/10.3390/microorganisms13061365
APA StylePark, Y., Kyung, S., Mun, S., Yu, B. S., Yun, K., Baek, C., Lee, D.-G., Kang, S., Kim, S. R., Kim, J.-H., Lee, Y., Park, B.-C., & Han, K. (2025). Comparative Analysis of Bacteriome in Hair Follicle Layers of Patients with Female Pattern Androgenic Alopecia. Microorganisms, 13(6), 1365. https://doi.org/10.3390/microorganisms13061365