
Flavonoid Derivatives as New Potent Inhibitors of Cysteine Proteases: An Important Step toward the Design of New Compounds for the Treatment of Leishmaniasis

 , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
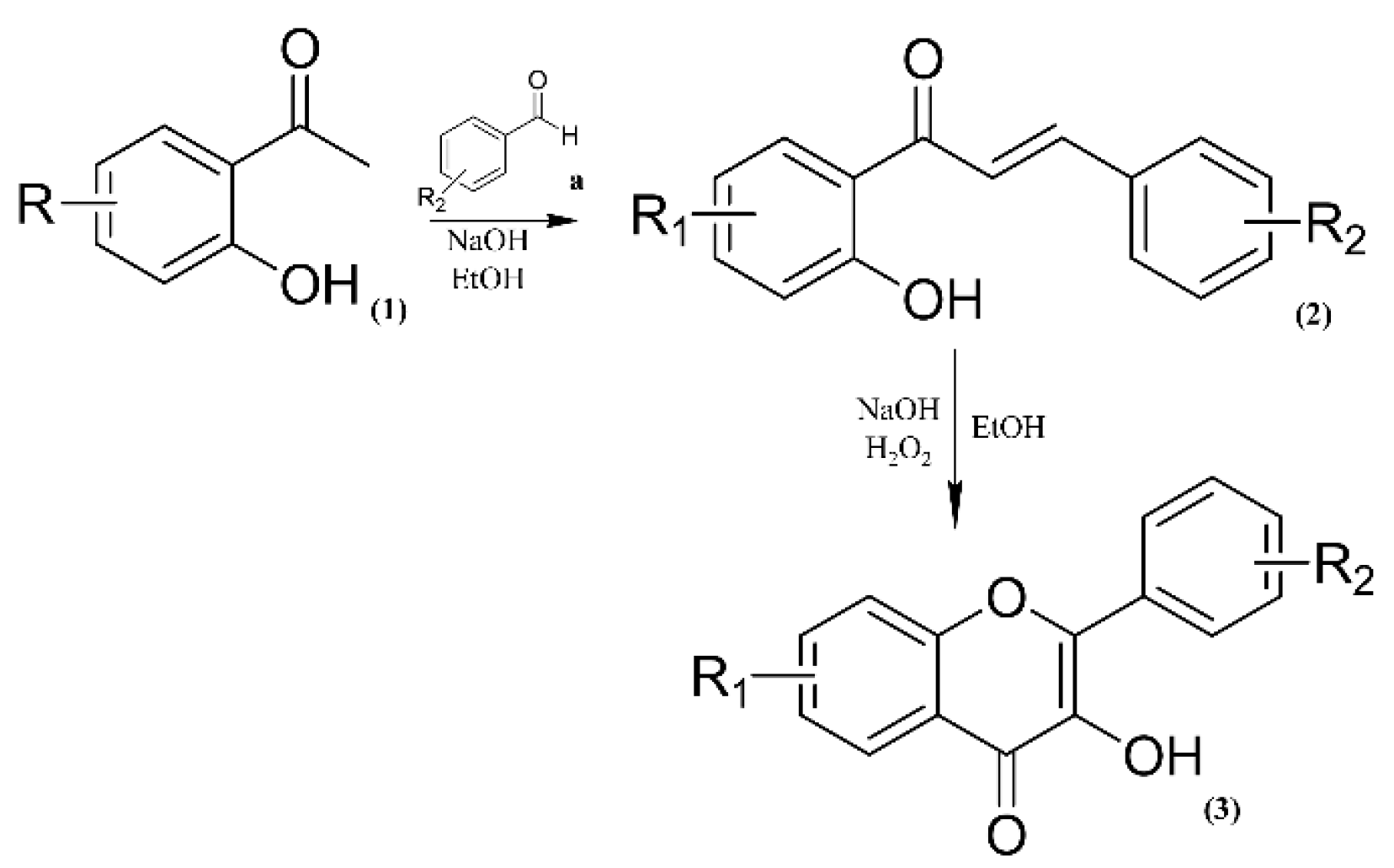
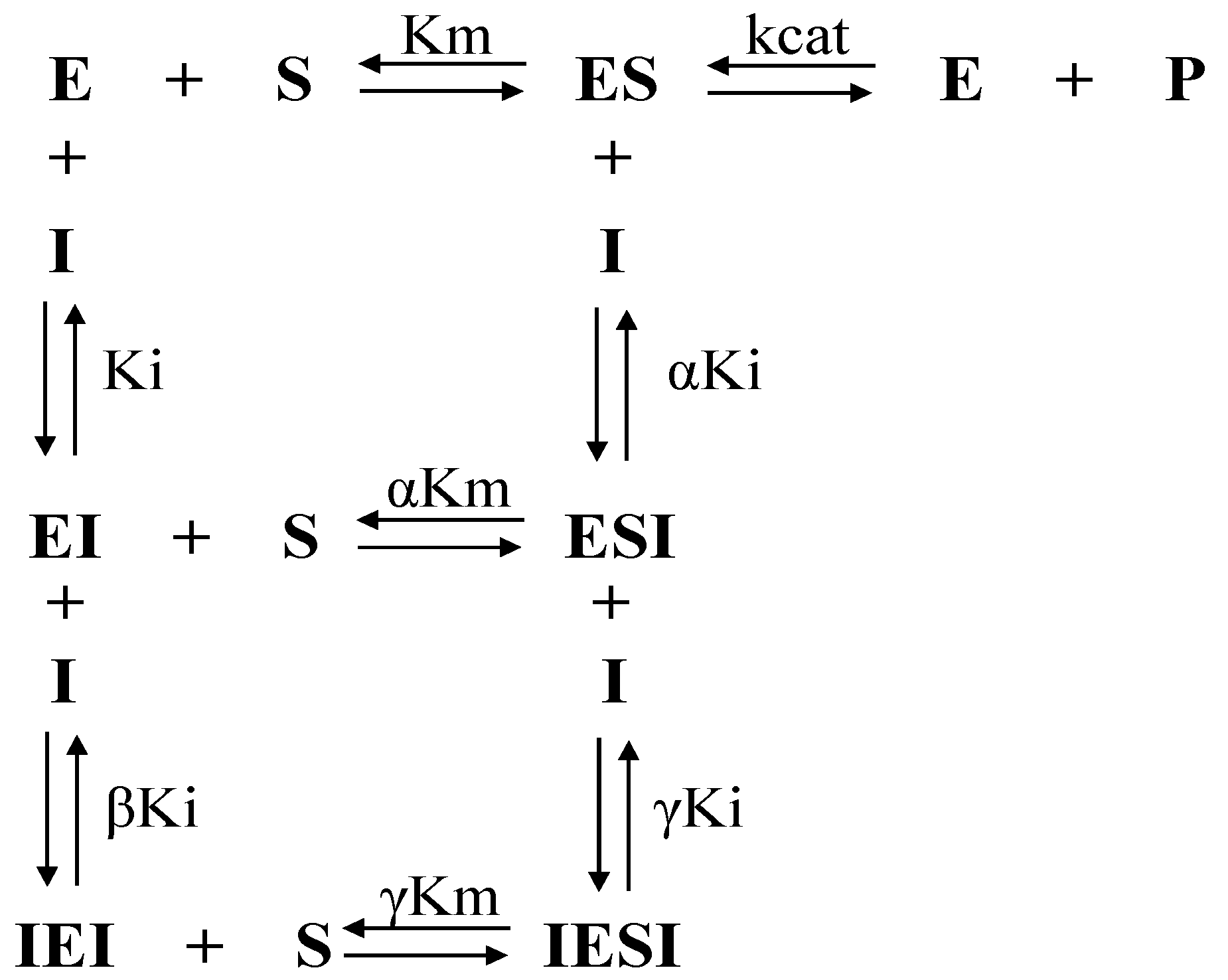
2.2. Determination of Inhibitory Potential (IC50), Mechanism of Cysteine Protease Inhibition and Evaluation of the Structure–Activity Relationship (SAR)
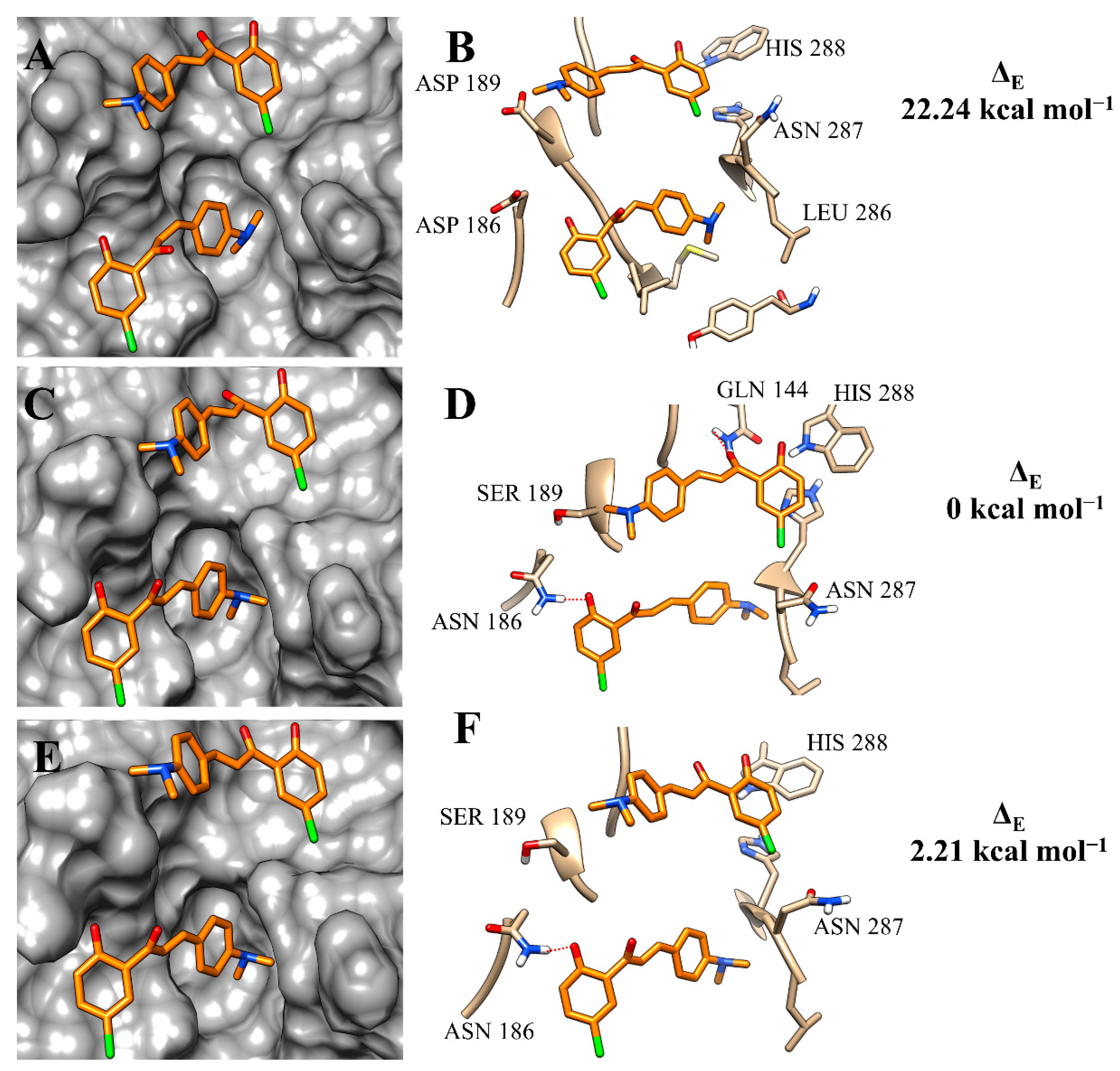
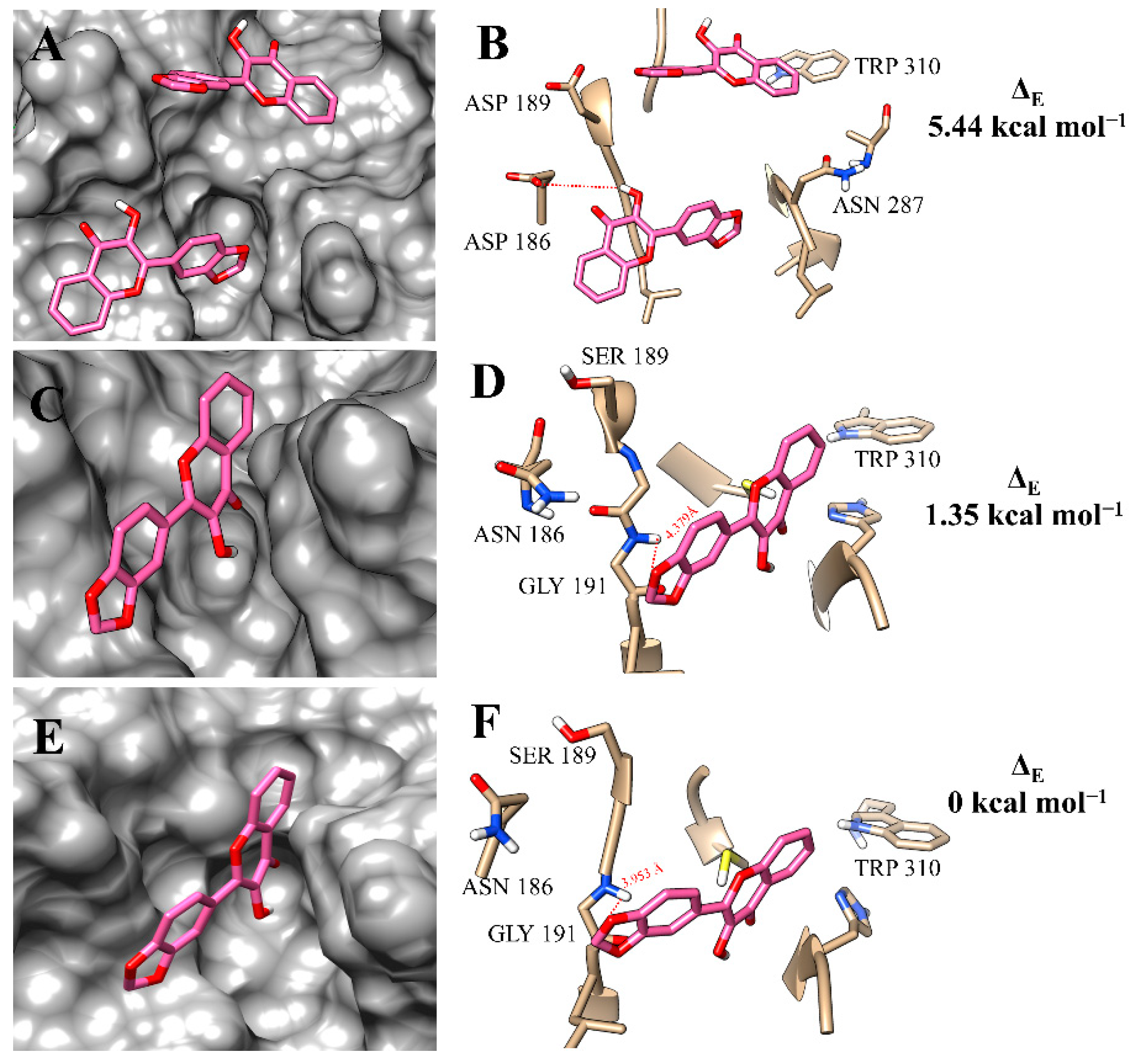
2.3. Molecular Modelling Study
2.4. Antipromastigote Assay and Cytotoxicity Elucidation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | LogPo/w 1 | LogS 2 | Promastigote IC50 (µM) 3 | NIH/3T3 Cells GL50 (µM) 5 | SI 6 |
|---|---|---|---|---|---|---|
| 1 | 1a | 3.97 | −4.59 | n.a 4 | 294.13 | n.d 7 |
| 2 | 1b | 3.87 | −5.03 | n.a 4 | 811.27 | n.d 7 |
| 3 | 1c | 4.04 | −4.91 | 1.14 | >874.83 | >767.39 |
| 4 | 2a | 3.10 | −4.05 | 0.71 | 940.14 | 1324 |
| 5 | 2b | 2.88 | −4.08 | n.a 4 | 910.58 | n.d 7 |
| 6 | 2c | 3.17 | −4.20 | n.a 4 | >935.21 | n.d 7 |
| 7 | 3a | 3.64 | −4.64 | 0.60 | >865.86 | >1443.10 |
| 8 | 3b | 3.46 | −4.67 | 0.96 | 59.13 | 61.59 |
| 9 | 3c | 3.65 | −4.79 | n.a 4 | >828.44 | n.d 7 |
| 10 | 4a | 3.42 | −4.20 | n.a 4 | 901.09 | n.d 7 |
| 11 | 4b | 3.24 | −4.23 | 1.95 | >873.36 | n.d 7 |
| 12 | 4c | 3.42 | −4.36 | 0.50 | >876.24 | >1752.48 |
| 13 | f12a | 2.83 | −4.09 | n.a 4 | >931.93 | n.d 7 |
| 14 | f12b | 2.68 | −4.11 | n.a 4 | >885.74 | n.d 7 |
| 15 | f12c | 2.85 | −4.23 | 0.73 | 759.13 | 1039.90 |
| 16 | f13a | 3.27 | −4.44 | 1.08 | >825.87 | >764.70 |
| 17 | f13c | 3.29 | −4.59 | n.a 4 | >791.76 | n.d 7 |
| Doxorubicin | --- | --- | --- | 0.05 | n.d 7 | |
| Pentamidine | --- | --- | 0.80 | --- | n.d 7 | |
| Amphotericin B | --- | --- | 0.10 | --- | n.d 7 |
3. Conclusions
4. Materials and Methods
4.1. Chemistry
4.2. General Procedure for the Synthesis of Chalcone-Like Compounds by the Claisen–Schmidt Reaction (1a–4c)
4.3. General Procedure for the Synthesis of Flavonol-Like Compounds by the Algar Flynn–Oyamada Reaction (f12a–f13c)
- (E)-1-(4-chlorophenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (1a): pale yellow crystal. Yield 67.11%. Mp 118–120 °C. Rf = 0.535. 1H-NMR (CDCl3, 300 MHz) δ ppm 7.96 (2H, d, J = 8.6, ArH), 7.81 (1H, d, J = 15.6 Hz, C=CH), 7.61 (2H, d, J = 8.7, ArH), 7.47 (2H, d, J = 8.6, ArH), 7.38 (1H, d, J = 15.6 Hz, C=CH), 6.95 (2H, d, J = 8.7, ArH), 3.85 (3H, s, –OCH3); 13C-NMR (CDCl3, 75 MHz) δ ppm 189.29, 161.97, 145.32, 139.05, 136.92, 130.45, 129.94, 128.98, 127.54, 119.27, 114.58, 55.54.
- (E)-3-(benzo[d][1,3]dioxol-5-yl)-1-(4-chlorophenyl)prop-2-en-1-one (1b): pale yellow crystal. Yield 86.19%. Mp 126–130 °C. Rf = 0.56. 1H-NMR (CDCl3, 300 MHz) δ ppm 7.96 (2H, d, J = 8.5, ArH), 7.76 (1H, d, J = 15.6 Hz, C=CH), 7.48 (2H, d, J = 8.5, ArH), 7.33 (1H, d, J = 15.6 Hz, C=CH), 7.15 (1H, d, J = 1.6 Hz, ArH), 7.13 (1H, dd, J = 8.1, ArH), 6.85 (1H, dd, J = 8.1, ArH), 6.03 (2H, s, –OCH2O); 13C-NMR (CDCl3, 75 MHz) δ ppm 188.69, 149.80, 148.16, 144.88, 138.74, 136.38, 129.52, 128.87, 125.13, 119.16, 108.41, 106.35, 101.40.
- (E)-1-(4-chlorophenyl)-3-(4-(dimethylamino)phenyl)prop-2-en-1-one (1c): yellow amorphous powder. Yield 73.26%. Mp 138–140 °C. Rf = 0.40. 1H-NMR (CDCl3, 300 MHz) δ ppm 7.96 (2H, d, J = 8.6, ArH), 7.82 (1H, d, J = 15.5 Hz, C=CH), 7.55 (2H, d, J = 8.9, ArH), 7.46 (2H, d, J = 8.6, ArH), 7.30 (1H, d, J = 15.5 Hz, C=CH), 6.70 (2H, d, J = 8.9, ArH), 3,04 (6H, s, –N(CH3)2); 13C-NMR (CDCl3, 75 MHz) δ ppm 189.94, 151.86, 146.08, 138.12, 137.10, 130.25, 129.43, 128.42, 122.13, 115.90, 111.50, 39.81.
- (E)-1-(2-hydroxyphenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (2a): yellow amorphous powder. Yield 76.53%. Mp 90–94 °C. Rf = 0.60. 1H-NMR (CDCl3, 300 MHz) δ ppm 12.96 (1H, s, OH), 7.92 (1H, dd, J = 2.3, 8.6 Hz, ArH), 7.91(1H, d, J = 15.5 Hz, C=CH), 7.62 (2H, d, J = 8.6 Hz, ArH), 7.55 (1H, d, J = 15.5 Hz, C=CH), 7.50–7.45 (1H, m, ArH), 7.02 (1H, d, J = 8.4 Hz, ArH), 6.95 (2H, d, J = 8.4 Hz, ArH), 7.00–6.90 (1H, m, ArH), 3,86 (3H, s, –OCH3); 13C-NMR (CDCl3, 75 MHz) δ ppm 193.35, 163.23, 161.72, 145.04, 135.82, 130.24, 129.22, 127.02, 119.80, 118.44, 118.25, 117.25, 114.20, 55.12.
- (E)-3-(benzo[d][1,3]dioxol-5-yl)-1-(2-hydroxyphenyl)prop-2-en-1-one (2b): yellow amorphous powder. Yield 71.43%. Mp 138–140 °C. Rf = 0.64. 1H-NMR (CDCl3, 300 MHz) δ ppm 12.89 (1H, s, OH), 7.90 (1H, 1.7, 8.1 Hz, ArH), 7.86 (1H, d, J = 15.5 Hz, ArH), 7.50 (1H, ddd, J = 1.7, 7.9, 8.1 Hz, ArH), 7.50 (1H, d, J = 15.5 Hz, ArH), 7.17 (1H, d, J = 1.7 Hz, ArH), 7.15 (1H, dd, J = 1.7, 8.1 Hz, ArH), 7.03 (1H, d, J = 8.1 Hz, ArH), 6.92 (1H, t, J = 7.2, 7.5, Hz, ArH), 6.86 (1H, d, J = 7.9 Hz, ArH), 6.03 (2H, s, –OCH2O); 13C-NMR (CDCl3, 75 MHz) δ ppm 193.23, 163.25, 150.0, 148.20, 145.02, 135.91, 129.21, 128.77, 125.44, 119.76, 118.48, 118.29, 117.69, 108.44, 106.44, 101.45.
- (E)-3-(4-(dimethylamino)phenyl)-1-(2-hydroxyphenyl)prop-2-en-1-one (2c): purple crystals. Yield 82.34%. Mp 178–181 °C. Rf = 0.71. 1H-NMR (CDCl3, 300 MHz) δ ppm 13.23 (1H, s, OH), 7.93 (1H, d, J = 15.4, C=CH), 7.90 (1H, d, J = 5.2 Hz, ArH), 7.57 (2H, d, J = 8.9 Hz, ArH), 7.48 (1H, d, J = 5.2 Hz, ArH), 7.46 (1H, d, J = 15.4, C=CH), 7.01 (1H, d, J = 7.9 Hz, ArH), 6.94 (1H, t, J = 7.7 Hz, ArH), 6.69 (2H, d, J = 8.9 Hz, ArH), 3.04 (6H, s, –N(CH3)2); 13C-NMR (CDCl3, 75 MHz) δ ppm 193.18, 163.16, 152.02, 146.25, 135.32, 130.55, 129.06, 121.97, 120.08, 118.26, 118.13, 113.90, 111.48, 39.76.
- (E)-1-(5-chloro-2-hydroxyphenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (3a): yellow amorphous powder. Yield 90.40%. Mp 108–109 °C. Rf = 0.79. 1H-NMR (CDCl3, 500 MHz) δ ppm 12.87 (1H, s, OH), 7.93 (1H, d, J = 15.5, C=CH), 7.85 (1H, s, ArH), 7.65 (2H, d, J = 8.1 Hz, ArH), 7.45–7.41 (2H, d, J = 15.5, C=CH, ArH), 6.98 (3H, m, ArH), 3.87 (3H, s, –OCH3); 13C-NMR (CDCl3, 125 MHz) δ ppm 192.53, 162.17, 161.87, 146.30, 135.77, 130.69, 128.57, 126.92, 123.27, 120.57, 120.03, 116.66, 114.45, 55.35.
- (E)-3-(benzo[d][1,3]dioxol-5-yl)-1-(5-chloro-2-hydroxyphenyl)prop-2-en-1-one (3b): yellow amorphous powder. Yield 86.70%. Mp 142–146. Rf = 0.72. 1H-NMR (CDCl3, 500 MHz) δ ppm 12.80 (1H, s, OH), 7.87 (1H, d, J = 15.3 Hz, C=CH), 7.83 (1H, d, J = 2.5 Hz, ArH), 7.43 (1H, d, J = 8.9, 2.5 Hz, ArH), 7.39 (1H, d, J = 15.3 Hz, C=CH), 7.19 (1H, d, J = 1.7 Hz, ArH), 6.98 (1H, d, J = 8.9 Hz, ArH), 6.87 (1H, J = 8.1 Hz, ArH), 6.05 (2H, s, –OCH2O); 13C-NMR (CDCl3, 125 MHz) δ ppm 192.43, 161.88, 150.50, 148.44, 146.27, 135.86, 128.65, 128.54, 126.02, 123.33, 120.52, 120.06, 117.08, 108.67, 106.71, 101.72.
- (E)-1-(5-chloro-2-hydroxyphenyl)-3-(4-(dimethylamino)phenyl)prop-2-en-1-one (3c): purple crystals. Yield 81.74%. Mp 161–163 °C. Rf = 0.68. 1H-NMR (CDCl3, 300 MHz) δ ppm 13.16 (1H, s, OH), 7.95 (1H, d, J = 15.1 Hz, C=CH), 7.85 (1H, d, J = 2.1 Hz, ArH), 7.58 (2H, d, J = 8.7 Hz, ArH), 7.41 (1H, dd, J = 8.8, 2.2 Hz, ArH), 7.35 (1H, d, J = 15.1 Hz, C=CH), 6.98 (1H, d, J = 8.8 Hz, ArH), 6.70 (2H, d, J = 8.7 Hz, ArH), 3.06 (6H, s, –OCH3); 13C-NMR (CDCl3, 75 MHz) δ ppm 192.05, 161.65, 152.25, 147.29, 135.00, 130.87, 128.75, 122.86, 121.75, 120.76, 119.70, 113.08, 111.48, 39.76.
- (E)-1-(5-fluoro-2-hydroxyphenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (4a): yellow amorphous powder. Yield 84.32%. Mp 124–125 °C. Rf = 0.68. 1H-NMR (CDCl3, 300 MHz) δ ppm 12.63 (1H, s, OH), 7.95 (1H, d, J = 15.3 Hz, C=CH), 7.65 (2H, J = 8.7 Hz, ArH), 7.60 (1H, dd, J = 9.2, 3.0 Hz, ArH), 7.44 (1H, d, J = 15.3 Hz, C=CH), 7.25 (1H, m, ArH), 7.00 (3H, m, ArH), 3.87 (3H, s, –OCH3); 13C-NMR (CDCl3, 75 MHz) δ ppm 192.46 (d, J = 2.7 Hz, ArH), 161.98, 159.38 (d, J = 1.0 Hz, ArH), 156.12 (d, J = 238.7 Hz, ArH), 145.94, 129.48, 126.80, 123.44 (d, (d, J = 23.6 Hz, ArH), 119.51 (d, J = 7.3 Hz, ArH), 119.4 (d, J = 6.3 Hz, ArH), 116.67, 114.28, 113.99, 56.17.
- (E)-3-(benzo[d][1,3]dioxol-5-yl)-1-(5-fluoro-2-hydroxyphenyl)prop-2-en-1-one (4b): yellow amorphous powder. Yield 76.51%. Mp 168–171 °C. Rf = 0.72. 1H-NMR (CDCl3, 300 MHz) δ ppm 12.61 (1H, s, OH), 7.89 (1H, d, J = 15.3 Hz, C=CH), 7.58 (1H, dd, J = 9.2, 2.8 Hz, ArH), 7.38 (1H, d, J = 15.3 Hz, C=CH), 7.27 (1H, m, 1H), 7.21–7.15 (2H, m, ArH), 7.01 (1H, dd, J = 9.1, 4.6 Hz, ArH), 6.88 (1H, d, J = 7.9 Hz, ArH), 6.05 (2H, s, –OCH2O); 13C-NMR (CDCl3, 75 MHz) δ ppm 192.38 (d, J = 2.7 Hz, ArH), 159.40 (d, J = 1.1 Hz, ArH), 156.12 (d, J = 238.2 Hz), 150.28, 148.29, 145.94, 128.54, 125.75, 123.57 (d, J = 23.4 Hz), 119.56 (d, J = 7.3 Hz), 117.10, 114.29 (d, J = 23.4 Hz), 108.51, 106.51, 101.55.
- (E)-3-(4-(dimethylamino)phenyl)-1-(5-fluoro-2-hydroxyphenyl)prop-2-en-1-one (4c): red crystals. Yield 95.69%. Mp 186–189 °C. Rf = 0.48. 1H-NMR (CDCl3, 300 MHz) δ ppm 12.96 (1H, s, OH), 7.94 (1H, d, J = 15.1 Hz, C=CH), 7.56 (3H, d, J = 8.7 Hz, ArH), 7.31 (1H, d, J = 15.1 Hz, C=CH), 7.22–7.15 (1H, m, ArH), 6.98 (1H, dd, J = 4.5, 9.1 Hz, ArH), 6.69 (2H, d, J = 8.5 Hz, ArH), 3.04 (6H, s, –OCH3); 13C-NMR (CDCl3, 75 MHz) δ ppm 192.10 (d, J = 2.8 Hz), 159.26, 156.04 (d, J = 237.0 Hz), 152.18, 147.11, 130.77, 122.77 (d, J = 23.7 Hz), 121.70, 119.65 (d, J = 6.1 Hz), 119.27 (d, J = 7.7 Hz), 114.15 (d, J = 23.3 Hz), 113.15, 111.46, 39.74.
- 3-hydroxy-2-(4-methoxyphenyl)-4H-chromen-4-one (f12a): yellow amorphous powder. Yield 71.34%. Mp 233–235 °C. Rf = 0.39. 1H-NMR (DMSO-d6, 500 MHz) δ ppm 9.24 (1H, s, OH), 8.20 (2H, dt, J = 8.9, 3.0, 2.1 Hz, ArH), 8.12 (1H, dd, J = 6.3, 1.5 Hz, ArH), 7.77 (1H, dt, J = 7.8, 1.0 Hz, ArH), 7.72 (1H, dd, J = 8.6, 1.0 Hz, ArH), 7.45 (1H, dt, J = 7.8, 1.0 Hz, ArH), 7.12 (2H, dt, J = 8.9, 3.0, 2.1 Hz, ArH), 3.85 (3H, s, –OCH3); 13C-NMR (DMSO-d6, 125 MHz)) δ ppm 172.48, 160.37, 154.35, 145.52, 137.95, 133.25, 129.24, 124.57, 124.26, 123.48, 121.23, 118.09, 113.91, 55.21.
- 2-(benzo[d][1,3]dioxol-5-yl)-3-hydroxy-4H-chromen-4-one (f12b): yellow amorphous powder. Yield 69.87%. Mp 213–215 °C. Rf = 0.39. 1H-NMR (DMSO-d6, 300 MHz) δ ppm 9.52 (1H, s, OH), 8.10 (1H, d, J = 8.1 Hz, ArH), 7.84 (1H, dd, J = 8.5, 1.6 Hz, ArH), 7.78–7.73 (3H, m, ArH), 7.47 (1H, td, J = 8.1, 6.4, 1.7 Hz, ArH), 7.11 (1H, d, J = 8.3 Hz, ArH), 6.12 (2H, s, –OCH2O); 13C-NMR (DMSO-d6, 75 MHz) δ ppm 172.68, 154.40, 148.54, 147.52, 145.10, 138.33, 133.54, 125.05, 124.69, 122.76, 121.25, 118.36, 108.43, 107.52, 101.67.
- 2-(4-(dimethylamino)phenyl)-3-hydroxy-4H-chromen-4-one (f12c): Orange crystals. Yield 79.40%. Mp 182–183 °C. Rf = 0.39. 1H-NMR (DMSO-d6, 500 MHz) δ ppm 9.16 (1H, s, OH), 8.12 (1H, d, J = 8.8 Hz, ArH), 8.09 (1H, dd, J = 7.8, 1.4 Hz, ArH), 7.69–7.75 (2H, m, ArH), 7.44 (1H, t, J = 7.3, 7.4 Hz, ArH), 6.83 (2H, d, J = 8.8 Hz, ArH), 2.99 (6H, s, –N(CH3)2); 13C-NMR (DMSO-d6, 125 MHz) δ ppm 171.98, 154.29, 151.03, 146.85, 137.29, 133.09, 128.99, 124.65, 124.32, 121.45, 118.15, 117.93, 111.40, 39.64.
- 6-chloro-3-hydroxy-2-(4-methoxyphenyl)-4H-chromen-4-one (f13a): yellow amorphous powder. Yield 86.42%. Mp 207–208 °C. Rf = 0.52. 1H-NMR (DMSO-d6, 500 MHz) δ ppm 9.44 (1H, s, OH), 8.19 (2H, dt, J = 9.2, 2.9 Hz, ArH), 8.02 (1H, t, J = 1.4 Hz, ArH), 7.79 (2H, d, J = 1.8 Hz, ArH), 7.12 (2H, dt, J = 9.2, 2.9 Hz, ArH), 3.85 (3H, s, –OCH3); 13C-NMR (DMSO-d6, 125 MHz) δ ppm 171.42, 160.54, 152.86, 146.13, 138.12, 133.11, 129.37, 128.78, 123.18, 122.39, 120.64, 113.94, 55.25.
- 6-chloro-2-(4-(dimethylamino)phenyl)-3-hydroxy-4H-chromen-4-one (f13c): Orange crystals. Yield 78.67%. Mp 235–238 °C. Rf = 0.57. 1H-NMR (DMSO-d6, 500 MHz) δ ppm 8.98 (1H, s, OH), 8.11 (2H, dt, J = 9.1, 3.0 Hz, ArH) 7.99 (1H, t, J = 1.7 Hz, ArH), 7.73–7.71 (2H, m, ArH), 6.83 (2H, dt, J = 9.1, 3.0 Hz, ArH), 7.11 (1H, d, J = 8.3 Hz, ArH), 3.01 (6H, s, –N(CH3)2); 13C-NMR (DMSO-d6, 125 MHz) δ ppm 170.51, 152.57, 151.01, 147.28, 137.03, 132.45, 128.75, 128.44, 123.14, 122.36, 120.20, 117.40, 111.12, 39.25.
4.4. Screening the Inhibitory Activity of Compounds
4.5. Determination of IC50 Values for Inhibitors
4.6. Enzyme Kinetics and Determination of the Mechanism of Inhibition
4.7. Molecular Modelling
4.8. Cytotoxicity on Mammalian Cells
4.9. Parasites
4.10. Antipromastigote Assay
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- BRASIL Doenças Tropicais Negligenciadas; Ministério da Saúde: Distrito Federal, Brazil, 2021.
- Brindha, J.; Balamurali, M.M.; Chanda, K. An Overview on the Therapeutics of Neglected Infectious Diseases—Leishmaniasis and Chagas Diseases. Front. Chem. 2021, 9, 622286. [Google Scholar]
- Alvar, J.; Yactayo, S.; Bern, C. Leishmaniasis and Poverty. Trends Parasitol. 2006, 22, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, C.; Engels, D. Leaving No One behind: A Neglected Tropical Disease Indicator and Tracers for the Sustainable Development Goals. Int. Health 2015, 8, i15–i18. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A Perspective on Multi-Target Drug Discovery and Design for Complex Diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Barbosa Gomes de Carvalho, Y.M.; Shanmugam, S.; Batista, M.S.; Serafini, M.R.; Araújo, A.A.D.S.; Quintans Júnior, L.J. Pharmaceutical Agents for Treatment of Leishmaniasis: A Patent Landscape. Expert Opin. Ther. Pat. 2020, 30, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Dias, M.C.; Pinto, D.C.G.A.; Silva, A.M.S. Plant Flavonoids: Chemical Characteristics and Biological Activity. Molecules 2021, 26, 5377. [Google Scholar] [CrossRef]
- Carter, N.S.; Stamper, B.D.; Elbarbry, F.; Nguyen, V.; Lopez, S.; Kawasaki, Y.; Poormohamadian, R.; Roberts, S.C. Microorganisms Natural Products That Target the Arginase in Leishmania Parasites Hold Therapeutic Promise. Microorganisms 2021, 9, 267. [Google Scholar] [CrossRef]
- Gervazoni, L.F.O.; Gonçalves-Ozório, G.; Almeida-Amaral, E.E. 2′-Hydroxyflavanone Activity in vitro and in vivo against Wild-Type and Antimony-Resistant Leishmania amazonensis. PLoS Negl. Trop. Dis. 2018, 12, e0006930. [Google Scholar] [CrossRef]
- Braga, F.C.; Ojeda, M.; Perdomo, R.T.; de Albuquerque, S.; Rafique, J.; de Lima, D.P.; Beartriz, A. Synthesis of cardanol-based 1,2,3-triazoles as potential green agents against neoplastic cells. Sustain. Chem. Pharm. 2021, 20, 100408. [Google Scholar] [CrossRef]
- Saba, S.; dos Santos, C.R.; Zavarise, B.R.; Naujorks, A.A.S.; Franco, M.S.; Schneider, A.R.; Scheide, M.R.; Affeldt, R.F.; Rafique, J.; Braga, A.L. Photoinduced, Direct C(sp2)−H Bond Azo Coupling of Imidazoheteroarenes and Imidazoanilines with Aryl Diazonium Salts Catalyzed by Eosin Y. Chem. Eur. J. 2020, 26, 4461–4466. [Google Scholar] [CrossRef]
- Galant, L.S.; Rafique, J.; Braga, A.L.; Braga, F.C.; Saba, S.; Radi, R.; da Rocha, J.B.T.; Santi, C.; Monsalve, M.; Farina, M.; et al. The Thiol-Modifier Effects of Organoselenium Compounds and Their Cytoprotective Actions in Neuronal Cells. Neurochem. Res. 2021, 46, 120–130. [Google Scholar] [CrossRef]
- Franco, M.S.; Saba, S.; Rafique, J.; Braga, A.L. KIO4-mediated Selective Hydroxymethylation/Methylenation of Imidazo-Heteroarenes: A Greener Approach. Angew. Chem. 2021, 1333, 18602–18608, Angew. Chem. Int. Ed. Engl.2021, 60, 18454–18460. [Google Scholar] [CrossRef]
- Veloso, I.C.; Delanogare, E.; Machado, A.E.; Braga, S.P.; Rosa, G.K.; de Bem, A.F.; Rafique, J.; Saba, S.; da Trindade, R.N.; Galetto, F.Z.; et al. A selanylimidazopyridine (3-SePh-IP) reverses the prodepressant- and anxiogenic-like effects of a high-fat/high-fructose diet in mice. J. Pharm. Pharmacol. 2021, 73, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Peterle, M.M.; Scheide, M.R.; Silva, L.T.; Saba, S.; Rafique, J.; Braga, A.L. Copper-Catalyzed Three-Component Reaction of Oxadiazoles, Elemental Se/S and Aryl Iodides: Synthesis of Chalcogenyl (Se/S)-Oxadiazoles. ChemsitrySelect 2018, 3, 13191–13196. [Google Scholar] [CrossRef]
- dos Santos, D.C.; Rafique, J.; Saba, S.; Grinevicuis, V.M.A.S.; Filho, D.W.; Zamoner, A.; Braga, A.L.; Pedrosa, R.C.; Ourique, F. IP-Se-06, a Selenylated Imidazo[1,2-a]pyridine, Modulates Intracellular Redox State and Causes Akt/mTOR/HIF-1α and MAPK Signaling Inhibition, Promoting Antiproliferative Effect and Apoptosis in Glioblastoma Cells. Oxid. Med. Cell. Longev. 2022, 2022, 3710449. [Google Scholar] [CrossRef] [PubMed]
- Frizon, T.E.A.; Cararo, J.H.; Saba, S.; Dal-Pont, G.C.; Michels, M.; Braga, H.C.; Pimentel, T.; Dal-Pizzol, F.; Valvassori, S.S.; Rafique, J. Synthesis of Novel Selenocyanates and Evaluation of Their Effect in Cultured Mouse Neurons Submitted to Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 5417024. [Google Scholar] [CrossRef]
- Rahman, A.F.M.M.; Ali, R.; Jahng, Y.; Kadi, A.A. A Facile Solvent Free Claisen-Schmidt Reaction: Synthesis of α,α’-Bis-(Substituted-Benzylidene)Cycloalkanones and α,α’-Bis-(Substituted-Alkylidene)Cycloalkanones. Molecules 2012, 17, 571–583. [Google Scholar] [CrossRef]
- Wang, P.; Li, H.F.; Zhao, J.Z.; Du, Z.H.; Da, C.S. Organocatalytic Enantioselective Cross-Aldol Reaction of o-Hydroxyarylketones and Trifluoromethyl Ketones. Org. Lett. 2017, 19, 2634–2637. [Google Scholar] [CrossRef]
- Wang, Z. Algar-Flynn-Oyamada (AFO) Reaction. In Comprehensive Organic Name Reactions and Reagents; Wiley: New York, NY, USA, 2010. [Google Scholar]
- Gopinath, V.S.; Pinjari, J.; Dere, R.T.; Verma, A.; Vishwakarma, P.; Shivahare, R.; Moger, M.; Kumar Goud, P.S.; Ramanathan, V.; Bose, P.; et al. Design, Synthesis and Biological Evaluation of 2-Substituted Quinolines as Potential Anti Leishmania Agents. Eur. J. Med. Chem. 2013, 69, 527–536. [Google Scholar] [CrossRef]
- Neuenschwander, A.; Rocha, V.P.C.; Bastos, T.M.; Marcourt, L.; Morin, H.; da Rocha, C.Q.; Grimaldi, G.B.; de Sousa, K.A.F.; Borges, J.N.; Rivara-Minten, E.; et al. Production of Highly Active Antiparasitic Compounds from the Controlled Halogenation of the Arrabidaea Brachypoda Crude Plant Extract. J. Nat. Prod. 2020, 83, 2631–2640. [Google Scholar] [CrossRef]
- Filarowski, A.; Koll, A.; Kochel, A.; Kalenik, J.; Hansen, P.E. The Intramolecular Hydrogen Bond in Ortho-Hydroxy Acetophenones. J. Mol. Struct. 2004, 700, 67–72. [Google Scholar] [CrossRef]
- Gomes, M.N.; Muratov, E.N.; Pereira, M.; Peixoto, J.C.; Rosseto, L.P.; Cravo, P.V.L.; Andrade, C.H.; Neves, B.J. Chalcone Derivatives: Promising Starting Points for Drug Design. Molecules 2017, 22, 1210. [Google Scholar] [CrossRef] [PubMed]
- Rai, P.; Chettri, P.; Kar, S.; Nagar, M.A.; Srivastava, S.; Golakoti, N.R. Synthesis, Characterization and Structure–Activity Relationship of Non-Linear Optical Response of Chalcone Derivatives with in silico Insights. Chem. Pap. 2021, 75, 2603–2615. [Google Scholar] [CrossRef]
- Montes-Avila, J.; Díaz-Camacho, S.P.; Sicairos-Félix, J.; Delgado-Vargas, F.; Rivero, I.A. Solution-Phase Parallel Synthesis of Substituted Chalcones and Their Antiparasitary Activity against Giardia Lamblia. Bioorg. Med. Chem. 2009, 17, 6780–6785. [Google Scholar] [CrossRef]
- Regenass, P.; Abboud, D.; Daubeuf, F.; Lehalle, C.; Gizzi, P.; Riché, S.; Hachet-Haas, M.; Rohmer, F.; Gasparik, V.; Boeglin, D.; et al. Discovery of a Locally and Orally Active CXCL12 Neutraligand (LIT-927) with Anti-Inflammatory Effect in a Murine Model of Allergic Airway Hypereosinophilia. J. Med. Chem. 2018, 61, 7671–7686. [Google Scholar] [CrossRef] [PubMed]
- Detsi, A.; Majdalani, M.; Kontogiorgis, C.A.; Hadjipavlou-Litina, D.; Kefalas, P. Natural and Synthetic 2′-Hydroxy-Chalcones and Aurones: Synthesis, Characterization and Evaluation of the Antioxidant and Soybean Lipoxygenase Inhibitory Activity. Bioorg. Med. Chem. 2009, 17, 8073–8085. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, H.J. Highly Activated Michael Acceptor by an Intramolecular Hydrogen Bond as a Fluorescence Turn-on Probe for Cyanide. Chem. Commun. 2010, 46, 9197–9199. [Google Scholar] [CrossRef] [PubMed]
- Mewett, K.N.; Fernandez, S.P.; Pasricha, A.K.; Pong, A.; Devenish, S.O.; Hibbs, D.E.; Chebib, M.; Johnston, G.A.R.; Hanrahan, J.R. Synthesis and Biological Evaluation of Flavan-3-Ol Derivatives as Positive Modulators of GABAA Receptors. Bioorg. Med. Chem. 2009, 17, 7156–7173. [Google Scholar] [CrossRef] [PubMed]
- Kamecki, F.; Knez, D.; Carvalho, D.; Marcucci, C.; Rademacher, M.; Higgs, J.; Žakelj, S.; Marcos, A.; de Tezanos Pinto, F.; Abin-Carriquiry, J.A.; et al. Multitarget 2′-Hydroxychalcones as Potential Drugs for the Treatment of Neurodegenerative Disorders and Their Comorbidities. Neuropharmacology 2021, 201, 108837. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.L.; Liu, C.L.; Huang, W.; Wang, Y.Z.; Yang, G.F. Synthesis and Fungicidal Evaluation of Novel Chalcone-Based Strobilurin Analogues. J. Agric. Food Chem. 2007, 55, 5697–5700. [Google Scholar] [CrossRef]
- Borsari, C.; Santarem, N.; MacEdo, S.; Jiménez-Antón, M.D.; Torrado, J.J.; Olías-Molero, A.I.; Corral, M.J.; Tait, A.; Ferrari, S.; Costantino, L.; et al. SAR Studies and Biological Characterization of a Chromen-4-One Derivative as an Anti- Trypanosoma Brucei Agent. ACS Med. Chem. Lett. 2019, 10, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Muller, B.M.; Mai, J.; Yocum, R.A.; Adler, M.J. Impact of Mono- and Disubstitution on the Colorimetric Dynamic Covalent Switching Chalcone/Flavanone Scaffold. Org. Biomol. Chem. 2014, 12, 5108–5114. [Google Scholar] [CrossRef] [PubMed]
- Gharpure, M.; Chaudhary, R.G.; Juneja, H.; Ingle, V.; Gandhare, N. Oxovanadium (IV) Complexes of 2-Aryl/Heteroaryl-3-Hydroxy-4H-Chromones: Synthesis, Spectral and Thermal Degradation Studies. J. Chin. Adv. Mater. Soc. 2013, 1, 257–267. [Google Scholar] [CrossRef]
- Karmakar, A.; Ambure, P.; Mallick, T.; Das, S.; Roy, K.; Begum, N.A. Exploration of Synthetic Antioxidant Flavonoid Analogs as Acetylcholinesterase Inhibitors: An Approach towards Finding Their Quantitative Structure–Activity Relationship. Med. Chem. Res. 2019, 28, 723–741. [Google Scholar] [CrossRef]
- Xiong, W.; Wang, X.; Shen, X.; Hu, C.; Wang, X.; Wang, F.; Zhang, G.; Wang, C. Synthesis of Flavonols via Pyrrolidine Catalysis: Origins of the Selectivity for Flavonol versus Aurone. J. Org. Chem. 2020, 85, 13160–13176. [Google Scholar] [CrossRef] [PubMed]
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and Challenges in Phenotypic Drug Discovery: An Industry Perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Olías-molero, A.I.; de la Fuente, C.; Cuquerella, M.; Torrado, J.J.; Alunda, J.M. Anti Leishmania Drug Discovery and Development: Time to Reset the Model? Microorganisms 2021, 9, 2500. [Google Scholar] [CrossRef]
- Buxbaum, L.U.; Denise, H.; Coombs, G.H.; Alexander, J.; Mottram, J.C.; Scott, P. Cysteine Protease B of Leishmania exicana Inhibits Host Th1 Responses and Protective Immunity. J. Immunol. 2003, 171, 3711–3717. [Google Scholar] [CrossRef]
- Siqueira-Neto, J.L.; Debnath, A.; McCall, L.I.; Bernatchez, J.A.; Ndao, M.; Reed, S.L.; Rosenthal, P.J. Cysteine Proteases in Protozoan Parasites. PloS Negl. Trop. Dis. 2018, 12, e0006512. [Google Scholar] [CrossRef] [PubMed]
- Gontijo, V.S.; Judice, W.A.S.; Codonho, B.; Pereira, I.O.; Assis, D.M.; Januário, J.P.; Caroselli, E.E.; Juliano, M.A.; de Carvalho Dosatti, A.; Marques, M.J.; et al. Leishmanicidal, Antiproteolytic and Antioxidant Evaluation of Natural Biflavonoids Isolated from Garcinia Brasiliensis and Their Semisynthetic Derivatives. Eur. J. Med. Chem. 2012, 58, 613–623. [Google Scholar] [CrossRef]
- Juliano, M.A.; Brooks, D.R.; Selzer, P.M.; Pandolfo, H.L.; Judice, W.A.S.; Juliano, L.; Meldal, M.; Sanderson, S.J.; Mottram, J.C.; Coombs, G.M. Differences in Substrate Specificities between Cysteine Protease CPB Isoforms of Leishmania exicana Are Mediated by a Few Amino Acid Changes. Eur. J. Biochem. 2004, 271, 3704–3714. [Google Scholar] [CrossRef]
- Denise, H.; McNeil, K.; Brooks, D.R.; Alexander, J.; Coombs, G.H.; Mottram, J.C. Expression of Multiple CPB Genes Encoding Cysteine Proteases Is Required for Leishmania exicana Virulence in vivo. Infect. Immun. 2003, 71, 3190–3195. [Google Scholar] [CrossRef] [PubMed]
- Raghav, N.; Kaur, R. Chalcones, Semicarbazones and Pyrazolines as Inhibitors of Cathepsins B, H and L. Int. J. Biol. Macromol. 2015, 80, 710–724. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.N.; Alcântara, L.M.; Neves, B.J.; Melo-Filho, C.C.; Freitas-Junior, L.H.; Moraes, C.B.; Ma, R.; Franzblau, S.G.; Muratov, E.; Andrade, C.H. Computer-Aided Discovery of Two Novel Chalcone-like Compounds Active and Selective against Leishmania infantum. Bioorg. Med. Chem. Lett. 2017, 27, 2459–2464. [Google Scholar] [CrossRef] [PubMed]
- de Novais, L.M.R.; de Arueira, C.C.O.; Ferreira, L.F.; Ribeiro, T.A.N.; Sousa, P.T.; Jacinto, M.J.; de Carvalho, M.G.; Judice, W.A.S.; Jesus, L.O.P.; de Souza, A.A.; et al. 4′-Hydroxy-6,7-Methylenedioxy-3-Methoxyflavone: A Novel Flavonoid from Dulacia Egleri with Potential Inhibitory Activity against Cathepsins B and L. Fitoterapia 2019, 132, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Kerr, I.D.; Lee, J.H.; Farady, C.J.; Marion, R.; Rickert, M.; Sajid, M.; Pandey, K.C.; Caffrey, C.R.; Legac, J.; Hansell, E.; et al. Vinyl Sulfones as Antiparasitic Agents and a Structural Basis for Drug Design. J. Biol. Chem. 2009, 284, 25697–25703. [Google Scholar] [CrossRef] [PubMed]
- Mendieta, L.; Picó, A.; Tarragó, T.; Teixidó, M.; Castillo, M.; Rafecas, L.; Moyano, A.; Giralt, E. Novel Peptidyl Aryl Vinyl Sulfones as Highly Potent and Selective Inhibitors of Cathepsins L and B. ChemMedChem 2010, 5, 1556–1557. [Google Scholar] [CrossRef]
- Brinen, L.S.; Hansell, E.; Cheng, J.; Roush, W.R.; McKerrow, J.H.; Fletterick, R.J. A Target within the Target: Probing Cruzain’s P1’ Site to Define Structural Determinants for the Chagas’ Disease Protease. Structure 2000, 8, 831–840. [Google Scholar] [CrossRef]
- Freeman, A.M.; Mole, B.M.; Silversmith, R.E.; Bourret, R.B. Action at a Distance: Amino Acid Substitutions That Affect Binding of the Phosphorylated CheY Response Regulator and Catalysis of Dephosphorylation Can Be Far from the CheZ Phosphatase Active Site. J. Bacteriol. 2011, 193, 4709–4718. [Google Scholar] [CrossRef]
- Judice, W.A.S.; Mottram, J.C.; Coombs, G.H.; Juliano, M.A.; Juliano, L. Specific Negative Charges in Cysteine Protease Isoforms of Leishmania exicana Are Highly Influential on the Substrate Binding and Hydrolysis. Mol. Biochem. Parasitol. 2005, 144, 36–43. [Google Scholar] [CrossRef]
- Santiago, A.S.; Pita, S.S.D.R.; Guimarães, E.T. Tratamento Da Leishmaniose, Limitações Da Terapêutica Atual e a Necessidade de Novas Alternativas: Uma Revisão Narrativa. Res. Soc. Dev. 2021, 10, e29510716543. [Google Scholar] [CrossRef]
- Cohen, A.; Azas, N. Challenges and Tools for in vitro Leishmania Exploratory Screening in the Drug Development Process: An Updated Review. Pathogens 2021, 10, 1608. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S. Nishi, Visceral Leishmaniasis: Experimental Models for Drug Discovery. Indian J. Med. Res. 2011, 133, 27–39. [Google Scholar] [PubMed]
- Trefzger, O.S.; das Neves, A.R.; Barbosa, N.V.; Carvalho, D.B.; Pereira, I.C.; Perdomo, R.T.; Matos, M.F.C.; Yoshida, N.C.; Kato, M.J.; de Albuquerque, S.; et al. Design, Synthesis and Antitrypanosomatid Activities of 3,5-Diaryl-Isoxazole Analogues Based on Neolignans Veraguensin, Grandisin and Machilin G. Chem. Biol. Drug Des. 2019, 93, 313–324. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Sander, T. OSIRIS Property Explorer; Organic Chemistry Portal: Basel, Switzerland, 2001. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals; Pergamon Press: Oxford, UK, 1988. [Google Scholar]
- James, J.P. Stewart, Stewart Computational Chemistry, Colorado Springs, CO, USA. MOPAC2016. Available online: http://OpenMOPAC.net (accessed on 1 November 2022).
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Spoel, D.V.D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A. Olivier Michielin SwissParam: A Fast Force Field Generation Tool ForSmall Organic Molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; Mcmahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-wolff, A.; et al. Feasibility of a High-Flux Anticancer Drug Screen Using a Diverse Panel of Cultured Human Tumor Cell Lines. J. Natl. Cancer Inst. 1991, 11, 757–766. [Google Scholar] [CrossRef] [PubMed]








 | |||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | R4 | Yield (%) |
| 1 | H | Cl | H | –OCH3 | 67.11 (1a) |
| 2 | H | Cl | H | –OCH2O | 86.19 (1b) |
| 3 | H | Cl | H | –N(CH3)2 | 73.26 (1c) |
| 4 | OH | H | H | –OCH3 | 76.26 (2a) |
| 5 | OH | H | H | –OCH2O | 71.43 (2b) |
| 6 | OH | H | H | –N(CH3)2 | 82.34 (2c) |
| 7 | OH | H | Cl | –OCH3 | 90.40 (3a) |
| 8 | OH | H | Cl | –OCH2O | 86.70 (3b) |
| 9 | OH | H | Cl | –N(CH3)2 | 81.74 (3c) |
| 10 | OH | H | F | –OCH3 | 84.32 (4a) |
| 11 | OH | H | F | –OCH2O | 76.51 (4b) |
| 12 | OH | H | F | –N(CH3)2 | 95.69 (4c) |
| 13 | - | H | H | –OCH3 | 71.34 (f12a) |
| 14 | - | H | H | –OCH2O | 69.87 (f12b) |
| 15 | - | H | H | –N(CH3)2 | 79.40 (f12c) |
| 16 | - | H | Cl | –OCH3 | 86.42 (f13a) |
| 17 | - | H | Cl | –N(CH3)2 | 78.67 (f13c) |
 | ||||
|---|---|---|---|---|
| Entry | Compound | IC50 (µM) | ||
| rCPB2.8 | rCPB3 | rH84Y | ||
| 1 | 1a | 13.66 ± 0.74 | 9.91 ± 0.59 | 29.74 ± 2.62 |
| 2 | 1b | 7.42 ± 0.25 | 5.60 ± 0.35 | 15.34 ± 0.60 |
| 3 | 1c | 2.75 ± 0.18 | 10.17 ± 0.52 | 11.70 ± 1.27 |
| 4 | 2a | 2.35 ± 0.19 | 7.14 ± 0.86 | 16.34 ± 0.46 |
| 5 | 2b | 20.63 ± 1.06 | 10.99 ± 0.40 | 4.67 ± 0.13 |
| 6 | 2c | 11.74 ± 0.38 | 8.96 ± 0.61 | 9.90 ± 0.48 |
| 7 | 3a | 7.11 ± 0.32 | 4.22 ± 0.81 | 6.84 ± 0.26 |
| 8 | 3b | 18.37 ± 1.22 | 11.56 ± 0.77 | 8.23 ± 0.21 |
| 9 | 3c | 3.97 ± 0.08 | 5.47 ± 0.59 | 4.81 ± 0.32 |
| 10 | 4a | 3.35 ± 0.17 | 4.51 ± 0.19 | 20.34 ± 1.78 |
| 11 | 4b | 6.61 ± 0.41 | 17.27 ± 1.46 | 7.77 ± 0.72 |
| 12 | 4c | 9.88 ± 0.73 | 37.97 ± 7.24 | 4.34 ± 0.43 |
 | ||||
|---|---|---|---|---|
| Entry | Compound | IC50 (µM) | ||
| rCPB2.8 | rCPB3 | rH84Y | ||
| 13 | f12a | 4.72 ± 0.38 | 7.71 ± 0.78 | 3.85 ± 0.27 |
| 14 | f12b | 5.23 ± 0.32 | 7.06 ± 0.63 | 8.85 ± 0.33 |
| 15 | f12c | 10.32 ± 0.39 | 16.67 ± 2.53 | 28.56 ± 1.62 |
| 16 | f13a | 1.88 ± 0.07 | 10.56 ± 1.11 | 11.71 ± 1.54 |
| 17 | f13c | 14.18 ± 0.65 | 29.75 ± 2.08 | 20.35 ± 1.02 |
| rCPB2.8 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Ki (µM) | αKi (µM) | βKi (µM) | γKi (µM) | α | β | γ | Mechanism |
| 3c | 100 ± 18 | 147 ± 28 | 0.55 ± 0.10 | 1.15 ± 0.21 | 1.47 | 0.0055 | 0.0115 | NCPC |
| f12a | 12.4 ± 1.4 | 10.7 ± 1.1 | 3.65 ± 0.39 | 0.25 ± 0.03 | 1 | 0.29 | 0.02 | NCPC |
| f12b | 39.2 ± 4.1 | 24.9 ± 2.3 | 1.17 ± 0.12 | 0.51 ± 0.09 | 0.634 | 0.029 | 0.013 | NCPC |
| rCPB3 | ||||||||
| 3c | 15.4 ± 3.0 | 141 ± 27 | 2.21 ± 0.16 | 0.77 ± 0.14 | 9.15 | 0.14 | 0.05 | NCPC |
| f12a | 7.47 ± 0.64 | 7.50 ± 0.66 | --- | --- | 1 | SLNC | ||
| f12b | 13.7 ± 1.4 | 13.7 ± 1.2 | --- | --- | 1 | SLNC | ||
| rH84Y | ||||||||
| 3c | 29.4 ± 4.7 | 127 ± 20 | 1.45 ± 0.27 | 1.06 ± 0.18 | 4.31 | 0.049 | 0.036 | NCPC |
| f12a | 11.2 ± 0.8 | 11.2 ± 0.5 | --- | --- | 1 | SLNC | ||
| f12b | 6.08 ± 0.37 | 6.06 ± 0.24 | --- | --- | 1 | SLNC | ||
| Enzyme | Binding-Free Energy | ΔE | Ki (µM) |
|---|---|---|---|
| 3c | |||
| rCPB2.8 | −2.46 | 22.24 | 100 ± 18 |
| rCPB3 | −24.70 | 0 | 12.4 ± 1.4 |
| rH84Y | −22.49 | 2.21 | 39.2 ± 4.1 |
| f12a | |||
| rCPB2.8 | −3.58 | 2.07 | 12.4 ± 1.4 |
| rCPB3 | −5.65 | 0 | 7.47 ± 0.64 |
| rH84Y | −5.24 | 0.42 | 11.2 ± 0.8 |
| f12b | |||
| rCPB2.8 | −0.21 | 5.44 | 39.2 ± 4.1 |
| rCPB3 | −4.30 | 1.35 | 13.7 ± 1.4 |
| rH84Y | −5.65 | 0 | 6.08 ± 0.37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lourenço, E.M.G.; Di Iório, J.F.; da Silva, F.; Fialho, F.L.B.; Monteiro, M.M.; Beatriz, A.; Perdomo, R.T.; Barbosa, E.G.; Oses, J.P.; de Arruda, C.C.P.; et al. Flavonoid Derivatives as New Potent Inhibitors of Cysteine Proteases: An Important Step toward the Design of New Compounds for the Treatment of Leishmaniasis. Microorganisms 2023, 11, 225. https://doi.org/10.3390/microorganisms11010225
Lourenço EMG, Di Iório JF, da Silva F, Fialho FLB, Monteiro MM, Beatriz A, Perdomo RT, Barbosa EG, Oses JP, de Arruda CCP, et al. Flavonoid Derivatives as New Potent Inhibitors of Cysteine Proteases: An Important Step toward the Design of New Compounds for the Treatment of Leishmaniasis. Microorganisms. 2023; 11(1):225. https://doi.org/10.3390/microorganisms11010225
Chicago/Turabian StyleLourenço, Estela Mariana Guimarães, Juliana Fortes Di Iório, Fernanda da Silva, Felipe Leonardo Bley Fialho, Melquisedeque Mateus Monteiro, Adilson Beatriz, Renata Trentin Perdomo, Euzébio Guimarães Barbosa, Jean Pierre Oses, Carla Cardozo Pinto de Arruda, and et al. 2023. "Flavonoid Derivatives as New Potent Inhibitors of Cysteine Proteases: An Important Step toward the Design of New Compounds for the Treatment of Leishmaniasis" Microorganisms 11, no. 1: 225. https://doi.org/10.3390/microorganisms11010225
APA StyleLourenço, E. M. G., Di Iório, J. F., da Silva, F., Fialho, F. L. B., Monteiro, M. M., Beatriz, A., Perdomo, R. T., Barbosa, E. G., Oses, J. P., de Arruda, C. C. P., de Souza Júdice, W. A., Rafique, J., & de Lima, D. P. (2023). Flavonoid Derivatives as New Potent Inhibitors of Cysteine Proteases: An Important Step toward the Design of New Compounds for the Treatment of Leishmaniasis. Microorganisms, 11(1), 225. https://doi.org/10.3390/microorganisms11010225